
Method Article
使用Jsubtomo从电子Cryotomography重构平均病毒包膜糖蛋白穗
摘要
An approach is presented for determining structures of viral membrane glycoprotein complexes using a combination of electron cryo-tomography and sub-tomogram averaging with the computational package Jsubtomo.
摘要
包膜病毒利用在其表面上的膜糖蛋白介导进入宿主细胞。这些糖蛋白'尖峰'的三维结构分析通常是在技术上具有挑战性,但重要的是了解病毒的发病机制和药物设计。在这里,一个协议提交病毒刺突结构的决心,通过低温电子断层扫描数据计算平均值。低温电子断层扫描是电子显微镜技术来获得三维断层容积重建,或断层,多形性生物样本,如在接近本机,冷冻水合状态的膜病毒。这些断层图像揭示的利益结构在三维空间中,虽然在低的分辨率。子体积,或子断层图像的计算平均值,是必要的,以获得重复的结构基序,如病毒糖蛋白的尖峰的更高分辨率的细节。阿里的详细计算方法gning和平均使用Jsubtomo软件包子断层概述。这种方法使病毒糖蛋白的尖峰结构的可视化高阶尖峰对尖峰上的病毒膜相互作用的研究在20-40范围内的分辨率和研究。典型的结果示于Bunyamwera病毒,从家庭布尼亚病毒科的包膜病毒。该系列是病原体威胁到人类和动物的健康构成威胁一个结构多元化的群体。
引言
电子冷冻断层扫描是一种电子低温显微成像技术允许一个三维(3D)重建的复杂生物样品的计算。合适的样本范围从纯化的大分子复合物1中 ,单丝2,被小泡3,和多形性膜病毒4至整个原核细胞5,甚至薄整个真核细胞6的区域。以下的倾斜系列,3D断层扫描卷或断层图像的数据收集,可以使用几种建立软件包,包括Bsoft 7和IMOD 8来计算。
固有的生物标本通过电子低温断层限制相应的断层卷的生物学解释的研究两个方面。首先,由于可应用到生物材料引入显著辐射损伤之前的有限的电子剂量,Signa的升噪比在断层数据通常是非常低的。其次,由于数据收集过程中有限样本的倾斜几何的结果,一些意见的对象仍然存在,导致在断层容积所谓的"缺少楔"神器。然而,无论这些限制可以如果断层容积包含重复相同的结构,如大分子复合物,可以成功地平均9-12来克服。
之前,均从断层图像的重建结构中,感兴趣的对象,必须找到并对齐,以相同的方向。定位这种结构可以通过一个模板结构中的断层容积的互相关使用通常被称为模板匹配13的方法来实现。在这个匹配过程中使用的模板可以从电子低温显微术或电子低温层析结合3D重建中得到,或者它可以是从模拟的密度图原子结构。一些计算软件包已经开发完成这些任务11。
平均膜病毒,如HIV-1糖蛋白尖峰,一直是特别成功的方法来研究它们的结构14-16。该结构的理解是积分揭示病毒 - 宿主相互作用的两个分子基础和指导抗病毒和疫苗的设计开发。而大分子晶体是首选的高分辨率个别病毒糖蛋白及其复合物的结构分析(通常大于4埃更好)的技术中,从该方法中产生的X射线结构是蛋白质的天然膜环境中分离的病毒颗粒的。因此,重要的信息,如病毒糖蛋白的高次结构,在病毒粒子的情况下,仍然缺乏。另一方面,电子低温显微术和单粒子重建的整个包膜病毒被限制到病毒粒子具有二十面体对称17,18。电子cryotomography结合分卷排列也因此成为一个互补的技术,允许形状不规则, 在原地多形病毒糖蛋白尖刺的研究。
我们已经开发了一个名为Jsubtomo(www.opic.ox.ac.uk/jsubtomo)的断层分卷的检测,校准和平均软件。 Jsubtomo已被用于一些细胞和病毒结构19-26的结构测定。在这里,我们勾勒出一个详细的协议,这使得测定病毒表面刺突结构。要通过相关的噪声规避过度细化的平均结构中,"黄金标准"细化方案采用10,27。最后,对于可视化的典型结果的解释策略进行了讨论。
研究方案
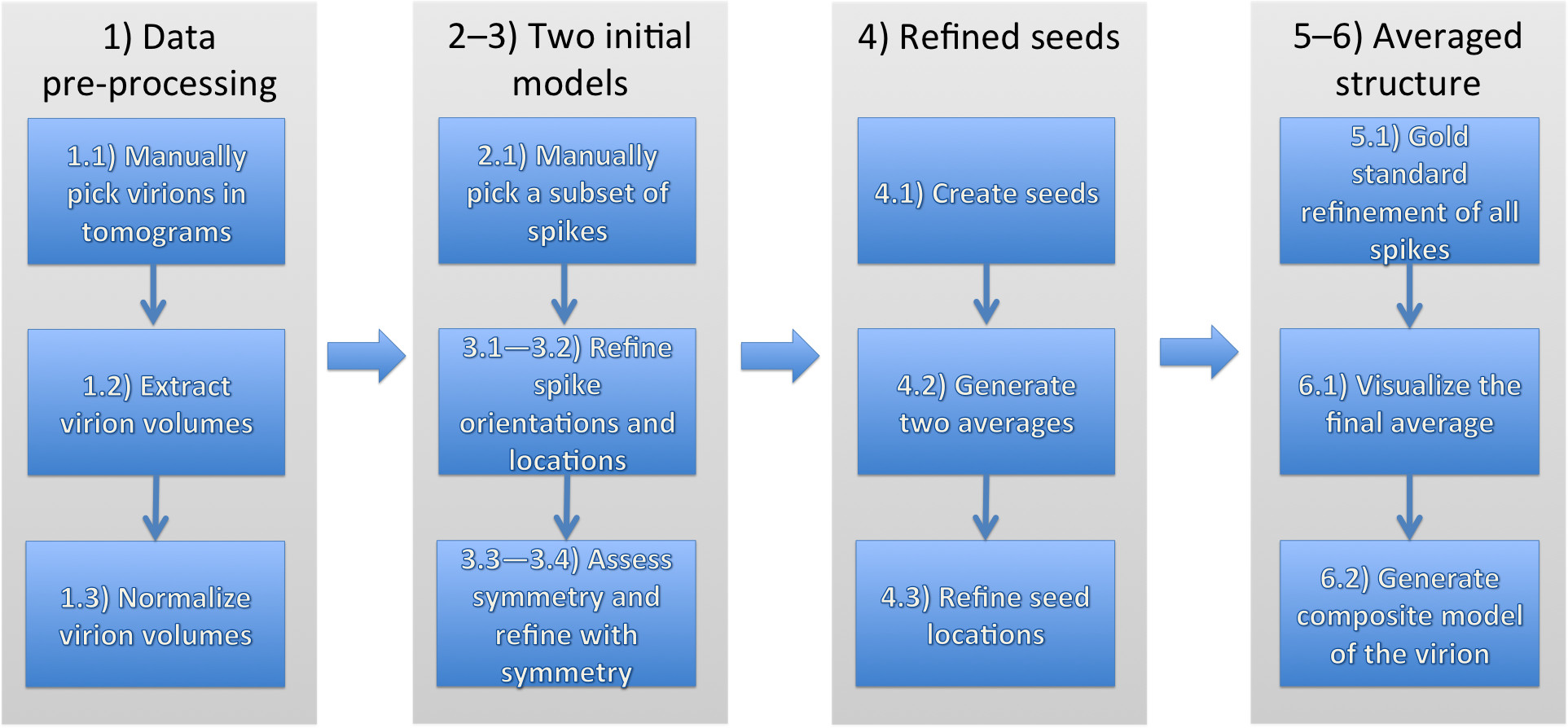
为计算路线和病毒糖蛋白的尖峰随后均得到详细的协议概述。协议如下图1中所示的工作流程,并结合一种自动化搜索使用初始模板结构和黄金标准结构细化的尖峰。
