
需要订阅 JoVE 才能查看此. 登录或开始免费试用。
Method Article
从胚胎脊髓连合神经的解剖和文化
* 这些作者具有相同的贡献
摘要
这个视频演示的方法来剖析和文化连合从E13大鼠脊髓背侧神经元。是有用的游离连合神经元轴突的生长和指导,研究细胞和分子机制。
摘要
连合神经元已被广泛用于研究的内在机制,在胚胎脊髓发展的轴突指导。这些神经元的胞体位于脊髓背侧,其轴突遵循胚胎发育过程中的定型的轨迹。最初连合轴突项目的腹部朝向floorplate。越过中线后,这些轴突转向对大脑前部和项目。上述每个步骤是受几个指导线索的行动。连合神经元高度浓缩的文化,非常适合解决许多轴突寻路机制,包括车削实验,免疫组织化学和生物化学实验。在这里,我们描述的方法来剖析和文化连合从E13大鼠脊髓背侧神经元。首先,是孤立的脊髓和背条被解剖了。胰蛋白酶和机械破碎成细胞悬液,然后背的组织是分离的。神经元被镀上聚- L -赖氨酸镀膜玻璃盖玻片或组织培养皿。 30小时后
研究方案
1。胚胎大鼠背脊髓的解剖
一般建议
置于冰上的L - 15中型和经常改变在清扫菜中保持胚胎的凉爽。这有助于维持组织的完整性。除非另有说明,所有步骤都与两个对杜蒙#5镊子。为了避免污染,所有的工具和工作表面喷用70%乙醇,并保持封闭夹层介质瓶。菜之间传输的胚胎中,使用切割塑料吸管或穿孔勺。这是至关重要的,不损伤脊髓(刻痕,拉伸)成功地完成了清扫。
制备
- 冷L - 15培养基
- 50毫升的L - 15 + 10%热灭活马血清(HiHS)。置于冰上。
脊髓夹层
- 安乐死E13怀孕上演大鼠(E0 =第一天交配天)与CO 2室根据机构指引。
- 喷雾70%的乙醇,上腹部。捏和拉起降低腹部手术剪钳和切割区域的皮肤。重复通过切断肌肉和腹膜层,以达到腹腔。创建一个V形切口,切割组织沿腹部两侧,胸部。升降机及拉回组织暴露腹腔。
- 子宫是连接三个地点:在较低的中心腹部,双上肢外侧角落。提起子宫的胚胎囊之间的组织敛。剪下的结缔组织切除子宫,并在培养皿中的L - 15在冰充满。
- 接下来的步骤是根据解剖镜下完成。胚外组织和胚胎膜分离胚胎,抢子宫囊之间的组织,与一对镊子。另一对,掐国资委更加透明的一面穿过肤浅膜(黑暗面是胎盘)。轻轻按国资委的胎盘端挤出胚胎。删除所有在培养皿中的L - 15在冰充满的胚胎和地方。
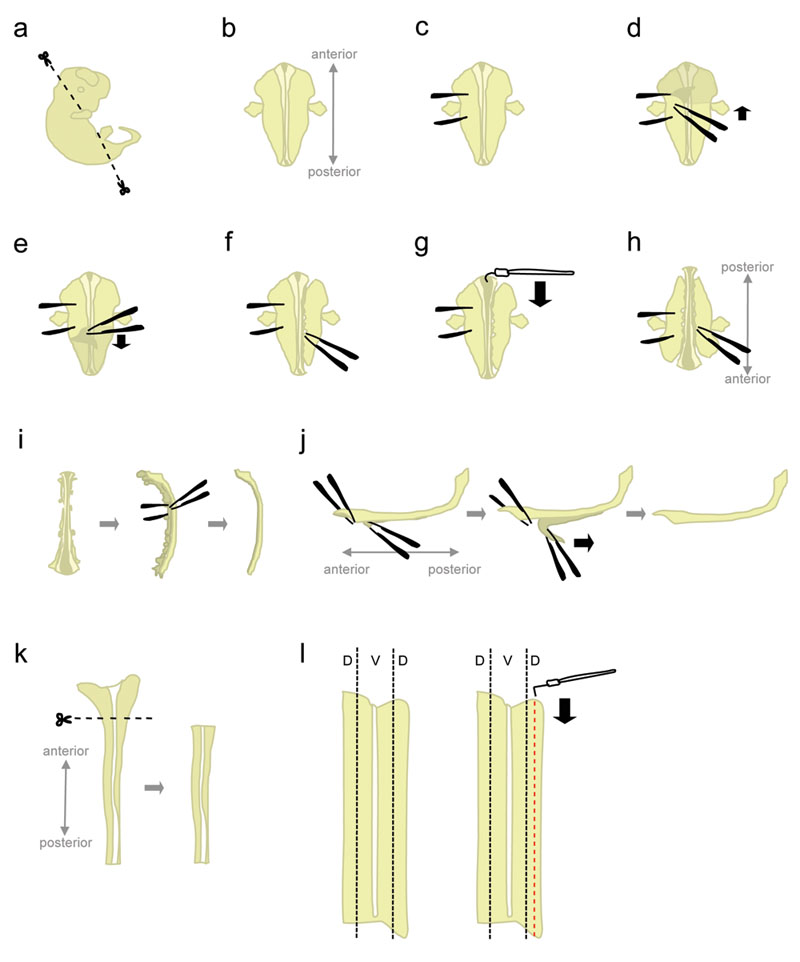
- 将一个胚胎在10厘米用冰冷的L - 15的Petri充满菜。使用microscissors,切去头图1a所示的角度和后部分。
在这些角度切割,将有助于位置放置时,“腹朝下”的胚胎。 - 胚胎的位置“腹正面朝下”(前背向后指向实验者)(图1B)。
- 牢牢把握胚胎从一个侧面,使用钳“牵制”的胚胎(图1C)。不要移动镊子,因为它会撕裂组织。随着一双镊子等,涵盖脱落,皮肤开始从地区之间的“控股”钳(图1D - E)在胚胎的背面。这将会使脊髓,在这一点上,包装膜(脑膜)。
