
Method Article
基于滴瓣的成年哺乳动物组织单细胞转录瘤
* 这些作者具有相同的贡献
摘要
该协议描述了为基于液滴、高通量的单细胞 rna-seq 制剂准备健康的成年哺乳动物单细胞所需的一般过程和质量控制检查。还提供了测序参数、读取对齐和下游单细胞生物信息学分析。
摘要
分析组织或微环境中数千个单个细胞的单个细胞基因表达是识别细胞组成、功能状态的判别以及观察到的组织背后的分子途径的宝贵工具功能和动物行为。然而, 分离完整的, 健康的单个细胞从成年哺乳动物组织, 以便随后下游单细胞分子分析可能是具有挑战性的。该协议描述了从神经系统或皮肤获得高质量成人单细胞制剂所需的一般过程和质量控制检查, 从而能够进行后续的无偏见单细胞 rna 测序和分析。还提供了下游生物信息学分析的指南。
引言
随着高通量单细胞技术 1、2 的发展和用户友好生物信息学工具在过去十年3的进步, 高分辨率基因表达分析的一个新领域已经出现--单细胞 rna 测序 (scrna-seq)。对单个细胞基因表达的研究最初是为了识别定义的细胞群 (如干细胞或癌细胞) 中的异质性, 或者是为了识别 4、5 细胞中罕见的细胞群, 这些细胞是无法使用传统的块状 rna 测序技术。生物信息工具使人们能够识别新的亚群 (seurat)2, 可视化沿 psuedotime 空间 (monocle)6的细胞顺序, 定义种群内或种群之间的主动信号网络 (场景)7, 预测在人工3d 空间 (seurat 等) 中组装单个细胞的情况.随着这些新的和令人兴奋的分析提供给科学界, scrna-seq 正在迅速成为基因表达分析的新标准方法。
尽管 scrna-seq 具有巨大的潜力, 但生成干净的数据集和准确解释结果所需的技术技能对新来者来说可能具有挑战性。这里提出了一个基本但全面的协议, 从从从整个主要组织分离单个细胞到可视化和表示用于发布的数据 (图 1)。首先, 健康的单个细胞的分离可以被认为是具有挑战性的, 因为不同的组织对酶消化和随后的机械离解的敏感性程度各不相同。此协议在这些隔离步骤中提供指导, 并在整个过程中确定重要的质量控制检查点。其次, 了解单个单元技术和下一代测序技术之间的兼容性和要求可能会造成混淆。该协议提供了实现用户友好的、基于液滴的单细胞条形码平台和执行测序的指南。最后, 计算机编程是分析单细胞转录数据集的重要前提。此协议提供了开始使用 r 编程语言的资源, 并提供了有关实现两个流行的 srna-seq 特定 r 包的指导。该协议可以共同指导新来者执行 scrna-seq 分析, 以获得清晰、可解释的结果。该协议可以根据小鼠的大多数组织进行调整, 重要的是可以修改为与包括人体组织在内的其他生物一起使用。根据组织和使用者的不同进行调整。
在遵循此协议时, 需要记住几个注意事项;包括, 1) 建议遵循本协议第1步和第2步中的所有质量控制准则, 以确保感兴趣样本中所有细胞的单细胞悬浮, 同时确保准确的细胞总数计数 (图2概述) ).一旦实现了这一点, 如果遵循了所有优化的条件, 质量控制步骤就可以放弃 (以节省时间--保持 rna 质量和减少细胞丢失)。在任何下游处理之前, 强烈建议您确认成功地从感兴趣的组织中分离出高活性的单细胞。2) 由于某些细胞类型对压力比其他细胞更敏感, 过度分离技术会无意中偏向人群, 从而混淆下游分析。在没有不必要的细胞剪切和消化的情况下, 温和的分离对于实现高细胞产量和组织组成的准确表示至关重要。剪切力发生在三化、流式细胞仪和再悬浮步骤过程中。3) 与任何 rna 工作一样, 在制备过程中, 最好在样品中引入尽可能少的额外 rnase。这将有助于保持高质量的 rna。