输入数据,该协议是一组病毒体的断层重建的。一个断层图像中包含的一个或多个病毒粒子。最初,尖峰的一小部分是手工采摘并用来平均水平,完善两个独立的模型。这些模型用于自动定位尖峰上的所有病毒颗粒。最后,两个独立的精炼进行运行,将所得的平均值进行比较,并结合以产生最终的结构。
细化方法证明了使用程序从Jsubtomo包。从Bsoft包28程序用于一般图像Processing任务和分子图形包UCSF嵌合体29是用于可视化的结果。个别课程以斜体字和文件格式,给出的名称标注有大写的文件扩展名。

图1:用于确定从形性膜病毒糖蛋白刺突复合物的结构总体战略中的数字对应的协议在不同的章节,请点击这里查看该图的放大版本。
{kind=link}
1,从全尺寸断层提取病毒分卷
- 在X线断层手动拾取的病毒颗粒。
- 打开bshow断层图像文件。定义一个病毒partic的中心坐标使用乐颗粒采摘工具。重复此步骤,直到所有的病毒粒子都被处理。保存病毒坐标在星形文件。
- 直到所有的断层图像进行了处理,重复步骤1.1.1。请记下病毒粒子直径的像素。
注:如果尖峰很难看到断层,使用bfilter到低通滤波器的断层图像,以80的分辨率(参数"-bandpass 80,10000")。
- 运行jsubtomo.py中提取模式(参数"--mode提取物")中提取病毒分卷到单个卷中的文件。使用保存在步骤1.1.1输入文件STAR文件。
- 给一大小(参数"--size")〜比在数据集的最大病毒体大25%。 例如 ,如果病毒是〜200个像素的直径,使用尺寸250×250×250像素。
- 病毒粒子体积的输出文件将在MAP格式。此外,创建一个伴随STAR文件为每个病毒粒子的体积(parame之三"--output")。
- 正常化病毒卷bimg(参数"-rescale 0,1")。
2,生成两个独立的初始模型
- 匹克在病毒分卷尖峰的一个子集。
- 打开bshow病毒体分卷映射文件。定义使用粒子采摘工具,通过工具箱窗口访问穗的中心坐标。重复此步骤,直到所有分明尖峰进行了处理。保存秒杀坐标在星文件。
- 如有必要,重复步骤2.1.1其他病毒体,直到大约200糖蛋白尖刺已被处理。注意像素秒杀尺寸。
注:〜200尖峰是一个粗略的指引,更可能需要的一些应用。如果可能的话,目的是挑选尖峰在不同的方位。避免只采摘俯视图。
- 通过运行jviews.p分配一个初始视图向量的尖峰年。使用步骤2.1.1输入文件生成的STAR文件。
注:此图矢量近似于参照病毒的刺突的方向。- 使用中央协调分配意见的球形病毒颗粒。例如,如果病毒体被集中(步骤1.2之后)在一个盒子具有250×250×250个像素的大小,中心坐标是125125125像素(选项"--Radial 125125125")。
- 要指定一个丝状病毒的看法,通过使用在步骤2.1.1作为输入文件中定义的文件之星打开bshow每个病毒分卷。限定使用灯丝拾取工具灯丝的两端点,并保存STAR文件。重复此步骤,直到所有明星的文件已被更新并运行jviews.py使用更新之星文件作为输入文件(--option段)。
- 使用jsubtomo_create_mask.py产生真正的空间屏蔽。确保在实际空间掩模是同一三维地球的离子作为将被用于平均的结构(参见2.6.2),并足够大以包含两个尖峰和底层膜的贴剂。
- 使用jsubtomo_create_wedgemask.py产生倒易空间的面具。倒数空间掩码用于排除的区域中的"缺失槽楔'从单一轴断层数据收集所得的区域。确保它是相同的尺寸,为将用于所述平均结构(见2.6.2)。
- 生成一个选择文件(SEL文件)限定属于设定"1"和"2"使用jsubtomo_evenodd.py并在步骤2.2中产生的STAR文件的病毒粒子。
- 使用jsubtomo_create_averages.py产生两个初始平均值。
- 使用步骤2.5作为输入文件生成的SEL文件。
- 举一个大小(参数"--size")至少32像素比秒杀的最大尺寸。 例如 ,如果秒杀〜40个像素长,使用尺寸72×72×72像素。
- 适用于高对称性( 如 C100)的平均值(参数"--symmetry C100"),近似围绕秒杀的长轴圆柱形的平均水平。使用对称,以减少在初始平均噪声。
- 项目(参数"--suffix")的输出文件提供一个唯一的名称。
