
Method Article
微操作系统技术允许分析骨架调节器的形态发生动力学和营业额
摘要
我们描述了微和 photomanipulation 技术, 如酶和 photoactivation 如何能够确定运动参数和蛋白质在迁移细胞内的时空动力学。实验读数包括亚细胞动力学和运动调节器的更替或底层肌动蛋白骨架。
摘要
研究蛋白质的时空动力学可以揭示它们在不同语境中的功能重要性。本文讨论了漂白 (酶) 和 photoactivation 技术后荧光恢复方法在亚细胞位置蛋白质时空动力学研究中的应用。我们还展示了这些技术如何能够直接确定与肌动蛋白骨架调节和细胞运动有关的各种参数。此外, 细胞的显微注射还被描述为一种替代疗法 (可能之前或补充上述 photomanipulation 技术), 以触发移位蛋白对细胞的瞬时影响形态和功能。微操作系统, 如蛋白质注射或局部应用血浆膜通透性药物或骨架抑制剂, 可以作为一个强有力的工具, 以记录特定治疗的直接后果的细胞行为在单细胞和亚细胞水平。这是在这里的例子, 立即诱导 lamellipodial 细胞边缘突出的注射重组 Rac1 蛋白, 建立了一个季度前。此外, 我们提供了一个协议, 以确定增强的绿色荧光蛋白 (EGFP)-虚拟的营业额, 肌动蛋白长丝聚合酶显著积累在 lamellipodial 提示 B16-F1 细胞, 使用酶和包括相关数据分析和曲线拟合。我们还提出了评估 lamellipodial 肌动蛋白网络聚合速率的指导原则, 如表达 EGFP 标记β肌动蛋白的细胞所示。最后, 给出了如何调查细胞胞浆内肌动蛋白单体流动率的指示, 其次是肌动蛋白在快速灯丝组装部位的 lamellipodia, 如突出的小贴士, 使用 photoactivation方法。这些协议都不限于肌动蛋白细胞骨架的成分或调节器, 但可以很容易地扩展到类似的方式探索蛋白质在不同亚细胞结构或功能中的时空动力学和功能上下文。
引言
在细胞和分子生物学的许多领域中, 监测蛋白质和其他分子的时空动力学已经成为一个必不可少的工具。先进的荧光显微技术包括荧光共振能量转移 (烦恼) 和焦虑荧光寿命成像 (烦恼-电影), 或酶, 荧光损失漂白 (翻转) 和 photoactivation 以及许多其他允许对于蛋白质-蛋白质相互作用的时空跟踪, 构象的变化, 以及在细胞1,2中确定不同蛋白质的扩散和定位的动力学。酶和 photoactivation 技术, 特别是, 广泛适用于检查调节肌动蛋白细胞骨架和细胞迁移。这些技术可以单独应用或与附加的微操作系统技术 (如显微注射 3) 结合使用, 并涉及荧光标记蛋白的表达.它们允许将蛋白质联想的动力学估计为参与细胞迁移的富含肌动蛋白的结构, 如丝状伪足或 lamellipodia、病灶粘连中蛋白质的更替4或分支肌动蛋白网络 5.它们还能确定 lamellipodial 肌动蛋白聚合速率, 单体肌动蛋白在细胞质内的分散度, 亚细胞肌动蛋白单体易位率在突出lamellipodia6和其他参数。
酶是一种可视化和量化在活细胞内蛋白质流动性的方法, 最初在二十世纪七十年代由阿克塞尔罗德7开发。一个在细胞内的感兴趣区域 (ROI), 用荧光标记的蛋白质填充, 瞬时暴露在高强度的激光中, 足以导致在给定的短时间内对该区域中的荧光分子进行漂白。在漂白过程中, 未漂白的、荧光标记的蛋白质, 会根据它们的时空动力学, 扩散和渗透到漂白的区域, 从而导致 photobleached 分子随着时间的推移而迁移。漂白地区荧光回收率的大小取决于各种因素, 包括给定分子的扩散率和传播速率, 以及在假定相关的漂白结构中的更替率。因此, 可溶性蛋白质将通过扩散迅速地在漂白的 ROI 中调节荧光的恢复, 而与结构紧密相关的蛋白质, 如焦粘连, 将有更长的周转时间, 因为它们的荧光恢复将依赖于蛋白质可溶性分数的扩散和结构相关分数的离解-联想动力学。荧光恢复通常是获得和量化, 直到初始水平的前漂白强度的荧光达到。但是, 如果初始荧光强度的一部分属于所谓的不动分数, 则无法通过扩散补充或以非常慢的速率补充, 而与包括移动的大多数分子相比, 这种情况不会发生。分数。为了确定蛋白质更替率, 酶曲线的产生, 代表了荧光恢复的程度随着时间的推移。从这些恢复曲线可以计算蛋白质回收率的平均半倍。通过创建平均酶数据的曲线拟合, 进而进行数学分析, 也可以推断流动分数的平均周转率是否构成一个分子的均匀种群的组合, 或者它是否由两个或更多的分子亚群在不同的速率翻转。除了通过定量方法估计蛋白质周转率外, 跟踪 lamellipodia 中 photobleached 区域的恢复也可以精确量化 lamellipodial 运动参数, 如逆行流、突起、和肌动蛋白聚合速率。因此, 酶构成了一种多功能工具, 用于评估活细胞结构中的各种参数。
Photoactivation 是一种用于跟踪来自指定细胞位置的蛋白质或分子的扩散和流动性的方法。例如, 该技术采用了一种野生型绿色荧光蛋白 (GFP) 的变体, 最初由帕特森和利平科特8开发, 这种变异方式使其荧光在接触到紫外线 (紫外线) 光 (大约400毫微米; 这里, 405 毫微米)。正如帕特森et al所描述的, 野生型 GFP 染色体作为中性酚类和阴离子 phenolates 的混合种群存在, 其产生的吸光度峰值约为 397 nm, 较小的是 475 nm。在紫外光照射下, 种群经历 photoconversion, 向阴离子形态转移。当兴奋 488 nm, photoconverted/光活化作用蛋白表现出3倍的荧光增加, 在实践中缺乏区分激活和非活化 GFP 由于高内在背景荧光。然而, 通过在 GFP 序列中引入单一氨基酸突变 (在位置 203), 降低了背景强度。由此产生的 T203H 突变体, 也称为 photoactivatable-gfp (PA-gfp) 的特点是显著降低了小峰的吸光度, 这在辐照与紫外线照射后增加近100倍, 随后激发 488 nm 光。因此, 过度表达的 PA-GFP 标记蛋白是一种广泛使用的方法, 它允许测定分子在细胞内的扩散和运动。我们以前应用了 PA-GFP 标记肌动蛋白, 以确定肌动蛋白单体分散的速率从胞浆区域, 不仅允许探索他们的流动性在细胞质, 而且他们的合并率到突出lamellipodial 肌动蛋白网络6。最近的文献还介绍了新颖的, 可转换的蛋白质, 原则上可以使用类似的方式, 但窝藏潜在的优势已经可见, 在照片转换之前。这组荧光蛋白的例子包括 Dendra2 和 mEos29,10,11,12。
在本文中, 我们解释了 microinjecting 细胞与蛋白质的方法。我们进一步解释了这项技术如何与酶结合, 通过漂白蛋白参与肌动蛋白细胞骨架调节和运动, 以及如何酶曲线和半时间恢复的移动分数可以获得。此外, 我们提供了一个例子, 酶技术如何可以用来确定肌动蛋白聚合速率的 lamellipodial 网络。我们还提供有关如何执行 photoactivation 实验的指示和提示, 可用于确定单体肌动蛋白的胞浆流动性和肌动蛋白在 lamellipodia 中的比例。当然, 这些技术不仅限于跟踪肌动蛋白骨架成分, 而且在可能需要适度的适应或优化的情况下, 可以广泛应用于其他细胞类型或研究不同的蛋白质、结构和参数。