使用核糖核酸酶抑制剂溶液与冲洗清洁工具和任何设备, 不是无 rnase, 但避免 depc 处理的产品。4) 尽快做好准备。这将有助于维持高质量的 rna 和减少细胞死亡。根据组织解剖长度和动物数量, 考虑同时开始多项解剖/制剂。5) 尽可能在冰上准备细胞, 以保持高质量的 rna, 减少细胞死亡, 减缓细胞信号和转录活动。尽管, 冰凉处理是大多数细胞类型的理想选择, 但某些细胞类型 (如中性粒细胞) 在室温下加工时表现更好。6) 在细胞制备过程中避免使用钙、镁、edta 和 depc 处理的产品。
研究方案
这里描述的所有协议都符合卡尔加里大学动物护理委员会的规定并得到其批准。
1. 分离组织 (第1天)
- 对过量服用戊巴比妥钠 (即 50 mg/kg) 或根据动物伦理协议酌情对小鼠进行安乐死。然后从老鼠的背部和腿部取出不需要的头发, 并对解剖区域进行乙醇消毒。
- 解剖感兴趣的组织或微环境。对于这个方案, 我们使用皮肤和神经组织来证明成人组织离解后, 液滴巴线基单细胞转录体的普遍性。
- 对于坐骨神经, 使用斯特拉顿等人发现的详细协议。9. 简单地说, 将皮肤从老鼠后腿的后部剪掉。用无菌手术刀刀片沿大腿长度做一个切口。使用精细的钳子和剪刀暴露和删除坐骨神经。
- 对于背部皮肤, 使用 biernaskie 等人的详细协议.10. 简单地说, 用细钳和剪刀从肩部到肩部, 穿过臀部和背部切口, 解剖背部背部皮肤。使用无菌手术刀片将皮肤切成薄片 (厚度0.5 厘米)。
- 用冰凉的 hbss 清洗组织 2次, 并在解剖显微镜下去除不需要的结缔组织、脂肪沉积物或碎片。
- 仅在皮肤真皮上, 在37°c 下将切片漂浮在 hbss 中的消示酶 (5 mgml, 5 uml) 中30-40分钟。手术将表皮与真皮分离。如果有兴趣, 丢弃表皮或使用胰蛋白酶进一步离解。
- 用无菌手术刀片将样品切成1-2 毫米, 并放入新鲜解冻的 2 mgml 冷胶原酶-iv 酶 (2 mg/ml, 125 cmu/mg, 在 f12 培养基中)。
- 对于神经, 每2个坐骨神经使用 ~ 500μl。对于皮肤, 使用 ~ 8 毫升每1x 鼠标背部皮肤。
注: 组织应完全淹没在胶原蛋白-iv 溶液中。至关重要的是, 任何消化酶都要得到适当的处理、储存和准备。如果酶长时间留在室温下, 单细胞分离将需要过度的机械三化, 并降低细胞的活力。胶原蛋白-iv 也可以在细胞活力最理想的细胞培养培养基中组成。然而, 这可能会改变酶活性或转录签名, 因此应该由用户进行优化。
- 对于神经, 每2个坐骨神经使用 ~ 500μl。对于皮肤, 使用 ~ 8 毫升每1x 鼠标背部皮肤。
- 在37°c 的浴缸中将酶中的样品加氢 30分钟, 每10分钟轻轻摇晃一次。放置在37°c 的振动台也是一个合适的选择。
- 在30分钟后酶添加时, 用 p1000 移液器进行触控20-30 次。
- 每30分钟重复一次三化, 直到溶液出现混浊, 组织块在很大程度上脱离。
注: 确保电池的完全释放 (图 2b, 2b)。为了确认完全释放, 与 blue (每1毫升2滴) 的板细胞和20分钟后, 在显微镜下进行检查, 以确保所有细胞核与单个细胞而不是碎片有关。对于每种组织类型或条件, 在给定的实验中检查细胞释放程度至关重要。在纤维化组织 (即慢性损伤) 或未受伤的成人组织中, 细胞的释放将因急性损伤或胚胎组织的不同而有很大差异。这一点尤其重要, 因为某些细胞类型从组织中释放的可能性低于其他细胞类型, 因此优先将这些细胞排除在下游分析之外。- 对于神经, 离解组织总时间 0.5-1.5 小时。对于皮肤, 分离组织总共 2小时 (在孵育的最后一个小时, 在皮肤样本中加入 dnase (1 mg/2))。