- 使用在步骤2.3中产生的真实空间掩模掩盖远背景(参数"--mask")。使用在步骤2.4中产生的倒易空间掩模占信号由丢失楔(参数"--mask")的存在下引入的损耗。
3,黄金标准迭代对齐和两个初始穗模型的平均
反复调整和平均在第2与jsubtomo_iterate_gold.py生成的两个初始模型。
- 阶段一,这个阶段的目的是改进观察向量的方向。输入文件是在步骤2.5生成的SEL文件。
- 使用一个8度角取样(参数"--angles 8,8,8")。
- 允许在穗视图向量的方向的16度的变化(参数"--thetaphilimit 16"),但保持周围的穗长轴线固定的(参数"--alphalimit 0")的角度。
- 让小平移的变化( 例如 ,5像素)来解释不准确的尖峰人工采摘(参数"--shiftlimit 5")。
- 适用于高对称性( 如 C100)的平均值(参数"--symmetry C100"),近似围绕秒杀的长轴圆柱形的平均水平。
注:对称化是用来减少在平均噪声。 - 使用真正的空间屏蔽和步骤2.3和2.4生成的倒易空间面具来掩盖周围走尖峰和账户失踪的楔形(参数"--M问"和"--mask",分别)。
- 使用在2.6生成的两个MAP文件(由标记"偶"表示,并在该文件名"奇")作为模板(参数"--Template1"和"--Template2",分别)。
- 应用低通滤波器50的分辨率(参数"--resolution"),以防止对准偏差。
- 应用2的仓因素,加快细化(参数"--bin 2")。
- 运行5次迭代调整,平均的(参数"--firstiter 1 --lastiter 5")。
- 第二阶段。这个阶段的目的是缩小视图四周矢量的角度。输入文件是步骤3.1.9输出SEL文件。
- 通过打开在步骤3.1.9最后一次迭代中产生的低通滤波的MAP文件测量尖峰的精确尺寸在bshow(由变量"_lp"中的文件名 表示)。产生一个新的现实空间屏蔽类似于步骤2.3定义秒杀,但不包括大部分的膜和邻近的尖峰。
注:面罩的最佳尺寸取决于大小和尖峰的特征。 - 用8度角取样(参数"--angles 8,8,8")作为之前。
- 允许在围绕鞋钉视图向量(参数" - alphalimit 180")的角180度的变化而保持的θ和披角度固定(参数"--thetaphilimit 0")。
- 不要让任何翻译班(参数"--shiftlimit 0")。
- 不适用的平均值(省略参数"--symmetry")的任何对称性。
- 使用在步骤2.4中产生的倒易空间掩模占缺失槽楔。
- 使用步骤3.1.9的最后一次迭代而不对称性所产生的两个Map文件作为模板(参数R(按标签"even_nosym",并在文件名"odd_nosym"表示)20; - template1的"和"--Template2",分别)。
- 在第一次迭代应用低通滤波器50的分辨率(参数"--resolution"),以防止对准偏差。两个独立的平均值之间自动调整,在后续的迭代滤波器参数的基础上的黄金标准傅立叶壳牌相关(FSC)(参数"--adaptivefilter")。用0.143,标准(参数"--fsccrit 0.143")。不允许细化过去的对比度传递函数(CTF)(参数"--minhires")的第一个零,除非CTF校正已被施加到X线断层图。
- 运行5-10重复排列和平均( 例如 ,参数"--firstiter 6 --lastiter 15")。监视中的变化报道的分辨率。当没有显著变化观察到的,该过程可以停止。
- 通过打开在步骤3.1.9最后一次迭代中产生的低通滤波的MAP文件测量尖峰的精确尺寸在bshow(由变量"_lp"中的文件名 表示)。产生一个新的现实空间屏蔽类似于步骤2.3定义秒杀,但不包括大部分的膜和邻近的尖峰。
- 评估对称度由examini结构纳克在嵌合体 。 例如所得的低通滤波后的MAP文件(文件名 中表示为变量"_lp")时,如果道钉是三聚体复合物,3次对称性应该是显而易见的。
- 第三阶段。这个阶段的目的是进一步缩小视图四周矢量的角度与正确的对称性。输入文件是步骤3.2.9输出SEL文件。的参数是相同的,在步骤3.2,只有少数例外:
- 让周围的秒杀观察向量的夹角适当的修改。例如,如果该结构具有3倍的对称性,允许以60度的α角的变化(参数"--alphalimit 60")。
- 适用于平均值(参数--symmetry C3")的正确对称( 如 C3)。