研究方案
1. 盖玻片洗涤和杀菌
- 浸泡15毫米 (直径) 盖眼镜 (1) 在500毫升瓶含有40毫升 37% HCl 和60毫升100% 乙醇 (不超过100盖玻片每100毫升洗涤解决方案) 的混合物。
注: 即使新鲜购买, 盖玻片必须严格清洗之前, 种子细胞到他们的表面。这是因为它们可能含有一些宏观上看不见的油脂薄膜, 但能有效地干扰活细胞的黏附力和适当的传播。而这样的电影可以有效地去除含有酸或碱的溶液 (参见 Fischer et 。13), 我们经常使用上面描述的酸/酒精混合物。 - 在旋转振动筛上摇动装有盖子玻璃的烧瓶30分钟。选择一个速度, 使盖眼镜可以自由旋转, 但足够慢, 以避免频繁打破。过滤解决方案, 以去除碎玻璃碎片, 如果重用。
- 将盖子玻璃转移到含有至少200毫升不育水的烧瓶中, 在旋转振动筛上孵化, 同时反复更换水, 直到酸性气味消失为止。建议在几个小时内进行多次清洗, 以彻底消除 HCL-乙醇的痕迹。
- 在一张滤纸上烘干单独的盖子玻璃。
- 将盖子玻璃放在10厘米 (直径) 培养皿的底部, 用滤纸覆盖, 热干杀菌。避免热处理, 因为这将导致盖眼镜粘在一起。
2. 细胞的治疗, 转染和播种到盖玻片
- 根据标准细胞培养条件生长 B16-F1 鼠黑色素瘤细胞 DMEM (4.5 克/升葡萄糖), 含10% 胎小牛血清, 2 毫米谷氨酰胺, 1% 青霉素-链霉素在37°c, 7% CO2。
- 根据标准细胞培养条件 (37 °c, 7% CO2) 在 DMEM (4.5 克/升葡萄糖) 中 NIH3T3 成纤维细胞为 microinjections 生长, 含10% 胎牛血清, 1 毫米丙酮酸钠, 1x 内存非必需氨基酸, 2 毫米谷氨酰胺, 1% 青霉素-链霉素。
- 对于 transfections, 生长 B16-F1 细胞到100% 汇合在一个10厘米的盘子和通道1:5 的比例成3厘米 (直径) 塑料碟。
- 同一天, 在 B16-F1 细胞被允许坚持至少6小时, 染与 500 ng/盘 photoactivatable PA-GFP 肌动蛋白或 EGFP 标记的β肌动蛋白质粒 DNA。对于 mCherry 编码载体的 PA-GFP 肌动蛋白的共 transfections, 每3厘米的皿中混合1µg 质粒 DNA。
- 用转染试剂 (材料表) 染 B16-F1 细胞。对于3厘米的菜, 混合200µL 150 毫米氯化钠含有 500 ng DNA 结构与200µL 的150毫米氯化钠含有1µL 的转染试剂 (即, DNA (µg): 试剂 (µL) 的比例1:2 被使用)。
- 在室温下 (RT) 和吸管上孵化出20分钟的转染混合物, 放到含有细胞的3厘米皿上。在37摄氏度, 7% CO2中, 轻轻地旋转盘子混合并孵化一夜。
- 制备含50毫米三、pH 7.4 和150毫米氯化钠的层粘连蛋白涂层缓冲液。
- 对于 B16-F1 细胞, 涂层15毫米覆盖眼镜通过传播150µL 的层粘连蛋白 (25 µg/毫升在粘连蛋白涂层缓冲) 和孵化1小时在 RT。对于 NIH3T3 细胞, 用纤维连接蛋白溶液 (25 µg/毫升在磷酸盐缓冲盐水 (PBS)) 上涂上盖子玻璃, 并在 RT 上孵化1小时。
- 用 pbs 冲洗层粘连蛋白--或纤连蛋白--孵化盖玻璃, 然后吸入 pbs 并添加2毫升转染细胞。
- 将转染的 B16-F1 细胞 (在一点半从汇合皿中的比例) 中播种后, 在染层粘连蛋白涂层的盖玻片上。种子 NIH3T3 成纤维细胞 (在1:20 比例从汇合盘) 到纤维连接蛋白涂层盖玻片。
- 允许细胞在层粘连蛋白-或纤维粘连蛋白涂层盖眼镜过夜在组织培养孵化器在37摄氏度之前, 显微镜。另外, 由于允许细胞传播至少 2–3 h, 所以可以在同一天启动显微实验。
3. 显微成像室组装
- 使用导热 RC-26 铝成像室进行显微术 (图 1a)。使用注射器在塑料封口机的轮廓周围涂抹硅脂 (图 1b)。
- 将盖子玻璃与单元格的侧向向上放置 (图 1c)。
- 将塑料封口机放在盖玻璃的顶部, 使盖玻片和燃烧室之间的安全密封。固定塑料封口机 (对角以避免盖玻片破损) 通过拧紧滑钳到房间, 以避免介质泄漏(图 1 d).
- 吸管37°c 预加热显微介质进入中心区域。对于中度降低的自体荧光, 从而优化显微镜, 使用相同的食谱作为培养基上所述, 但与 F12-HAM 而不是 DMEM, 另外包含20毫米 HEPES 的细胞培养在没有 CO2 (图1e)。
- 将热探测器插入会议厅的指定插槽中, 并将该腔室的电极链接到保持恒温37摄氏度的 TC-324B 自动温度控制器 (图 1f)。
- 把一小滴浸入油放到目标上, 把房间放在上面。
- 用细胞孵化室至少10–30分钟, 使其从安装过程中的温度下降中恢复, 并适应显微镜介质。
- 在显微镜开始之前, 更换室中央储层中的培养基 (大约800µL), 以避免中度成分和血清中蒸发的不适当浓度。长时间的显微镜会话与开放室将需要例行改变蒸发介质。
4. 显微注射程序
- 涂上盖玻片, 准备细胞, 并按照上面描述的那样组装成像室。
- 解冻被注射的纯化蛋白的整除 (通常为10µL 或更少), 并用适当的微注射缓冲剂稀释。
注: 缓冲成分可能因蛋白质和细胞类型而异, 但要注意使用6.95 和8.00 之间的 pH 值, 避免使用 pbs, 因为大多数细胞不喜欢注射 pbs。 - 对于 Rac1 显微注射, 准备缓冲含有100毫米氯化钠, 50 毫米三 HCl pH 7.5, 5 毫米氯化镁2, 1 毫米。Mg2 +离子对于小 GTPase 稳定性是必不可少的。
注: 蛋白质浓度通常在0.1–1毫克/毫升 (最大2毫克/毫升) 之间变化, 这取决于蛋白质、实验类型和细胞类型。 - 如果适用, 添加荧光染料, 如惰性葡聚糖 (0.5 µg/毫升, 70 kDa) 的蛋白质溶液, 这可以确认在注射前针流存在, 并允许在实验后成功注射的文件。
注意: 这里的实验不是针对注射 Rac1 的动力学, 只有在蛋白质的直接荧光标记后才有可能。蛋白质与荧光染料或融合到荧光蛋白的耦合是可能的, 但在这里避免, 因为它有干扰信号功能的风险, 特别是小蛋白, 如 GTPase Rac1 (20 kDa)。 - 离心蛋白溶液在 1万 x g 至少30分钟, 以去除可能导致针堵塞, 如果在微注射毛细管中的蛋白质聚集。
- 使用一个灵活的吸管尖端/microloader 提示, 用1µL 的注射混合物从后端加载一针 (显微注射毛细管)。
- 如果针尖中存在气泡, 请轻轻地轻敲针头底座以将其移除。迅速进行, 以避免针尖的干燥, 这可能导致针堵塞。
- 仔细调整微操作系统装置上的针座。如果使用倒置显微镜进行相衬成像, 在针装前, 确保有足够的空间在不妨碍显微镜冷凝器的情况下上下移动针头。
- 将 microcapillary 拧紧后, 将压力 (20–50的背景压力) 用微注射压力装置施加于针头上, 然后转运针尖进入细胞培养培养基。
注: 当针处于介质中时, 激活压力会导致介质被毛细管力吸干, 从而禁止注射感兴趣的溶液。 - 在视野中定位针头 (使用低放大目标促进)。在这里使用了40X 干燥目标的显微注射实验。