- 使用40μm 滤清器过滤两次。用冷冻滤芯冲洗过滤器 1% bsa/hbss。
- 以 260 x g 离心8分钟。然后去除上清液。
- 使用宽孔尖端在 hbss 中重新注入含有 1% bsa 的细胞颗粒, 并将其放置在冰上。再悬浮体积是根据组织体积 (800 毫克皮肤湿重 = 800μl 体积; 10 毫克湿重的神经 = 100μl 体积)。
- (可选) 从较低的重新悬挂音量开始, 然后根据流速度 (每秒事件) 在流单联分选器上根据需要进行调整。收集的最有效的排序密度 (在最小化时间的同时最大限度地增加收集的单元格数) 为3000-7000 事件/秒。
- 如果使用活力染料, 取出一个亚光剂进行无染色控制。然后使用宽孔尖端将 1:15 000 活力染料 (库存:20, 000 nm/μl) 添加到样品中 (1.3 nmμμl 最终浓度), 以减少剪切。
注: 对于每种组织类型或条件, 在给定的实验中检查细胞死亡程度是至关重要的。样品中的某些细胞类型比其他类型更容易死亡, 因此优先被排除在下游分析之外。- 在黑暗中的冰上用活力染料将样品加在盐中5-10。然后在样品中加入4毫升的冰凉 1% bsa/hbss。离心以 260 x g 8分钟去除多余的活力染料。以同样的方式对待没有活力染料的亚夸特。
2. 隔离可活健康的细胞 (第1天)
- 确保流式细胞仪设施遵循适当的荧光活化细胞分选 (流式细胞仪) 参数。
- 提前准备好 facs 机器, 以确保在步骤1中的最终离心机完成后将其准备就绪, 并确保收集隔间使用冰块保持冷。
- 使用以下参数: 流量: 1.0 (大致对应于 10μlmin);过滤器: 1.5 nd;喷嘴尺寸: 100μm;正向散射: 80-180 v (根据需要进行更改, 以区分事件的大小);侧面散射: 150-220 v (根据需要进行更改, 以区分事件的粒度/形状);激光: 100-400 v (根据需要进行更改, 以区分活力染料正与负事件 & 检查这是否不控制活力染料);门: 必要的变化, 以确保所有细胞被收集。参见图 2d-2g。
注: facs 参数高度依赖于所使用的单元格类型和分拣器, 因此需要由用户进行优化。
- 准备15毫升的窄底管, 8 毫升的冰凉 1% bsa/hbss 用于样品采集。管内静态和表面张力会影响收集效率。在收集前将管反转, 以确保液体表面与管内的界面湿润。
注: 如果使用的单元格数非常低, 请根据需要调整到小型收集容器。 - 一旦收集到所有细胞, 离心机样品在 260 x 克8分钟。
注: 离心前, 将1% 的 bsa/hbss 加入从侧面冲洗细胞, 并在流式细胞仪后立即将管混合。 - 在 1% bsa/hbss 中再置细胞颗粒, 并保持在冰上。与步骤3处理兼容的每个样品的最大体积为 33.8μl, 因此请确保最终的细胞稀释/再悬浮体积适合于获得33.8μl 中的理想细胞数。此步骤的其他稀释介质选项 (以及 1% bsa/hbss 中的所有先前稀释) 包括 dmem, 以及高达40% 的血清, 但避免使用含有钙、镁或 edta 的试剂。
- 将细胞留在冰上的时间最少。理想情况下, 在步骤2的最后一步中, 同事应准备好以下步骤 (步骤 3) 的所有设备和试剂。
- 细胞制剂关键检查
- 确认从流式细胞仪获得的细胞编号的估计。根据组织类型和离解长度, 碎片和细胞的大小和形状可能非常相似。因此, 除非使用荧光记者, 否则流式细胞仪不能排除所有碎片。建议在 facs 集合后执行最终单元格计数, 以了解给定制剂的事件百分比 (根据 facs) 实际上是多少个单元格 (图 2g)。使用血细胞计或自动细胞计数器执行细胞计数 (重复两次), 并计算由根据流式细胞仪收集的总事件表示的活细胞的百分比。