- 使用最后一次迭代生成的两个MAP文件步骤3.2.9(文件名记为标签",甚至"与"奇")作为输入模板文件(参数"--Template1"和 --Template2)。
- 运行5-10重复排列和平均( 例如 ,参数"--firstiter 16 --lastiter 25")。监视中的变化报道的分辨率。当没有显著变化观察到的,该过程可以停止。
- 第四阶段。这个阶段的目的是要精确地同时缩小所有三个角度。输入文件是步骤3.4.4输出SEL文件。的参数是相同的,在步骤3.4,只有少数例外:
- 允许在围绕鞋钉视图向量(参数" - alphalimit 8")的角8度的改变和在穗视图向量的方向8度的变化(参数"--thetaphilimit 8")。完善的方向,以4级精度(参数"--angles 8,8,8 --iterate 2")。
- 允许较小的变化( 比如 ,5个像素),来解释在先前的校准步骤的误差(参数"--shiftlimit 5")。
- ü硒在步骤3.4.4最后一次迭代生成的两个MAP文件(通过标签",甚至"表示,并在文件名"奇")作为输入模板文件(参数"--Template1"和--Template2)。
- 运行5-10重复排列和平均( 例如 ,参数"--firstiter 26 --lastiter 35")。监视中的变化报道的分辨率。当没有显著变化观察到的,该过程可以停止。
四代种子的病毒表面的模板匹配和对齐
- 位于生成均匀地涂在病毒表面的模板匹配的种子,并通过运行jviews.py分配一个初始视图向量的种子。使用步骤1.2.2输入文件生成的STAR文件。产生比尖峰的期望数目约1.5倍以上的种子。
注:这一观点矢量接近最接近每个种子点秒杀的方向。- 要产生均匀分布的种子在近似球形病毒颗粒(参数"--Even"),给半径(参数"--radius"),角距( 如 20度)的种子(参数"--angle 20" )和中央的病毒体的坐标。 例如 ,如果病毒体被集中(步骤1.2)中的箱具有250×250×250个像素的大小之后,中央坐标是125125125像素(选项"--origin 125125125") 。
- 要生成的丝状颗粒均匀分布的种子,使用参数"--Filament",并同时指定半径和螺旋对称参数(上升和扭曲)。
注:该螺旋对称的参数在这里只是作为界定均匀分布的种子位置的一种便捷方式,他们并不需要反思灯丝尖峰的实际顺序。
- 产生的病毒表面explaine两个独立的平均值d在第2节。
- 产生SEL文件中定义属于集合"1"和"2"使用jsubtomo_evenodd.py和步骤4.1生成的星病毒体文件。
注:为确保数据集的独立性,以前设置的病毒颗粒成团"1"和"2"必须保持不变。
- 产生SEL文件中定义属于集合"1"和"2"使用jsubtomo_evenodd.py和步骤4.1生成的星病毒体文件。
- 完善种子的位置。
- 按照3.1的步骤说明,除非另有说明。使用步骤4.2.1作为输入文件生成的SEL文件。
- 让种子沿法线仅转移至膜(参数"--zshiftlimit")。调节移位的容许量取决于病毒颗粒从理想球面几何多少偏离( 例如 ,参数--zshiftlimit 25")。
- 使用步骤4.2生成的两个MAP文件作为输入模板文件(参数"--Tem(由标签"偶",并在文件名"奇"表示)板1"和--Template2)。
- 采用独特的后缀,以避免覆盖原输入之星文件( 例如 ,参数"--suffix _seeds")。
- 使用的4像素合并以加速计算(参数"--bin 4")和一个低通滤波器( 例如 ,参数"--resolution 50")。
- 使用jviews.py(参数"--cmm")产生精种子之星档案奇美拉标记文件(CMM)。检查种子在嵌合体打开了CMM文件(以及相关的病毒MAP文件)。确保成品种子正确相对于病毒膜对齐。
注意:不同的颜色可以被用于区分标记物的单独套(参数"--color")。备选地精制标记可以基于它们的交叉相关系数( 例如 ,参数"--fomcolor 0.1,0.3")进行着色。
5,黄金展位ARD迭代对齐和穗粒结构的平均
自动找到附近使用精致的瓜子本地模板匹配的病毒分卷的所有尖峰和对齐和平均位于尖峰。使用从手工采摘尖峰初始模板的一个子集所产生的平均水平。