抓斗从两侧的皮肤,而不是从上面的脊髓,以避免刻痕的脊髓。 - 部分分离组织的脊髓(图1F)的右侧。在持物钳的水平开始,封闭钳戳的组织,尽可能接近到脊髓。然后,慢慢地打开镊子撕组织。这应该分离脊髓背根神经节(病种付费),而且还应该破坏腹侧机关。离开。 离开组织增加重量胚胎的前部和后部两端附加了一些组织,从而防止在下一步向上拉。此外,该组织是用来装在后面的步骤中的胚胎。
- 由前年底开始,用勾形的钨针削减脑膜和脊髓沿roofplate(图1G)打开。
- 胚胎旋转180度(后指出,前对实验者的指向)(图1H)。
这允许实验者用同一只手从另一侧的胚胎组织分离。 - 保持到以前分离的方抓住胚胎,并从剩余方使用相同的方法(见1.8)。的组织破坏。完成后,完全分离的脊髓两侧的组织。
另一种方法:彻底分离组织余下的左侧,但离开的前部和后部端连接的右侧上一些组织。有时,这可以帮助脊髓维持在1.12的一步位置。 - 在其一侧脊髓和删除其余的间叶组织和背根节(图1I)。
- 在这一步,脑膜和脊髓是两个“表”到对方apposed组织。牵制较大,脊髓的最前部分,并剥离过短节段脊髓COR的脑膜D(图1J,左)。现在,持有抓住钳跨越两地分居段,平稳,恒定的运动(图1J)脑膜剥离。
不均“剥离”将导致脊髓和/或脑膜层破损。它通常是不可能的,收回部分仍然附着在脑膜脊髓。 - 用塑料吸管,隔离脊髓转移到培养皿中含有的L - 15 + 10%HiHS留在冰上。通常情况下,所有胚胎的脊髓前收集解剖背部分。
脊髓背侧夹层
- 放置一个脊髓中含有的L - 15 10%HiHS,平趴在一个“开卷”配置一个Petri菜。切去更宽,前部(其中包括后脑的一部分)(图1K)。
- 背侧组织是位于最外侧部分的脊髓(图1L,左)。虽然寄希望于采用了直板的钨针的电源线,使用“L”形的钨针,切出一个地带,是1/5th宽度的一半脊髓。 (图右)1L,。将在15毫升的塑料管,在冰中含有的L - 15 + 10%HiHS背条。
背〜12 E13胚胎神经管段将产生解离电镀后〜3.5-4亿个细胞。可以得到更多的细胞进行脊髓背侧(切割更广泛的背条)粗清扫,但连合神经元的纯度会降低。
2。连合神经元文化
一般建议
应除非另有说明,组织培养罩在无菌条件下进行的所有步骤。使用新鲜培养液和新鲜解冻的补充和试剂 。 在Ca 2 + / Mg 2 +的免费的HBSS,以尽量减少的Ca 2 + / Mg 2 +的依赖性粘附的解离和trituration步骤进行。
制备
- 涂层的盖玻片(使用德国Desag玻璃)或组织培养板(见下文涂层过程)。
- 温暖Neurobasal电镀媒体(见下文),在组织培养皿和在组织培养孵化器的CO 2平衡电镀前至少为1.5 h 。
- 2.5%胰蛋白酶在37℃水浴
- 2瓶HBSS中的Ca 2 + /镁2 +,在4 ° C时,在37 ° C.