- 将针尖从宏观上定位在相对于物镜中间的垂直位置 (这将加速找到针尖)。用相衬光学显微镜在水平平面上相对于视场移动针尖, 在光学平面上, 在细胞层之上。
注: 针最初将作为阴影出现在视野中, 然后可以调整焦点平面以可视化笔尖。一旦发现针尖, 逐渐降低光学平面后跟针尖向下的位置接近细胞层。 - 使用荧光葡聚糖时, 通过切换到荧光通道来检查针流, 并用压力装置调整流量以获得恒定的 "背景" 流。
注: 在本文中, 我们描述手动注射, 这是介导的通过接触细胞膜的细胞表面和在恒定的针流中的针尖的温和运动, 通过等离子膜突破。这必须区别于自动注射设备, 伴随着编程的针降低和针压增加在注射事件, 这是更适合注射更高的细胞数后, 随后的细胞数量分析。本文所描述的方法是在显微注射前、期间和之后通过时间推移显微镜对单细胞分析进行优化。 - 找到一个感兴趣的细胞, 并逐渐降低细胞上方的针。
- 当准备 microinject 时, 用机器人操纵杆的细小齿轮逐渐降低针向细胞核周区域, 同时保持细胞集中。
- 对于微注射, 轻轻地触摸细胞的等离子膜, 这可能足以穿透细胞, 或帮助瞬变膜破裂由一个非常温和的水龙头在显微镜设置。
注: 针尖上的白点会指示与等离子膜接触的时间;膜破裂后, 针尖将重新封装, 伴随着注入溶液的温和流动进入细胞。 - 一旦进入细胞可见 (理想的0.3 秒内), 通过将针尖向上移动到介质中, 停止注射过程。当使用荧光葡聚糖, 成功的注射可以立即记录荧光。
- 如果需要, 在微注射之前或之后启动时间推移图像采集。
注: 药物或抑制剂的局部应用可以在这里的所有步骤执行, 除了微注射事件本身. 对于局部应用, 活性分子的扩散可以通过流动压力控制, 并通过荧光记录, 针尖可以定位在所需的高度。有关本地应用程序实验的示例, 请参见例如小型和 Rottner14或 Kaverina等。15 - 在显微注射之后, 等待蛋白质的作用发生。对于不同的蛋白质, 取决于预期的结果, 潜伏期可能会有所不同。对于小 GTPase Rac1, lamellipodium 组的响应可以在1分钟或更短的时间内启动, 但平均需要大约10-15 分钟才能完全开发 (图 1g, h)。
- 在显微注射后判断细胞的生存能力。
注: 不适当或有害的注射可能导致细胞损伤, 这往往伴有非特异的细胞边缘退缩或血浆膜破裂。- 避免通过顶部和底部的等离子膜, 这可能发生在扁平细胞区注射。
注: 注射量应保持在最低限度 (理想的 < 5% 的细胞体积), 通常会在 femtoliter 范围内。所需的注射量也可以由浓度的变化控制, 但请注意, 对于蛋白质, 浓度 > 2 毫克/毫升可能变得不切实际由于频繁的针头堵塞。然而, 这也取决于纯化蛋白的质量和行为;例如, 注射荧光耦合肌动蛋白是复杂的浓度依赖性和不可避免的聚合在针尖, 所以它很少执行今天 (见小et 等。16)。
- 避免通过顶部和底部的等离子膜, 这可能发生在扁平细胞区注射。
- 在显微注射的效果之前、期间或之后, 酶或 photoactivation 可以在同一个细胞上进行 (见5和6节)。
5. 酶程序
- 染细胞类型的兴趣 (这里 B16-F1 细胞) 与质粒 DNA 编码的荧光标记的蛋白质的兴趣 (这里, 一个 EGFP 标签版本的β肌动蛋白被使用)。将细胞种子到层粘连蛋白涂层的盖玻片 (步骤 2.10)。
- 组装成像室 (3 节)。
- 使用以下设置 lamellipodial 区域漂白:65 兆瓦激光功率 (根据实验设置和激光源的变量);10像素激光束直径;1 ms 漂白剂停留时间/像素;500毫秒的 GFP 曝光时间;1500毫秒时间间隔。本文采用 100X 1.4NA apochromatic 目标进行了实验研究。
- 执行激光校准以确保 photobleached 区域的尺寸精度。在校准之前, 将视野移动到缺少任何细胞/荧光信号的区域, 并观察显示器上的图片。
- 通过单击相应的放大按钮并减少 "面板 |" 中的激光功率 (3–5兆瓦), 选择目标放大倍数。强度 "菜单。要启动 Visiview 软件 (v2.1.4) 的手动校准, 请选择 "配置 |酶 "菜单并点击" 校准 |调整手动菜单。确保激光可以分辨为尖锐点。如果没有, 可以重新调整激光硬件。
- 通过手动引导激光到预先确定的软件 X Y 坐标来执行校准。这将指示软件如何针对当前放大器将激光具体定位到用户定义的区域。
- 在触发激光之前, 切换到 GFP 通道并启动图像/时间失效采集。
- 手动绘制在 GFP 通道上 photobleached 的区域, 同时查看显示器。
- 启动漂白由手动触发的 405 nm 激光, 至少3–4帧后开始的图像采集。在以后的数据分析中, 需要在漂白之前获取帧来进行图像的规范化。
6. Photoactivation 程序
注: 软件、显微镜设置和设置, 除了激光功率, 是类似于酶。在 photoactivation, 与酶相比, 一个重要的区别是, 405 nm 激光功率显著低于漂白使用, 以激活 PA-GFP 没有同时漂白它。
- 染细胞类型的兴趣 (这里 B16-F1 细胞; 参见步骤 2.5) 用质粒 DNA 编码 PA-GFP 肌动蛋白和另一种荧光标记蛋白 (例如, mCherry 或 mCherry Lifeact)。
注意: 在大多数情况下, mCherry 阳性细胞也将是阳性的 PA-gfp 肌动蛋白的载体, 后者通常不会在 GFP 通道在 photoactivation 之前。为了提高 mCherry 阳性细胞对 PA gfp 的阳性几率, 请使用 1:2 mCherry 的转染率: pa-gfp。根据该协议, 超过90% 的细胞表达 mCherry 显示成功激活的 PA-GFP 肌动蛋白。 - 将 B16-F1 细胞种子到层粘连蛋白涂层的盖玻片 (步骤 2.10)。
- 组装成像室 (3 节)。
- 在启动 photoactivation 实验之前, 如果需要, 对所选目标 (步骤 5.4–5.6) 执行激光校准。
- 将 GFP/488 纳米图像捕获设置为500毫秒曝光率和1500毫秒时间间隔 (取决于实验设计)。
-
通过标记 "波长系列" 正方形并选择所需的频道数来调整软件设置, 以获取双通道或三重通道的定时播放影片 "获取 |波长 "菜单。建议使用相位对比和 GFP 通道获取时间失效电影。
- 另外, 还包括 mCherry 通道;然而, 暴露太多光线的细胞可能会诱发光损伤。这可以避免与氧气清道夫如 Oxyrase17, 虽然有效的治疗需要细胞室密封。
- 在 mCherry 通道上找到转染的细胞。
- 在触发激光之前, 启动图像/时间失效采集, 并手动绘制在相位对比通道上光活化作用的区域, 同时查看显示器。
- 启动 photoactivation 由手动触发的 405 nm 激光 (强度设置之间的5–15兆瓦从 "面板 |强度 "菜单), 至少3–4帧后图像采集启动。
7. 数据分析和酶结果的表述
注: 所提出的方法用于调查动态肌动蛋白组装部位的蛋白质的周转情况, 在这种情况下, 虚拟与粘附部位和突出 lamellipodia 的提示相关联。我们正在分析其在 lamellipodium 尖端的营业额, 但同样的分析原则可以用于调查虚拟或任何其他蛋白质和其他亚细胞室的营业额。