- 围绕种子进行局部模板匹配以平均使用的所有程序jsubtomo_iterate_gold.py的尖峰。按照步骤3.5的说明,除非另有说明。
- 输入文件是在步骤4.3生成的种子提炼的SEL文件。
- 为视图矢量角用一个足够大的限制。 例如 ,如果种子产生每20度(步骤4.1),使用的角度大于该值的一半稍大(参数"--thetaphilimit 12")。让周围的秒杀观察向量的夹角适当的修改。
- 让种子在所述膜的平面内移动(参数"--xyshiftlimit")。 例如 ,如果种子到种子的距离是25个像素,使用总变速比一半的( 例如略多,参数"--shiftlimit 6 --xyshiflimit 10")。
可能需要额外的翻译转移,如果在步骤3.5生成的地图文件都集中在离中心病毒比种子不同的距离:注意。 - 使用步骤3.5生成的两个MAP文件(通过标签",甚至"表示,并在文件名"奇")作为输入模板文件(参数"--Template1"和--Template2)。
- 使用步骤3.5(参数--resolution)生成映射文件的当前的分辨率。
- 运行5-10重复模板匹配,对齐和平均( 例如 ,参数"--firstiter 1 --lastiter 10")。监视中的变化报道的分辨率。当没有显著变化观察到的,该过程可以停止。
- 每次迭代后,排除重叠的点击率。 例如 ,如果尖峰的宽度为30像素,排除命中是接近了30个像素,以其他的打击(参数"--mindist 30")。
- 排除安打与低互相关合作效率。例如,包括最好的75%的尖峰对于每个病毒粒子(参数"--topp 75")。
注:点击数与低互相关合作效率很可能代表假阳性命中。
6,可视化的结果
- 在嵌合体可视化开启秒杀的精致结构和装修的原子结构。
- 使用在最后一次迭代产生的低通滤波后的MAP文件(用标记"LP"),在步骤5.1.6作为输入文件。
- 使用基于在嵌合体装修交相关步骤5.1.6所示的决议"适合的地图"的工具。
- 创建virio的复合模式N使用jsubtomo_create_model.py。
- 使用在最后一次迭代产生的低通滤波后的MAP文件(用标记"LP"),在步骤5.1.6作为输入的MAP文件(参数"--template")。
- 在步骤5.1.6作为输入之星文件使用在最后一次迭代产生之星文件。
- 使用步骤3.2.1生成的模板文件占的复合模式(参数"--mask")重叠的密度。
- 表明病毒粒子MAP文件的大小,以产生相同的大小的复合模型(参数"--size")。
- 打开复合模型进行可视化的嵌合体 。
结果
我们证明上面使用此前公布的数据Bunyamwera病毒(Orthobunyavirus, 布尼亚病毒科 )的包膜糖蛋白复合物中列出的子断层扫描平均工作流程设置24应用程序。数据收集和精化参数列于表1中 ,其中一个代表断层图像示于图2。
| 参数(单位) | 价值 |
| 数据收集 | |
| 电压(kV) | 300 |
| 校准放大倍数(倍) | 111,000 |
| 像素尺寸() | 5.4 |
| 估计量(E - / 2) | 100 |
| Underfocus(微米) | 4.0-4.5 |
| 首先周大福零(一) | 26-30 |
| 倾斜范围(°) | -60-60 |
| 倾斜采样(°) | 3 |
| 数据和细化 | |
| 断层 | 11 |
| 病毒体 | 29 |
| 每个病毒种子 | 106 |
| 种子总数 | 3074 |
| 尖峰检测 | 1,346 |
| 除去重叠后,尖峰 | 1,401 |
| 互相关为基础的选择后的尖峰 | 1,022 |
| 包括在最终的平均尖峰 | 1,022 |
| 对称 | C3 |
| 角度采样(°) | 8 |
| 分辨率在细化使用范围(A) | 42-334 |
| 最后的解决估计(A)B | 35 |
表1:Bunyamwera数据收集和细化的统计数据。
à周大福,对比传递函数。
b。使用在0.143阈值两个独立的精致结构之间的傅立叶壳的相关计算。

图2:通过Bunyamwera病毒体的断层图像切片中的每个病毒粒子的外周明显若干尖峰的侧视图来表示带箭头。的断层图像已经经过低通滤波,以60埃。比例尺为100nm。