- 两个火抛光的玻璃巴斯德吸液管直径通常大小的一半,和一个直径略超过一半。使用消毒的巴斯德吸液管。消防波兰的移液器,使用本生灯(淡蓝色火焰上方)融化尖的直径略有下降。因为这一步是进行组织培养罩外,喷用70%乙醇的组织培养罩下配售的移液器。为更有效的恢复神经元,大衣的含药血清之前trituration一步的离解或(填写与媒体的移液器,并保持30秒)开始前的媒体巴斯德移液器。这将防止细胞粘在trituration巴斯德吸管内。
- milliQ无菌水洗涤PLL涂层的菜肴或盖玻片
- 12.5%的硫酸镁溶液中的HBSS
聚- L -赖氨酸涂层
如果使用盖玻片,24小时的酸清洗和消毒前电镀(见Kaech和银行家,2006年)。使用德国Desag盖玻片。
聚- L -赖氨酸盖玻片或塑料组织培养皿大衣:
- 下一个组织文化的引擎盖,覆盖了100微克/毫升的PLL解决方案为1.75-2小时的小圆顶曲面。
- 洗两次澡的milliQ水,至少每洗5分钟(可以在细胞分解步骤如下进行)。
- 在水店,直至使用。不要让PLL涂层表面的干燥。
为了减少盖玻片涂层的PLL浪费,在无菌细菌培养皿中的盖玻片 。 外套和洗涤的液体的圆顶上的盖玻片盖玻片 。 细菌的培养皿中的疏水性,使液体应保持在玻片上 。 电镀前的细胞,组织培养皿中的盖玻片 。
对于塑料组织培养皿上镀的神经元,附着力通常较高,从而突起伸长率可能会减少。
分离和电镀
- 验证背条落户试管底部。删除最巴斯德吸管的L - 15的10%HiHS。快速洗背神经管带,一旦加入3毫升冷(4℃)的HBSS。
- 让背神经管带定居2分钟,然后取出用巴斯德吸管的HBSS。
- 温暖(37℃)的HBSS体积为4.7毫升。 T母鸡添加0.3毫升2.5%胰蛋白酶给予终浓度为0.15%胰蛋白酶。
- 在37℃,7分钟的水浴。通过孵化轻轻搅拌一次中途。
- 加入终浓度为150 U / mL的30μLDNA酶(25万U /毫升)。加入60μl和硫酸镁混合简要终浓度为0.15%,。
对于组织由粗解剖,一个额外的1分钟37 ° C的水浴。
在这个阶段,背神经管段应支离破碎。如果背神经管段还没有开始的片段,它通常是指2.5%的胰蛋白酶股票是旧的,新的股票应解冻,或用冷的HBSS洗没有有效地去除样品HiHS。 - 离心机组织碎片,200克4分钟。
- 巴斯德吸管取出上清液,留在〜50-100μL液体的试管底部。
- 滑动管轻轻地松开颗粒,然后加入5毫升的温暖的HBSS洗细胞。让我们在室温下2分钟定居。
- 在200克离心5分钟。
- 巴斯德吸管取出上清液,留在〜50-100μL液体的试管底部。
- 滑动管,轻轻地松开颗粒和部分悬浮细胞。再加入2毫升温暖的HBSS。
- 使用小(半直径),火抛光玻璃巴斯德吸管游离于细胞慢慢地移液和4-6倍。避免气泡,吸液管对管端的液体。不要过度磨碎。
- 用最小的火抛光玻璃巴斯德吸管进一步游离于细胞慢慢移液和向下的3-4倍。避免气泡,吸液管对管端的液体。不要过度磨碎。
从粗糙的解剖组织,添加额外的1毫升温暖的HBSS管解离年底 。
当游离的细胞,它是没有必要的分解所有的细胞团块和聚合。停止吹打和向下或改变一个直径较小的巴斯德吸管,如果您没有看到进一步吹打细胞聚集规模进一步减少 。 - 让任何剩余的组织碎片,1分钟,在管内定居。这是没有必要的细胞转移到新的试管。
- 取20μL细胞悬液,加入5μL台盼蓝。一个血球计数细胞。
神经元应≥95%可行的台盼蓝拒。 - 板的神经元在Neurobasal电镀媒体(见下面的食谱) 。
- 建议电镀密度为获得分离出神经元(低密度文化,以避免结块或相互重叠的神经元):