- 打开从 Metamorph 软件 Visiview 派生的时间推移电影。在本文中, 使用了 Metamorph v7.8.10。
- 通过手动概述 Metamorph 上的各个区域, 导出 photobleached 区域的强度值。在覆盖整个或部分 photobleached 区域的 lamellipodium 顶端绘制一个形状, 并在需要时手动调整其在后续帧上的位置 (即,如果边缘突出), 以跟踪 lamellipodial 强度的变化。在末端位移期间各自组分。
- 为了矫正背景和漂白的获取, 分析细胞内外的区域。有关测量强度的代表性区域, 请参见图 2a 。
- 当选择 ROI 时, 使用菜单 "度量" (Metamorph) 提取其强度值。区域测量 "。确保在 "配置" 菜单中选择 "经过的时间" 和 "平均强度" 选项。单击 "打开日志" 并选择 "动态数据交换"。单击 "确定" 打开 excel 电子表格, 然后再次单击 "打开日志" 按钮, 将 Metamorph 值粘贴到 excel 中。
注意: 这些值用于生成荧光恢复曲线。 - 为在 photobleached 区域的 lamellipodium 尖端生成荧光恢复曲线 (在漂白之前正常化为区域强度), 请应用以下公式:
 等式1
等式1
其中: 酶Tn是漂白后每个感兴趣帧的 photobleached 区域强度;Tn是在漂白之后为每个感兴趣的帧以外的单元格 (背景) 所采取的区域强度;InsTn是漂白之后的每个感兴趣帧的两个平均内部区域强度 (用于在一段时间内为购置漂白正常化);酶T-1是漂白前的 photobleached 区域强度;T-1是在漂白之前在单元格 (背景) 外采取的区域强度;而 InsT-1是漂白前每个感兴趣帧的两个平均内部区域强度。 - 对于每一次感兴趣的帧, 请使用等式 1获取包含要调查的所有时间帧的荧光恢复曲线。时间长短严格取决于所调查的蛋白质。未知时, 进行初步实验, 获取蛋白质的更替率。
- 要计算恢复的半时间, 请将荧光恢复曲线的值与相应的时间 (以秒为单位) 粘贴到西格玛图 (v 12) 中, 并使用 "动态匹配向导" 来执行曲线拟合 |指数上升到最大 "工具。选择单指数 (单, 3 参数) 或双指数 (双, 4 参数) 函数, 取决于最佳曲线适合。
- 对单指数函数使用以下公式:
 等式2
等式2
- 对双指数函数使用以下公式:
 等式3
等式3
- 将从西格玛绘图 (等式 2或等式 3) 派生的参数 "b" 和 "d" 粘贴到 Excel 中, 以计算恢复的半时间。应用以下等式:
 等式4
等式4
或 等式5
等式5
- 当单指数函数产生精确的曲线拟合时, 仅应用等式 4。
- 当单指数函数不能产生良好的曲线拟合时, 应通过求解等式 4和等式 5来应用双指数公式。考虑结果两个半倍的恢复作为代表两个不同的蛋白质组分: 快速和缓慢交换分数, 分别。
8. 酶测定 Lamellipodial 肌动蛋白聚合速率
- 为了确定 lamellipodial 肌动蛋白聚合速率, 染 B16-F1 细胞与 EGFP 标记β肌动蛋白, photobleach lamellipodial 区域 (步骤 5.9) 使用1.5 的时间间隔和 500 ms GFP 暴露。
- 在 Metamorph 中, 打开从 Visiview 获取的定时电影, 并根据 "测量 |" 的目标校准像素/µm 比例校准距离 "工具。
- 播放时间推移电影, 并停止它的框架时, lamellipodial 荧光恢复, 它向后流向薄板作为一条线, 已经达到了薄片, 没有进一步的向后流可以跟踪。
- 测量 lamellipodium 尖端与恢复荧光的背面之间的µm 距离。此距离对应于逆行流和凸出距离的总和。
- 或者, 要将突出项与逆行流分开, 请在漂白前用一条线标记 lamellipodial 尖。使用该行作为后续框架中的参考点, 以引用漂白时 lamellipodium 笔尖的原始位置;参考点可用于测量凸出距离和逆行流。
- 记下漂白后荧光恢复所需的时间 (以秒为单位)。时间可以手动计算从帧速率或可视化通过 Metamorph 通过 "测量 |区域测量 "工具。
- 利用以下方程推导出肌动蛋白聚合速率 (与步骤8.4 和8.6 中基于 Metamorph 测量的一些方程参数):
 等式6
等式6
当肌动蛋白聚合速率为µm/分钟时, 逆行流动距离为µm, lamellipodial 突出距离为µm, 时间为秒。
9. Photoactivation 对蛋白质扩散和流动性的分析
注: 本文介绍的方法是通过使用 photoactivation 的肌动蛋白融合到 PA-GFP 来分析肌动蛋白单体的移动性, 并通过细胞质的可视化和量化来说明蛋白的扩散。
- 为了测量 photoactivatable 肌动蛋白从胞浆区域的扩散, 以及 lamellipodial 区域内的积累, 使用 Metamorph 来确定以下区域的强度 (如图 3a): 胞浆光活化作用区域 (PA);lamellipodial 区域, 光活化作用蛋白质预计在一段时间内积累 (林);用于背景荧光正常化的细胞外的区域。
- 在确定细胞质中肌动蛋白的移动性时, 测量不同的胞浆区域 (请参见图 3a、区域 R1-R5)。请注意, 漂白的收购不能以类似的方式确定为酶, 因为在激活时焦距和最终细胞范围的荧光增加。
- 如步骤7.4 所述, 将所有区域的强度值从 Metamorph 转移到 Excel 电子表格中。
- 检查 photoactivatable 肌动蛋白在 photoactivation 的胞浆区内的位移率, 或其在 lamellipodial 区域内的加入率 (两者均表示为胞浆光活化作用区域的百分比强度, 时间为 0), 从步骤9.3 中的数据生成荧光曲线。应用以下等式:
 等式7
等式7 等式8
等式8
其中: PATn是 photoactivation 后每个感兴趣框架的胞浆光活化作用区域的强度;林Tn是 photoactivation 后每个 lamellipodial 区域的强度;Tn是在 photoactivation 之后为每个感兴趣的框架在单元格外 (背景) 之外所采取的区域的强度;PAT-1是 photoactivation 前胞浆光活化作用区域的强度;林T-1是 photoactivation 前 lamellipodial 区域的强度;T-1是在 photoactivation 之前在单元格外部 (背景) 外进行的区域的强度;PAT0是胞浆光活化作用区域的强度, 时间为 0(即 photoactivation 后的第一帧);和出T0是在单元格外部 (背景) 在时间 0 (即photoactivation 之后的第一帧) 外所采取的区域的强度。 - 或者, 为了更好地可视化数据, 将强度曲线正常化为 0, 方法是在每个后续帧 photoactivation 后减去第一帧的强度。
注意: 下面的分析方法 (步骤 9.6–9.8) 也允许计算光活化作用肌动蛋白在细胞质内的胞浆弥散。 - 测量胞浆区域的强度, 这是从活化区远端连续定位的。