首先,我们使用精制205人工采摘尖峰初始模型( 图3)。最中心的尖峰的三次对称性是不施加任何对称性( 图3B)明显和被强加在随后的几轮细化( 图3C)。对于病毒粒子表面上自动地检测所有的尖峰,我们产生106种对每个病毒粒子在43 nm和20度( 图4A)的间距半径和迭代细化它们的位置相对于所述膜( 图4B)。

图3:起始模板结构的细化。 (一)圆柱的平均尖峰手动定义的位置建造(C100)模板(B)平均为密度经过五轮的细化,没有任何对称(C)施加显示器秒杀了三倍对称的特点。该模型的分辨率为48。穗(三)平均在41后的五轮细化了三次对称性解决。

图4:将种子细化AB)的种子之前(A的一个子集)和之后(B)的细化是从图2所示的一个病毒粒子密度在(B)的种子已经颜色编码基于各个交叉。相关系数(蓝色,低相关性;红色,高的相关性)。
最优相关糖蛋白刺突补丁(顶部75%的去除重叠后;〜1,000尖峰)被用于计算最终的平均值。平均得到解决,以35的分辨率( 图5)。它揭示了三聚尖峰结构的中间,在除了从六个邻近的尖峰一定的贡献。的病毒粒子,通过将结构中的已知位置来计算的复合模型,显示在病毒粒子表面上( 图6A)中的尖峰的位置。偶尔,尖峰局部有序的补丁是很明显的( 图6B)。

图5:模板匹配后的糖蛋白刺突层的贴片精致结构。 (AB)两地图"偶"和"奇",从数据的两半重建示。这两个图显示一个显着的相似程度,验证了该方法的合法性验证。围绕穗长轴线的方向是不同的两个映射已经重建完全彼此独立。这两个地图(C)平均显示在35埃的最终解决。

图6:在Bunyamwera病毒尖刺的放置。 (一)病毒体的复合模式。查看向量(支)表示尖峰的方向。颜色表示每个穗和模板结构(蓝色,低相关性;红色,高相关)之间的互相关(B)一个特写尖峰的有序贴剂。
讨论
知识的病毒糖蛋白的尖峰结构对病毒体膜是用于理解病毒的复制和显影疗法,治疗和预防感染是必不可少的。电子低温显微术结合单个颗粒的平均已成为使用最多的方法来解决包膜病毒粒子,包括糖蛋白的尖峰的结构。然而,该方法仅限于icosahedrally对称的病毒。在这里,通过低温电子断层扫描和subtomogram平均在Jsubtomo的应用程序,我们已经提出了一个通用协议,用于确定糖蛋白尖峰形性包膜病毒是不适合其他流动结构生物学的方法。我们的代表性结果证明,该方法的分辨率足以揭示见解域架构,低聚,并在完整的病毒颗粒的糖蛋白尖峰高阶组织。
jove_content">这个协议中最关键的一步是兴建两座可靠的启动模式,在统计上是相互独立的。成功执行此步骤假定糖尖峰足够大,不装反目成仇得太紧,使个人尖峰在视觉上识别和手动拾取的断层图像,以及两个独立的模式取平均。如果这是不可行的,两个修饰的协议可以尝试。首先,两个独立的随机模型可通过首先定义subtomograms两个随机子集,然后构造这些子集30内平均该subtomograms。其次,如果孤立穗的结构是来自通过其他手段,例如通过X射线晶体结构,它可以被用作初始模型,然而,必须小心到低使用低分辨率截止(50-70)带通滤波器这种模型中,如产生的两个模型在下一回合的细化将是统计独立只有超过这个分辨率。由于这个警告,前一种方法的建议。从该协议中获得的分辨率取决于四个主要因素:1。数据收集策略和所述输入数据的质量,ⅱ。该subtomograms的数目,三。该subtomograms的对准精度,以及iv。异质性的结构。而第一和第二限定可以在很大程度上克服通过使用高的信号 - 噪声直接电子检测器结合的CTF校正成像和自动数据采集,对准精度进一步受到关注自身结构的大小和形状。当应用该协议上缺乏显着的特征的小尖峰,它可能是有利的Fab片段结合于尖峰提高对准精度,因而分辨率31。最后,如果结构被平均展出多个构象,子tomogr时的分类方法可用于分别平均不同的构象。为此,Jsubtomo集成了发电机包,提供强大subtomogram分类9。
上述协议是互补的分离的病毒糖蛋白的X射线晶体结构。晶体结构可以被嵌入到子断层图像的平均值,得到的糖蛋白的精确取向相对于所述病毒体膜。这种方法的应用,无疑将继续脱落到包膜病毒的结构和病理学的光。
披露声明
The authors have nothing to disclose.
致谢