- 120 000 - 180 000个细胞/孔为6孔板
- 60 000 - 75 000个细胞/孔12孔板
- 16-18小时后,改变媒体Neurobasal生长介质(见下面的食谱)
不要使用真空泵吸从培养皿中的媒体变化的媒体时,轻轻地用吸管 。 这就避免了撞出的神经元。
代表性的成果:
电镀后4小时,应坚持以聚- L -赖氨酸(PLL)涂层表面的神经元。在相衬照明,坚持胞体通常是相对平坦的,椭圆形(图2a)。没有很好地坚持细胞出现的领域,移动时,菜是非常轻轻地放在一边挖略有。许多因素可能阻碍细胞粘附(见讨论)。
大多数神经元在体外培养 30小时后,已经扩展了一个可见的生长锥(图2C,D)一个轴突。如果穷人的轴突生长观察,验证,Neurobasal生长介质已与新鲜培养基和补充。在这些条件下至少有6天的神经元保持健康。此程序已被证明是可靠的产量连合神经元高度浓缩的准备工作,与〜90%的神经元表达DCC(荫等人,2009年)。脊髓背侧地带,准备细胞悬液的宽度会影响文化的纯洁性,具有更大的纯度时使用更薄条。示例应用程序是显示在图3(免疫)。荫等人的文章。 (2009年),为更多的例子。

图1。脊髓清扫步骤示意图。 D =背,V =腹。 点击这里查看大图。
{kind=link}

图2代表孤立连合的结果PLL的镀膜玻璃盖玻片上镀的神经元。 A,B)电镀后4小时,神经一直坚持到表面。酒吧= 20微米。 C,D)电镀后的30小时,大多数神经元轴突与一个可见的生长锥延长。酒吧= 20微米。

图3。一个连合神经细胞免疫染色为γ-微管蛋白(绿色),与F - actin的鬼笔环肽(F - actin的,红色)和细胞核的DAPI(蓝色)标记。
配方和意见
Neurobasal电镀媒体
- Neurobasal
- 10%热灭活胎牛血清(HiFBS)
- 2毫米L -谷氨酰胺(200毫米原液)
可选:青霉素/链霉素抗生素(使用正常浓度的一半)
Neurobasal生长介质
- Neurobasal
- B27(1 / 50稀释股票)
- 2毫米L -谷氨酰胺(200毫米原液)
- 可选:青霉素/链霉素抗生素(使用正常浓度的一半)
一旦媒体,它可以被保存在4 ° C为两个星期。电镀前至少1.5小时,在组织培养箱内培养的细胞,在培养皿中进行组织培养的地方媒体和地方媒体的温度和pH值达到平衡。
Neurobasal
Neurobasal培养基瓶已被打开后,它可以保留一个月,在4 ° C黑暗环境中。 Neurobasal已超过一个月打开,否则细胞存活率会降低处置。
热灭活胎牛血清(HiFBS)或马血清(HiHS)
热灭活胎牛血清或HS,热量在56 ° C在一个30分钟的水浴。摇动一瓶大约每隔10分钟左右。 (使用同样大小的瓶子装满水的准确性,一个放置温度计一瓶水,到56℃达到。开始在这一点上的时间。)热灭活胎牛血清可能需要离心清除沉淀物,并可以分装和重新冻结在-20 ° C
L -谷氨酰胺
始终解冻每个实验的L -谷氨酰胺的新鲜等分。
B27
可以保持在20 ° C的长期储存,或4 ° C的长达一个月的B27的等分。
讨论
我们已经描述了一个方法来剖析和文化连合从胚胎大鼠脊髓神经元。这个程序已经在我们的实验室经常使用可靠准备的神经元的轴突导向的研究细胞和分子机制。细胞生物学和免疫组织化学实验,解剖一窝产生足够的神经元。当需要更多的细胞,如在许多生物化学实验,解剖两个窝可能是必需的。对于一个受过训练的人,可以进行清扫和分解〜20胚胎在不到4个小时。更长的时间范围内将导致一个?...