- 要将这些区域的强度表示为光活化作用区域在时间0的强度百分比, 请应用等式 8, 其中 lamellipodial 强度被每个胞浆 ROI 的强度替换。区域的大小和数量可能根据要测量的细胞大小和扩散距离而变化。
- 为了获得光活化作用蛋白浸润每个胞浆区域的速率的可量化值, 将每个区域的荧光强度增加曲线的时间和值粘贴到西格玛图中 (类似于第7节中的酶分析), 请使用等式 2和等式 4推导出到达高原的荧光强度的半时间。比较不同实验组之间的 t1/2值。
结果
图 1g, h显示 NIH3T3 成纤维细胞的相对比图像, 前和10分钟后显微注射 Rac1, 这是一个小的细胞家族 GTPase 能够诱导 lamellipodia 形成通过与波复合体的相互作用。在微注射 (图 1g) 之前, 该单元格首先被可视化, 以确认其生存能力和形态学,例如,缺少 lamellipodia。在注射后10分钟后, 细胞已经明显改变了它的形态学, 这是预期从这种治疗, 并表示成功注射 (图 1h)。
为了简单和清晰起见, 我们接下来为细胞中的酶和 photoactivation 分析提供了模范的结果, 而这些研究还没有杏仁。
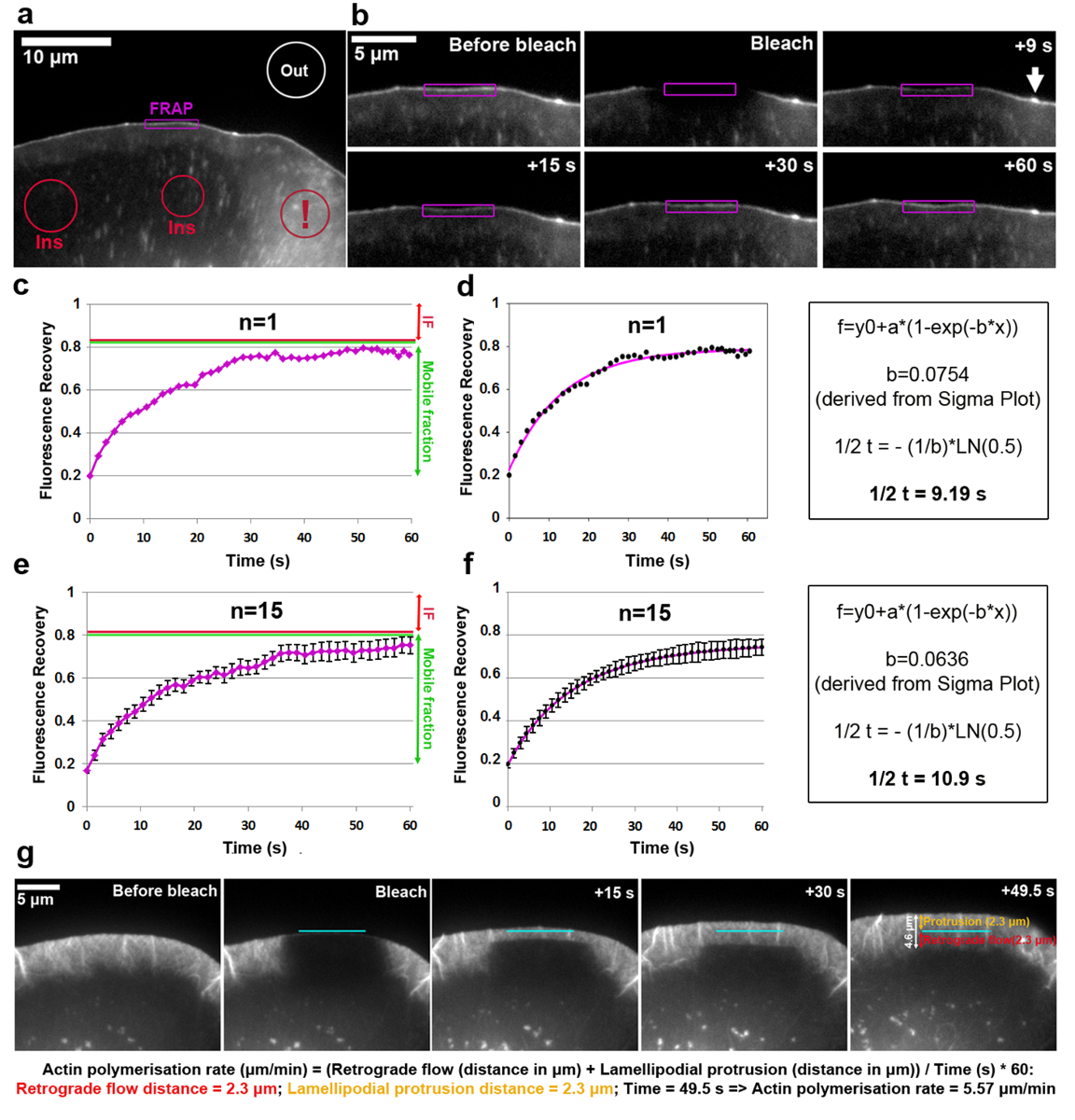
在图 2EGFP 中显示了在 lamellipodium 提示处标记的虚拟的营业额的分析.请注意, 虚拟另外目标到新生和焦点粘连, 小和拉长点在细胞内部18,19。lamellipodial 区域的荧光强度在尖端有明确的虚拟积累被漂白和测量每一个时间框架, 通过遵循 ROI 的轮廓之前, 期间, 漂白后, lamellipodium 突出向前。由于漂白的 EGFP-虚拟蛋白在这些部位被非漂白分子回收, 观察到荧光的逐渐恢复 (图 2b)。在图 2c中可以看到以这种方式获得的酶恢复曲线, 并将其规范化为前漂白强度 (表示为 1)。漂白效率可以变化, 约20% 的价值之前漂白在这个例子, 从价值确定从 t0 (第一帧后漂白)。荧光的增加到达一个高原在例子显示在大约80% 荧光在漂白之前。在静态结构中, 在实验的时间过程中, 如焦粘度, 恢复后达到的前漂白强度和高原荧光的差异定义为不动分数 (如果,图 2中的红色箭头)c、e), 而在漂白和完全恢复时间之间恢复的荧光量被定义为移动分数 (图 2c、e中的绿色双头箭头)。请注意, 在动态变化的结构, 如在这里分析的 lamellipodium 尖端, 其范围可能不仅代表不动分子, 但也从降低的凸出速度, 因为 EGFP 虚拟强度是众所周知的依赖于此参数18。要计算恢复的半时间, 在斯格码绘图 (图 2d) 上创建了一个拟合曲线。在这种情况下, 从解决等式 2中提取的 "b" 参数值等于 0.0754, 当应用到对数函数 (等式 4) 时, 将导致估计的恢复时间为9.19 秒 (图 2d, 远右面板), 在这个特定的单元格中相对较快, 这与以前发布的平均5相比。必须指出的是, 在同一人群中, 恢复半衰期有时会有很大差异。因此, 为了获得具有代表性的结果, 我们建议将此参数确定为至少15-20 个单元格的平均值。为了说明方差的程度, 生成了每个时间点平均从15个单元 EGFP 虚拟恢复的算术方法 (图 2e), 并以类似的方式创建和显示平均曲线匹配 (图 2f)。
lamellipodial 肌动蛋白网络的聚合速率包括正向网络凸出和逆行流的总和。酶可用于测量肌动蛋白聚合速率的转染细胞 (在这种情况下 B16-F1) 与 EGFP 标记β肌动蛋白和漂白一个突出的 lamellipodial 区域 (图2 g).为分析 lamellipodial 肌动蛋白网络聚合, EGFP 标记的β肌动蛋白漂白后的荧光恢复是随着时间的推移进行评估的。由于肌动蛋白单体的聚合进展在 lamellipodial 肌动蛋白花丝的有刺的末端 (所有指向前面20), 网络经常移位后方和进步批转, 速度可以容易地通过漂白上的荧光极化恢复获得。当漂白带到达 lamellipodium 后部与薄板的过渡区后, lamellipodium 的荧光恢复就完成了, 其特征是水平排列的纤维束密度较低。比 lamellipodium 中观察到的要慢得多。如图 2g所示, 荧光恢复可以被可视为水平线向边缘的直线, 向后流向薄板, 这允许测量凸出和逆行流的距离 (单独表示在图 2g的最右侧面板中, 分别为橙色和红色双头箭头。
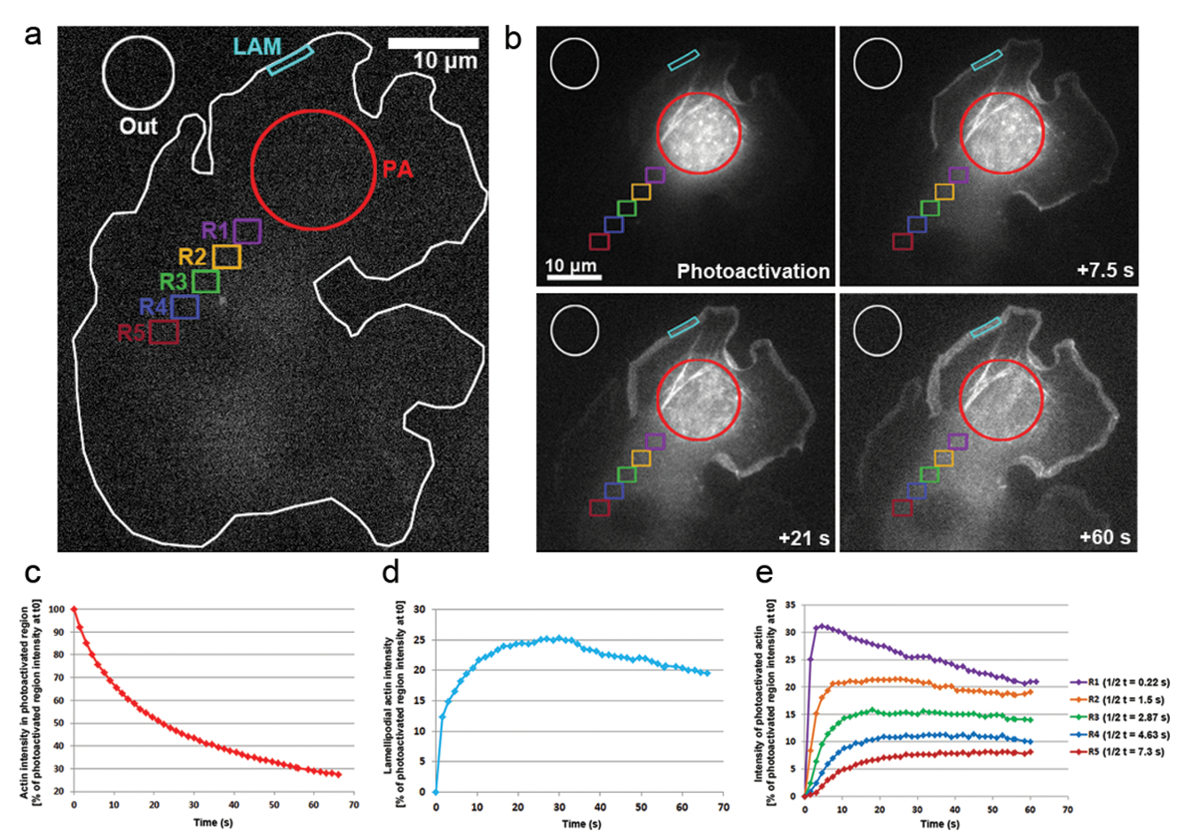
我们还应用 photoactivation 在 B16-F1 细胞转染的 PA-GFP-肌动蛋白, 以跟踪肌动蛋白单体在细胞质的流动性和其在突出 lamellipodia 的比例。如图 3a、b所示, 一个胞浆区域通过暴露于 405 nm 激光光活化作用, 而在 gfp 通道上每1.5 秒获取图像, 以可视化 gfp 标记的光活化作用肌动蛋白的分布。光活化作用 GFP-肌动蛋白在图 3 b 中的胞浆区域中可见扩散.光活化作用胞浆区的荧光强度下降率表现为 t0 (photoactivation 后第一帧) 初始强度的百分比;图 3c). 光活化作用肌动蛋白也集成在 lamellipodia 的提示, 其中新的肌动蛋白单体添加到增长的刺状的两端拉长肌动蛋白花丝在突出。为了估计 lamellipodial 的速率, 我们测量了一个二维轮廓/区域约5µm 宽度和1µm 高度的荧光强度;该地区在 lamellipodium 的尖端不断被重新定位。肌动蛋白在 t0 (图 3d) 中代表了光活化作用胞浆区域的荧光强度百分比。随着肌动蛋白纤维的伸长, 新的肌动蛋白单体在 lamellipodial 前被纳入。这些肌动蛋白单体的一小部分是随机衍生的胞浆池, 其中单体被光活化作用。这导致 lamellipodia 在 photoactivation 后的第一二十年代, 荧光的快速增加。随着新的单体被添加到 lamellipodial 前面, 以前加入的肌动蛋白单体流动与花丝向薄板逆行流动。随着时间的推移, ROI 是完全充满了荧光单体和一个高原在荧光达到 (图 3d)。然后观察到, 当光活化作用单体在整个细胞中扩散后, 荧光的逐渐下降, 非光活化作用肌动蛋白单体越来越多地被重新添加到 lamellipodial 的前方。荧光的减少将发现一个新的高原, 当平衡在光活化作用和非光活化作用单体之间的整个细胞被到达 (没有显示的数据) 时, 将很快到达。
肌动蛋白单体在整个细胞质的流动性是通过测量在光活化作用区域远端位置相同大小的区域的荧光强度而得出的 (举例说明在图 3 a 上的颜色编码区域标记为R1-R5)。如图 3e所示, 这些区域中的荧光强度逐渐减少, cytosolically 光活化作用区域, 因为光活化作用肌动蛋白单体的分数变得越来越稀释非活化 (即,非荧光) 单体。此外, 荧光的峰值是在以后达到的: 测量的区域从光活化作用区域的距离越远, 肌动蛋白单体扩散到这些区域所需的时间就越长。通过对到达荧光高原的半时间进行量化, 可以得出一个代表肌动蛋白单体渗入每个区域的程度的代表性值。该区域越远, 光活化作用肌动蛋白扩散到它所需的时间越长, 因此要达到的荧光平台需要更多的时候, 最终导致更高的t 1/2 值(图 3 e).