This work was supported by the Academy of Finland (130750 and 218080 to J.T.H.), the Wellcome Trust (090532/Z/09/Z; 089026/Z/09/Z to T.A.B.), and by the MRC (MR/J007897/1 to J.T.H and T.A.B; MR/L009528/1 to T.A.B.).
材料
| Name | Company | Catalog Number | Comments |
| Jsubtomo (ver 1.3.1) | University of Oxford | n/a | www.opic.ac.uk/jsubtomo |
| Bsoft (ver 1.8.7) | NIAMS, NIH | n/a | bsoft.ws |
| UCSF Chimera | UCSF | n/a | www.cgl.ucsf.edu/chimera |
参考文献
- Nickell, S., Mihalache, O., Beck, F., Hegerl, R., Korinek, A., Baumeister, W. Structural analysis of the 26S proteasome by cryoelectron tomography. Biochemical and biophysical research communications. 353 (1), 115-120 (2007).
- Goldie, K. N., Wedig, T., Mitra, A. K., Aebi, U., Herrmann, H., Hoenger, A. Dissecting the 3-D structure of vimentin intermediate filaments by cryo-electron tomography. Journal of structural biology. 158 (3), 378-385 (2007).
- Cheng, Y., Boll, W., Kirchhausen, T., Harrison, S. C., Walz, T. Cryo-electron tomography of clathrin-coated vesicles: structural implications for coat assembly. Journal of molecular biology. 365 (3), (2007).
- Grünewald, K., et al. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science. 302 (5649), 1396-1398 (2003).
- Murphy, G. E., Leadbetter, J. R., Jensen, G. J. In situ structure of the complete Treponema primitia flagellar motor. Nature. 442 (7106), 1062-1064 (2006).
- Medalia, O., Weber, I., Frangakis, A., Nicastro, D., Gerisch, G., Baumeister, W. Macromolecular architecture in eukaryotic cells visualized by cryoelectron tomography. Science. 298 (5596), 1209-1213 (2002).
- Heymann, J. B., Cardone, G., Winkler, D. C., Steven, A. C. Computational resources for cryo-electron tomography in Bsoft. Journal of structural biology. 161 (3), 232-242 (2008).
- Kremer, J., Mastronarde, D., McIntosh, J. Computer visualization of three-dimensional image data using IMOD. Journal of structural biology. 116 (1), 71-76 (1996).