- 验证细胞的制备。验证是否不存在大颗粒 (和 gt;100 微米), 因为它们可能会堵塞下游步骤中的设备。碎片清除不足可能有可能堵塞单细胞微流体芯片。板材剩余的细胞与蓝色 (如上), 以确保大碎片片段不存在。这也将允许确认细胞是奇异的 (即不粘在一起), 让人相信下游的单细胞遗传分析代表的是单个细胞, 而不是多个细胞。
- 确定细胞编号的顺序: 每个样本有大量的成人组织衍生细胞数, 可以加载到系统中, 最多可以同时运行8个样本。作者在每个样本中加载了 500-50, 000个单元格, 并获得了高质量的 scrna-seq 数据集。有关要加载的最适当的单元号的更多讨论, 请参阅 "讨论" 部分。测序细胞数量的最终输出在很大程度上取决于分离的单细胞的质量。装载 10, 000个成人组织衍生细胞可以返回 1, 000 至 4, 000个测序细胞 (10-40% 的回报)。如果对高细胞数 (约 10, 000个细胞, 此系统推荐的最大数量) 有兴趣, 则需要加载 25, 000-100, 000个单元格。
3. 创业板 (乳化中的凝胶珠) 生成和条码 (第1天)
注: 本协议的步骤3-6 设计用于与最常见的基于微液的单细胞平台结合使用, 该平台由10倍基因组学公司制造。制造商的协议中概述了步骤3和4的详细指南 (请参阅铬单细胞 3 ' 协议)11、12, 并且必须与此协议一起遵循。为了获得最佳效果, 必须在离解 (步骤 1) 和本协议第1天的单元隔离 (步骤 2) 步骤后立即完成步骤3。
- 根据制造商的协议11、12准备芯片。这种基于微液的单细胞平台使用的技术, 采样约 750, 000个条形码, 分别索引每个细胞的转录组。这是通过在 esmul队 (gem) 中将细胞划分为凝胶珠来实现的, 在这些环境中, 生成的 cdna 共享一个共同的条形码。在 gem 生成过程中, 传递单元格的目的是使大多数 (90-99%) 生成的 gem 不包含任何单元格, 而其余的单元格在很大程度上包含单个单元格。
- 将芯片放在芯片支架上。
- 准备细胞主混合在冰上。
- 在未使用的油井中加入50% 甘油, 并将90μl 的细胞主混合物添加到 1, 90μl 的凝胶珠中, 加入到2井中, 将油分成3口。
- 用垫圈覆盖芯片。
- 加载芯片并在单细胞控制器中运行。
- 弹出纸盒, 将芯片放入托盘中, 收回托盘, 然后按"播放"。创业板中的单个细胞 3 ' 凝胶珠包括含有部分 illumina r1 序列 (读为1测序引物)、16个核苷酸 (nt) 10x 条形码、10x 唯一分子标识符 (umi) 和多 dt 引物序列的引物。在运行过程中, 控制器中的凝胶珠被释放, 并与细胞裂解液和主混合混合。
- 收集100μl 的样品并放置在 pcr 管中。
- 将 pcr 管放置在预先设定的 pcr 机器中, 并根据试剂盒运行 pcr。孵育后, gm 将包括来自多腺化 mrna 的全长、条形码 cdna。
- 运行后, 放置在-20°c 过夜, 最长可达 1周, 然后再执行下一步。
4. 清理、放大、图书馆建设和图书馆量化 (第2天之前)
注: 制造商的协议11、12中概述了步骤4的详细准则, 并且必须与此协议一起遵循。
- 利用硅烷磁珠去除创业板反应混合物中剩余的生化反应物。
- 放大全长、条形码 cdna, 以产生足够的质量用于图书馆建设。
- 评估 dna 产量。在图书馆建设之前, 评估样本的 dna 产量。这将确定在下游 pcr 步骤中使用的周期数 (库构建过程中的样本索引 pcr)。根据给定样本的 rna 含量 (可能因激活状态 (例如, 对照与受伤等)、细胞类型和细胞产量而异, 推荐的周期数量可能会发生变化。