图 1: 成像室组件和显微注射过程.(a) 成像室组件。(b) 硅脂在塑料封口机的开口周围小心涂抹。(c) 盖玻片位于成像腔开口中心的单元侧。(d) 通过将塑料封口机放置在盖玻片的顶部并拧紧侧面夹具来建立安全密封。(e) 显微镜介质被 pipetted 到腔槽中。(f) 成像室位于显微镜阶段, 热检测器和电极连接到加热单元预设置为37°c, 并且允许细胞在显微镜开始前至少适应30分钟。在这个例子中, 显微镜阶段也配备了机器人执行 microinjections, 和注射针被浸入介质覆盖细胞层在成像室。(g) NIH3T3 成纤维细胞在显微注射前通过相衬显微术进行可视化。红十字在核周车厢表明未来微注射的地点, 对应于高细胞质区域由于接近大核。(h) 10 分钟后, 微注射与 Rac1, 细胞的反应由 lamellipodia 的显著形成周围的整个细胞周围 (由绿色箭头指示)。请单击此处查看此图的较大版本.
{kind=link}

图 2:酶允许确定蛋白质周转率或 lamellipodial 肌动蛋白聚合.(a) 代表在 lamellipodial 区域漂白前表示 EGFP 虚拟的 B16-F1 细胞的典型例子。不同颜色的轮廓/形状被标记, 以表明哪些区域被考虑为荧光强度测量随着时间的推移。注意标记有感叹号的红色轮廓, 它标签的胞浆区域位于包含多个囊泡和细胞表面褶边的区域内。在选择荧光参考区域时应避免这样的动态区域, 因为它们的特点是荧光的短期波动很强, 可能导致不准确的结果。(b) EGFP 虚拟在漂白前后表达细胞的 Lamellipodial 区域。在紫色标记的区域内漂白后的荧光信号恢复是随着时间的推移而可视化的。箭头表示 microspike 的尖端, 虚拟可能由于肌动蛋白花丝聚合的高密度而富集19。(c) 酶恢复曲线的一个示例, 它从量化 photobleached lamellipodium (紫色轮廓) 的荧光强度到 b. 右侧红色和绿色线条分别表示不动和移动分数。(d) c (左面板) 中的酶恢复曲线的适当性, 以及用于派生恢复半时间 (右面板) 的计算方法的示例。(e) 以15个细胞的荧光恢复曲线为平均值的酶恢复曲线的一个例子, 用 SEM 条表示样本种群内的变异性程度。(f) 从平均酶恢复曲线适合15个单元格 (左面板) 的曲线拟合, 以及用于推导恢复半时间 (右面板) 的计算方法示例。(g) 显示 EGFP 标记β肌动蛋白的 B16-F1 细胞在 lamellipodial 区域漂白之前和之后 lamellipodium 的时间推移板, 随后在 lamellipodium 随着时间的推移荧光恢复。在最右边的面板上, 为凸出和逆行距离测量的值 (分别为橙色和红色)。在图像面板下进行计算, 揭示了 lamellipodial 肌动蛋白网络的聚合速率与凸出和逆行距离的总和。请单击此处查看此图的较大版本.
{kind=link}

图 3:Photoactivation 在整个细胞内进行单体跟踪的 PA-GFP 肌动蛋白.(a) 一个代表 B16-F1 细胞的典型例子, 表达 pa-GFP 肌动蛋白, 在触发 photoactivation 之前, 在胞浆区域 (pa) 表示。不同颜色的轮廓标记, 以表明哪些区域被考虑为荧光强度测量随着时间的推移。(b) 说明 photoactivation 后的 PA-GFP 肌动蛋白的时间分布。注意在光活化作用、胞浆区 (红圈) 中荧光的逐渐减少, 因为光活化作用肌动蛋白扩散远离它。由于其扩散到前端和组装到网络, 光活化作用肌动蛋白单体逐渐增强, 在 lamellipodia (青色地区) 和整个细胞质 (不同的颜色编码区域) 的距离和时间依赖的方式。(c) 代表, 光活化作用胞浆区内荧光的时间下降 (b 中的红色轮廓)。(d) lamellipodial 区域荧光强度的时间变化 (b 中青色轮廓)。(e) 曲线代表了胞浆区域荧光强度的时间变化 (在 b 中的颜色编码), 因为定位于 photoactivation 区域的可变距离。注意到达荧光高原的一半时间 (在右侧的图例中表示) 随着给定区域与 photoactivation 区域的距离增加, 可能与肌动蛋白单体扩散所需的增加时间相关。各自区域。请单击此处查看此图的较大版本.
{kind=link}
讨论
在这里, 我们讨论了本文所描述的技术中的关键步骤, 以及如何在不同的实验条件下优化它们的应用。
显微注射是一种方法, 可用于监测细胞的即时效应, 从引进外源蛋白, 抑制剂, 或药物。它可以特别有利于确定蛋白质的功能在困难的染细胞类型或在情况下, 长期表达不需要。必须指出的是, 某些细胞类型的存活取决于它们所播种的细胞外基质。大多数内皮, 上皮, 或成纤维细胞类, 甚至小的像鱼基质 (见当et 等人21和安德森和交叉22) 可以成功地注入。然而, 也有一些例外, 例如在层粘连蛋白上播种的 B16-F1 细胞, 它构成了一个优良的细胞迁移模型系统, 但与这种类型的基质注射不相容, 原因不明。对于 NIH3T3 成纤维细胞, 我们通常在纤维粘连蛋白基底上进行注射, 以及其他 photomanipulation 技术, 如酶 (即使有 photoactivation; 在此显示的 B16-F1 细胞) 可以同样良好地执行在这些成纤维细胞 (见例如,Köstler et 。3). 还必须考虑到, 不同的蛋白质, 根据其功能特性和实验目标, 可能需要不同的时间来引起变化, 从几秒钟到几小时不等。该技术的优点是, 在使用质粒转染时, 在单细胞水平比例如更精确地控制外源剂的用量/浓度。此外, 荧光标记的蛋白质是不必要的, 以保证其存在的细胞, 这可以增加灵活性, 如果同时多通道可视化其他荧光标签的蛋白质是必需的。显微注射对于分析特定蛋白质或蛋白质混合物对细胞形态或细胞骨架的动态变化的即时影响特别有用 (例如,. 21 Arp2/3 复合抑制剂 Arpin 对迁移的即时影响示例)。技术的缺点是它的侵袭性, 可能导致细胞损伤或影响细胞形态学。因此, 在执行 microinjections 时, 一个重要的考虑因素是监测细胞的生存能力。此处介绍的方法依赖于手动操作。在测试的条件, 以配合成功的注射, 如成纤维细胞生长在纤维连接层, 这里所描述的手动注射协议允许近100% 成功率;这是必要的, 当结合这种方法与复杂和耗时的后续实验, 包括视频显微镜或酶, 如以前发布的3。这并不排除偶尔, 个别细胞可能会遭受微注射事件, 这可以被安全地识别的突然变化, 细胞核和细胞质的对比, 其次是细胞边缘退缩。这样罕见的实验性案例被排除并且因而不考虑为进一步分析。
然而, 半自动方法也通常被使用, 例如使用快速 (< 300 ms) 机器控制的针降低与射入压力增量重合, 因此针只必须在每个细胞之上被安置在各自之前注射。半自动注射的成功率低于上面描述的人工方法, 这仅仅是因为它对速度进行了优化, 接着分析了成功地在这种治疗中幸存下来的多个细胞;因而它不依靠成功的射入单独细胞。因此, 相对于单细胞分析, 半自动注射更适合分析数个百细胞的注射效果,例如,通过视频显微镜在低放大率或在细胞固定和染色。无论采用何种详细的方法, 显微注射都不构成终点化验, 但可以结合多种技术, 包括酶或 photoactivation3。
当通过酶确定蛋白质周转率时, 必须优化激光强度, 具体取决于显微镜的设置和成像条件 (放大、目标、等, 以及细胞类型、结构和荧光蛋白等。漂白)。请注意, 在最佳激光功率下, 高效漂白与最小可能的光损伤相结合, 以避免在分析 (例如, lamellipodia 或丝状伪足) 中的结构收缩或完全收缩, 甚至在细胞层面上造成损害。理想的情况是, 至少70–80% 的漂白效率应该达到, 虽然完全漂白可能会受到极快的蛋白质周转的阻碍, 在这种情况下, 任何50% 以上的东西也可以接受。对给定结构和荧光染料的最佳漂白功率应进行实验测试, 从低激光功率开始, 然后逐渐增加。当然, 任何荧光染料可以根据定义被漂白与激光接近其峰值的励磁 (488 毫微米为常用的绿色染料, 如 FITC 或 EGFP)。然而, 波长较短的激光, 如近紫外激光器, 提供了更高的功率, 因此也可用于有效漂白常用染料。我们通常使用405纳米二极管激光器 (120 兆瓦) 漂白的 EGFP 和红色荧光染料 (如 mCherry), 虽然其效率略低的情况下 (不显示数据)。由于405纳米二极管也可以用于 photoactivation (见下文), 它赋予这个系统最大的灵活性。
对于 B16-F1 细胞结构和荧光蛋白 photobleached, 在65–100兆瓦之间应用了405纳米激光功率。在分析 photobleached 区域时, 重要的是要考虑给定的结构是否保留在其原始形状上。例如, 在分析 lamellipodia 尖端蛋白质的周转时, 应注意 lamellipodia 的曲率是否随着时间的推移而显著改变, 因为如果分析的区域/轮廓不完全包含整个结构在每个被测量的框架。此外, 应该指出, 嵌入到 lamellipodia 中的捆绑, 如 microspikes, 可能会导致荧光强度的偏差。如图 2b (在9的时间帧中为白色箭头) 所示, 一个类似 microspike 的结构位于测量的 photobleached 区域的旁边, 但在整个测量期间仍处于外部, 因此不会导致任何误差。对于蛋白质周转的分析, 在选择分析区域的位置和大小时, 重要的考虑因素是它们的荧光随时间变化不应受到细胞形态学或其它因素的改变的显著影响, 而不是很难避免购置漂白。例如, 对所分析结构提供大量定量贡献的结构, 在分析过程中不应脱离实测区域;此外, 不相关的, 荧光实体, 如水泡结构, 吸引蛋白质不应该进入兴趣领域的分析。为确定 lamellipodial 肌动蛋白聚合的速率, 应注意不要对缩回或梳理 (即,向上折叠) lamellipodia 进行分析, 因为这将强烈影响结果的准确性。此外, lamellipodial 地区的退缩可能会出现快速向后移位, 有可能导致高估 lamellipodial 肌动蛋白聚合速率。另外考虑的是细胞内正常化区域的距离 (作为获取漂白校正的参考位置) 从漂白的实际位置, 应该足够大, 以避免直接photobleached 地区的影响。
在建立 photoactivation 绿色荧光标记结构的最佳条件时, 应注意避免 photoactivation 中的瞬时漂白。在我们的工作中, 获得的最佳结果是激光功率低于通常使用的 EGFP 漂白的5-10 倍。对于光活化作用分子的图像获取, 应通过考虑要光活化作用和分析的区域和结构的大小以及光活化作用的潜在移动性来优化帧间的曝光时间和时间间隔。蛋白质到其他亚细胞位置。对于所有类型的荧光成像, 维持细胞的生存能力是至关重要的获得生理相关的结果。