- Castaño-Díez, D., Kudryashev, M., Arheit, M., Stahlberg, H. Dynamo: a flexible, user-friendly development tool for subtomogram averaging of cryo-EM data in high-performance computing environments. Journal of structural biology. 178 (2), 139-151 (2012).
- Hrabe, T., Chen, Y., Pfeffer, S., Cuellar, L. K., Mangold, A. -V., Förster, F. PyTom: a python-based toolbox for localization of macromolecules in cryo-electron tomograms and subtomogram analysis. Journal of structural biology. 178 (2), 177-188 (2012).
- Fernández, J. J. Computational methods for electron tomography. 43 (10), Micron (Oxford, England). 1010-1030 (2012).
- Briggs, J. A. G. Structural biology in situ--the potential of subtomogram averaging). Current opinion in structural biology. 23 (2), 261-267 (2013).
- Frangakis, A. S., et al. Identification of macromolecular complexes in cryoelectron tomograms of phantom cells. Proceedings of the National Academy of Sciences of the United States of America. 99 (22), 14153-14158 (2002).
- Zanetti, G., Briggs, J. A. G., Grünewald, K., Sattentau, Q. J., Fuller, S. D. Cryo-electron tomographic structure of an immunodeficiency virus envelope complex in situ. PLoS pathogens. 2 (8), (2006).
- Liu, J., Bartesaghi, A., Borgnia, M. J., Sapiro, G., Subramaniam, S. Molecular architecture of native HIV-1 gp120 trimers. Nature. 455 (7209), (2008).
- Meyerson, J. R., et al. Determination of molecular structures of HIV envelope glycoproteins using cryo-electron tomography and automated sub-tomogram averaging. Journal of visualized experiments : JoVE. (58), (2011).
- Baker, T. S., Olson, N. H., Fuller, S. D. Adding the third dimension to virus life cycles: three-dimensional reconstruction of icosahedral viruses from cryo-electron micrographs. Microbiology and molecular biology reviews : MMBR. 63 (4), 862-922 (1999).
- Huiskonen, J. T., Butcher, S. J. Membrane-containing viruses with icosahedrally symmetric capsids. Current opinion in structural biology. 17 (2), 229-236 (2007).
- Liljeroos, L., Huiskonen, J. T., Ora, A., Susi, P., Butcher, S. J. Electron cryotomography of measles virus reveals how matrix protein coats the ribonucleocapsid within intact virions. Proceedings of the National Academy of Sciences of the United States of America. 108 (44), (2011).
- Arranz, R., et al. The structure of native influenza virion ribonucleoproteins. Science. 338 (6114), 1634-1637 (2012).
- Karotki, L., et al. Eisosome proteins assemble into a membrane scaffold. Journal of Cell Biology. 195 (5), 889-902 (2011).
- Pietilä, M. K., et al. Virion architecture unifies globally distributed pleolipoviruses infecting halophilic archaea. Journal of virology. 86 (9), 5067-5079 (2012).
- Huiskonen, J. T., et al. Electron cryotomography of Tula hantavirus suggests a unique assembly paradigm for enveloped viruses. Journal of virology. 84 (10), 4889-4897 (2010).
- Bowden, T. A., Bitto, D., McLees, A., Yeromonahos, C., Elliott, R. M., Huiskonen, J. T. Orthobunyavirus ultrastructure and the curious tripodal glycoprotein spike. PLoS pathogens. 9 (5), (2013).
- Maurer, U. E., et al. The Structure of Herpesvirus Fusion Glycoprotein B-Bilayer Complex Reveals the Protein-Membrane and Lateral Protein-Protein Interaction. Structure. 21 (8), London, England. 1396-1405 (1993).
- Gan, L., Ladinsky, M. S., Jensen, G. J. Chromatin in a marine picoeukaryote is a disordered assemblage of nucleosomes. Chromosoma. 122 (5), 377-386 (2013).
- Scheres, S. H. W., Chen, S. Prevention of overfitting in cryo-EM structure determination. Nature. 9 (9), 853-854 (2012).
- Heymann, J. B., Belnap, D. M. Bsoft: image processing and molecular modeling for electron microscopy. Journal of structural biology. 157 (1), 3-18 (2007).
- Goddard, T. D., Huang, C. C., Ferrin, T. E. Visualizing density maps with UCSF Chimera. Journal of structural biology. 157 (1), 281-287 (2007).
- Faini, M., et al. The structures of COPI-coated vesicles reveal alternate coatomer conformations and interactions. Science. 336 (6087), 1451-1454 (2012).
- Wu, S., et al. Fabs enable single particle cryoEM studies of small proteins. Structure. 20 (4), London, England. 582-592 (2012).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。