致谢
这项工作是支持的加拿大卫生研究院(CIHR),彼得Lougheed医学研究基金会,在Neuroengineering麦吉尔方案的补助金,全宗的RECHERCHE EN桑特魁北克(FRSQ),加拿大创新基金会(CFI )。塞巴斯蒂安D.朗格卢瓦是硕士培训奖全宗支持DE LA RECHERCHE EN健康魁北克(FRSQ)和卫生研究所(CIHR)加拿大研究所加拿大研究生奖学金硕士奖由弗雷德里克班廷和查尔斯。我们感谢与数字援助杰西卡吨范。
材料
胚胎大鼠背脊髓的解剖(参见表我)
- E13怀孕-上演大鼠
- 乙醇70%
- 手术剪刀,精细的科学工具
- 杜蒙#5,精细的科学工具钳,
- 培养皿
- L - 15培养基
- 解剖镜下,徕卡
- 塑料转移移液器
- Microscissors,精细的科学工具
- 钨针和针持有人(一个勾形针,一个L形状针,直针),精细的科学工具
- 热灭活马血清(HiHS)
- 15毫升的塑料管
连合神经元文化(参见表我)
- 组织培养孵化器(37 ° C,5%CO 2,湿度控制)
- 德国Desag盖玻片和/或塑料组织培养皿
- 聚- L -赖氨酸,西格玛
- 无菌milliQ H 2 O
- Neurobasal,Invitrogen公司
- 热灭活胎牛血清(HiFBS)
- L -谷氨酰胺
- B27,Invitrogen公司
- 青霉素/链霉素抗生素
- 37 °彗星水浴
- 无菌巴斯德滴管
- 本生气体刻录机
- HBSS中,钙2 + /镁2 +自由,Invitrogen公司
- DNA酶,沃辛顿
- 硫酸镁
- 离心分离
- Hemacytometer
- 台盼蓝溶液
参考文献
- Charron, F., Stein, E., Jeong, J., McMahon, A. P., Tessier-Lavigne, M. The morphogen sonic hedgehog is an axonal chemoattractant that collaborates with netrin-1 in midline axon guidance. Cell. , 113-1111 (2003).
- Fabre, P., Shimogori, T., Charron, F. Segregation of ipsilateral retinal ganglion cell axons at the optic chiasm requires the Shh Receptor Boc. Journal of Neuroscience. 30, 266-275 (2010).
- Helms, A. W., Johnson, J. E. Progenitors of dorsal commissural interneurons are defined by MATH1 expression. Development. 125, 919-928 (1998).
- Kaech, S., Banker, G. Culturing hippocampal neurons. Nature Protocols. 1, 2406-2415 (2006).
- Okada, A., Charron, F., Morin, S., Shin, D. S., Wong, K., Fabre, P. J., Tessier-Lavigne, M., McConnell, S. K. Boc is a receptor for sonic hedgehog in the guidance of commissural axons. Nature. 444, 369-373 (2006).
- Yam, P. T., Langlois, S. D., Morin, S., Charron, F. Sonic hedgehog guides axons through a noncanonical, Src-family-kinase-dependent signaling pathway. Neuron. 62, 349-362 (2009).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。