原则上, 荧光蛋白 (如 mEos 或 Dronpa 变体) 的绿-红 photoconversion 是一种同样强大的方法, 跟随 lamellipodium 的亚细胞结构的动态和更替 (见 例如,Burnette et 。23). 后一种方法相对于 PA-GFP 的优势是在转换前后采用两种不同颜色的蛋白质动力学的可能性, 而不需要共同表达额外的红色荧光蛋白。然而, 在我们的初步实验中, 与 photoconverted 探针相比, 在 photoactivation 上实现的荧光信号的对比度变化和强度更大, 可能是由于绿色与红色的优越光谱特征。荧光探针 (未显示数据)。在任何情况下, 对肌动蛋白长丝周转的详细研究, 如 lamellipodia 或痘苗病毒诱导的肌动蛋白的尾巴迄今只发表了使用 PA-GFP 衍生物5,6,24。
在考虑哪一个细胞区域来分析以下 photoactivation 时, 应该考虑几个因素, 这是用这里所示的具体例子来讨论的 (在细胞质激活后, 在细胞边缘加入肌动蛋白单体), 但当然可以推断出各种类似的科学问题。首先, 当测量 cytosolically 光活化作用蛋白的 lamellipodial 合并率时, 例如, 在不同的实验条件下 (如Dimchev et 所示)。6), 胞浆区域的大小及其与 lamellipodial 边缘的距离应在实验组之间比较。同样重要的是, 当 photoactivating 胞浆区域时, 细胞厚度在靠近细胞核的位置更大。激活较厚的细胞区域可能导致更多的活化蛋白, 因为在细胞质中 homogenously 分布的蛋白质的分布。最后, 被激活的蛋白质的表达水平在个体细胞肯定是高度可变的。由于所有这些可变性的考虑, 重要的是要比较在细胞中其他地方的 cytosolically 活化蛋白相对于在特定区域激活后获得的总荧光的合并水平。
我们已经描述了如何使用显微注射作为一个工具, 以调查蛋白质对细胞形态学的影响, 并表明了这一点, 证明了有效诱导 lamellipodial 结构在 NIH3T3 成纤维细胞杏仁与小 GTPase Rac1。我们以前应用这一技术来干扰细胞中的 Arp2/3 功能杏仁与 C 终端 WCA 领域的疤痕/波3。杏仁细胞中的各种参数可以通过其他的化验方法来分析, 如酶或 photoactivation。我们描述了酶和 photoactivation 如何用于研究肌动蛋白单体的亚细胞动力学和流动性。我们的小组以前使用过酶5来调查定位到 lamellipodia 的蛋白质的更替情况, 如虚拟、Abi、cortactin、cofilin 和上限蛋白, 或用于阐明在存在中的病灶粘连中成分的更替情况。和没有 Rac 信号4。此外, 测量肌动蛋白聚合速率可以通过漂白 EGFP 标记的β肌动蛋白5来完成, 但存在替代方法。跟踪荧光不均匀性的实时细胞成像-兼容探针标记细胞肌动蛋白花丝,如Lifeact25, 也可以使用6, 26.这里的优点是可以避免β肌动蛋白的过度表达, 它能够增加细胞边缘突起和迁移, 从而可能干扰特定的化验或实验问题 (参见例如,凯奇et 。26;Peckham et 。27). 然而, Lifeact 探针的一个明显的缺点是它的快速的开/关动力学与肌动蛋白花丝结合, 因此, 在细胞中标记的肌动蛋白长丝结构的漂白只提供探针更替的信息,但不是肌动蛋白花丝的营业额, 它绑定25。以前使用的荧光不均匀性的跟踪6, 26 确实提供了一个实际的折衷, 类似于广泛使用的跟踪荧光斑点纳入丝状骨架结构 (请参见例如鲑鱼和酶28), 但可能不像 EGFP 标记的 F 肌动蛋白结构那样直接使用和精确。Photoactivation 已经被我们应用于估计单体肌动蛋白在凸 lamellipodia 中的比例, 以及它在细胞质的移动, 在实验性地调整胞浆 F 肌动蛋白水平6。该技术在检查从相对较大的区域 (如胞浆区域) 衍生的蛋白质的流动性和分布方面是有用的。然而, 研究从相对较小的光活化作用结构中提取的蛋白质的分布;例如,由于激活的荧光分子数量少, 信号微弱, 因而缺乏灵敏度, 因此生长锥可能具有挑战性。荧光 photoactivation 或 photoconversion 的潜在替代技术 (见上文) 可能包括逆酶, 它依赖于漂白整个细胞, 除了 ROI, 然后跟踪荧光分子的流动性远离这个地区。该技术不需要 overexpressing photoactivatable 版本的蛋白质, 但将始终涉及暴露在异常高剂量的激光功率, 可能导致不必要的副作用, 如光损伤。
显然, photoactivation 和酶不能区分蛋白质是否作为单体、脂肪酸、甚至小的低聚物移动, 以及它们是否与附加的结合性伙伴相结合。这类信息可以通过荧光相关光谱学技术 (29 ) 来获得, 或者是电影-烦恼30。尽管如此, 酶和 photoactivation 是直接评估细胞内局部和全球蛋白质动态的简单方法, 而不管感兴趣的蛋白质、亚细胞位置或研究的类型。
披露声明
作者没有什么可透露的。
致谢
我们感谢德国研究基金会 (DFG) 提供财政支持 (RO2414/5-1 给 KR)。
材料
| Name | Company | Catalog Number | Comments |
| B16-F1 mouse skin melanoma cells | American Type Culture Collection, Manassas, VA | CRL-6323 | |
| NIH-3T3 cells | American Type Culture Collection, Manassas, VA | CRL-1658 | |
| DMEM 4.5g/L glucose | Life Technologies, Thermno Fisher Scientific, Germany | 41965-039 | |
| Ham’s F-12 medium | Sigma-Aldrich | N8641 | |
| Fetal calf serum (FCS) | PAA Laboratories, Linz, Austria | A15-102 | |
| Fetal bovine serum (FBS) | Sigma-Aldrich, Germany | F7524 | Lot054M3396 |
| MEM Non essential amino acids | Gibco, ThermoFisher Scientific, Germany | 11140035 | |
| L-Glumatine 200mM (100x) | Life Technolgies | 25030-024 | |
| Pen-Strep 5000 U/mL | Life technologies | 15070063 | |
| Sodium Pyruvate (100 mM) | Gibco, ThermoFisher Scientific, Germany | 11360-039 | |
| Laminin | Sigma-Aldrich | L-2020 | |
| Laminin coating buffer | Self-made: 50mM Tris ph7.4, 150mM NaCl | ||
| Fibronectin from human plasma | Roche Diagnostics, Mannheim, Germany | 11 051 407 001 | |
| Jetpei | Polyplus Transfection, Illkirch, France | 101-10N | |
| JetPei buffer | Polyplus Transfection, Illkirch, France | 702-50 | 150mM NaCl |
| PA-GFP-actin plasmid DNA | described in Koestler et al.2008 | ||
| pEGFP-actin plasmid DNA | Clontech, Mountain View, CA, USA | ||
| Rac1 protein for microinjection | Purified as GST-tagged version, and cleaved from GST prior to injection | ||
| Microinjection buffer | Self-made: 100mM NaCl, 50mM Tris-HCl ph7.5, 5mM MgCl2, 1mM DTT | ||
| Dextran, Texas Red, 70,000 MW, Lysine Fixable | Molecular Probes, Thermno Fisher Scientific, Germany | D1864 | |
| Microscope circular cover glasses 15mm, No.1 | Karl Hecht, Aisstent, Sondheim, Germany | 1001/15 | |
| Eppendorf Femtotips Microloader Tips | Eppendorf, Hamburg, Germany | 5242 956 003 | |
| Eppendorf Femtotip Microinjection Capillary Tips | Eppendorf, Hamburg, Germany | 930000035 | |
| Silicone Grease | ACC Silicones, Bridgewater, England | SGM494 | |
| Aluminium Open Diamond Bath Imaging Chamber | Warner instruments | RC-26 | |
| Automatic temperature controller | Warner Instruments | TC-324B | |
| Microscope: Axio Observer | Carl Zeiss, Jena, Germany | ||
| CoolSnap-HQ2 camera | Photometrics, Tucson, AZ | ||
| Lambda DG4 light source | Sutter Instrucment, Novato, CA | ||
| Laser source | Visitron Systems | ||
| Eppendorf FemtoJet microinjector | Eppendorf, Hamburg, Germany | With built-in compressor for pressure supply | |
| Nikon Narishige Micromanipulator system | Nikon Instruments, Japan | ||
| Visiview software v2.1.4 | Visitron Systems, Puchheim, Germany | ||
| Metamorph software v7.8.10 | Molecular Devices, Sunnyvale, CA | ||
| Sigma Plot v.12 | Systat Software Inc. |
参考文献
- Day, R. N., Davidson, M. W. The fluorescent protein palette: tools for cellular imaging. Chem Soc Rev. 38 (10), 2887-2921 (2009).
- Ishikawa-Ankerhold, H. C., Ankerhold, R., Drummen, G. P. Advanced fluorescence microscopy techniques--FRAP, FLIP, FLAP, FRET and FLIM. Molecules. 17 (4), 4047-4132 (2012).
- Koestler, S. A., et al. Arp2/3 complex is essential for actin network treadmilling as well as for targeting of capping protein and cofilin. Mol Biol Cell. 24 (18), 2861-2875 (2013).
- Steffen, A., et al. Rac function is crucial for cell migration but is not required for spreading and focal adhesion formation. J Cell Sci. 126, 4572-4588 (2013).
- Lai, F. P., et al. Arp2/3 complex interactions and actin network turnover in lamellipodia. EMBO J. 27 (7), 982-992 (2008).
- Dimchev, G., et al. Efficiency of lamellipodia protrusion is determined by the extent of cytosolic actin assembly. Mol Biol Cell. 28 (10), 1311-1325 (2017).
- Koppel, D. E., Axelrod, D., Schlessinger, J., Elson, E. L., Webb, W. W. Dynamics of fluorescence marker concentration as a probe of mobility. Biophys J. 16 (11), 1315-1329 (1976).
- Patterson, G. H., Lippincott-Schwartz, J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 297 (5588), 1873-1877 (2002).
- McKinney, S. A., Murphy, C. S., Hazelwood, K. L., Davidson, M. W., Looger, L. L. A bright and photostable photoconvertible fluorescent protein. Nat Methods. 6 (2), 131-133 (2009).
- Gurskaya, N. G., et al. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol. 24 (4), 461-465 (2006).
- Lippincott-Schwartz, J., Patterson, G. H. Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging. Trends Cell Biol. 19 (11), 555-565 (2009).
- Kremers, G. J., Piston, D. Photoconversion of purified fluorescent proteins and dual-probe optical highlighting in live cells. J Vis Exp. (40), (2010).
- Fischer, A. H., Jacobson, K. A., Rose, J., Zeller, R. Preparation of slides and coverslips for microscopy. CSH Protoc. 2008, 4988 (2008).
- Small, J. V., Rottner, K., Carlier, M. F. . Actin-based Motility. , (2010).
- Kaverina, I., et al. Enforced polarisation and locomotion of fibroblasts lacking microtubules. Curr Biol. 10 (12), 739-742 (2000).
- Small, J., Rottner, K., Hahne, P., Anderson, K. I. Visualising the actin cytoskeleton. Microsc Res Tech. 47 (1), 3-17 (1999).
- Mikhailov, A. V., Gundersen, G. G. Centripetal transport of microtubules in motile cells. Cell Motil Cytoskeleton. 32 (3), 173-186 (1995).
- Rottner, K., Behrendt, B., Small, J. V., Wehland, J. VASP dynamics during lamellipodia protrusion. Nat Cell Biol. 1 (5), 321-322 (1999).
- Svitkina, T. M., et al. Mechanism of filopodia initiation by reorganization of a dendritic network. J Cell Biol. 160 (3), 409-421 (2003).
- Small, J. V., Isenberg, G., Celis, J. E. Polarity of actin at the leading edge of cultured cells. Nature. 272 (5654), 638-639 (1978).
- Dang, I., et al. Inhibitory signalling to the Arp2/3 complex steers cell migration. Nature. 503 (7475), 281-284 (2013).
- Anderson, K. I., Cross, R. Contact dynamics during keratocyte motility. Curr Biol. 10 (5), 253-260 (2000).
- Burnette, D. T., et al. A role for actin arcs in the leading-edge advance of migrating cells. Nat Cell Biol. 13 (4), 371-381 (2011).
- Humphries, A. C., et al. Clathrin potentiates vaccinia-induced actin polymerization to facilitate viral spread. Cell Host Microbe. 12 (3), 346-359 (2012).
- Riedl, J., et al. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 5 (7), 605-607 (2008).
- Kage, F., et al. FMNL formins boost lamellipodial force generation. Nat Commun. 8, 14832 (2017).
- Peckham, M., Miller, G., Wells, C., Zicha, D., Dunn, G. A. Specific changes to the mechanism of cell locomotion induced by overexpression of beta-actin. J Cell Sci. 114, 1367-1377 (2001).
- Salmon, E. D., Waterman, C. M. How we discovered fluorescent speckle microscopy. Mol Biol Cell. 22 (21), 3940-3942 (2011).
- Machan, R., Wohland, T. Recent applications of fluorescence correlation spectroscopy in live systems. FEBS Lett. 588 (19), 3571-3584 (2014).
- Becker, W. Fluorescence lifetime imaging--techniques and applications. J Microsc. 247 (2), 119-136 (2012).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。