- 对于约3000个组织源性细胞 (与活化状态无关) 的测序, 作者发现14个周期 (样本: ~ 10-100 ng nna) 是标准的。
- 使用生物分析仪进行 dna 分析。请参阅《用户指南》 13。
- 片段样本, 并选择 dna 的大小。在库构建之前, 使用酶碎片和大小选择协议来获得适当的 cdna 放大图标大小。
- 为图书馆建设准备样品。而 r1 (读为1引物序列) 在创业板孵化过程中被添加到分子中;在库建设过程中添加 p5、p7 (样本索引) 和 r2 (读为2引物序列)。
- 评估 dna 产量。大多数测序设施都需要提交最终的文库, 其中包括 dna 产量和质量信息。因此, 在完成整个协议后, 在运送到测序设施之前, 运行生物分析仪。
- 将样品存放在-80°c 下, 最长可存放2个月。
- 在测序之前, 使用 dna 定量试剂盒对样本进行量化。这可以在测序设施完成。
5. 图书馆测序 (第3天开始)
注: 本协议中使用的单细胞转录组条形码平台生成与光照兼容的付费端库, 以 p5 和 p7 序列开头和结尾。虽然解决细胞类型身份所需的最小深度可以是 10000–50000 endscyp15,16, ~100, 000 读数 sell-细胞, 作为成人体内细胞的最佳成本覆盖率权衡 (请记住一些细胞类型或最小激活的细胞状态将在 30000-50, 000 读元细胞时达到饱和度。
- 将干冰上的 cdna 文库输送到配备了适当的 ilumina 测序器的测序设施。
- 向测序设施提供以下信息:
- 提供示例详细信息: 与每个库相对应的示例索引 id;种;初级组装的基因组数据库 (即鼠标的 grcm38);显示生物分析仪碎片大小的电泳图 (200 至 9, 000 bp);cdna 浓度 (ng/μl) 和库总浓度 (总产量从 200–1400 ng 不等);样品的体积 (μl)。
- 提供测序请求: 使用 dna 定量试剂盒对样品进行定量处理;适应指数类型 (truseq dna);板型 (Eppendorf twint. tec, 全裙-推荐 dna);测序技术库类型 (10倍, 完整的测序指令和循环建议)17。
- 运行浅层排序 (可选): 分析多个生物样本的研究将受益于汇集样本 (聚合) 生成一个包含所有样本数据的单一基因条形码矩阵。为了最大限度地减少池时样本之间的批处理影响, 应标准化不同库之间的读取深度。为了做到这一点, 需要精确地逼近单个细胞数。miseq 测序器将允许浅测序, 是获得准确细胞估计的一种经济高效、实用的方法。
注: 一次使用 miseq sr50 排序器运行可提供足够的覆盖范围, 以准确估计约 20, 000个细胞。此运行将近似恢复每个唯一条形码的 umi 数。在图3a 中, 显示了示例 (示例 1.6) 输出 (. csv) 的标头, 其中列出了条形码及其相应的 umi 计数, 这些标记由自信映射的读取确定。- 咨询生物信息专家, 熟悉 r 编程语言。有关详细信息,请参阅 datacamp 教程。
- 使用提供的 r 脚本作为模板 19, 评估从排序器中获得的原始数据.原始数据是指映射到每个唯一单元格条形码的 umis 数。脚本读取. csv 文件, 其中第一列是条形码列表, 第二列是其相应的 umi 计数。此脚本将提供一个绘图 (图 3b) 以及每个示例中条形码单元的估计数量。调整脚本以确保给定样本的输入的 umi 计数数位于第一个陡峭下降的三分之一点。在图 3b中, 此肘部落在 225个 umis 附近, 相当于 3 480个条形码细胞。
- 与使用 hiseq 进行的全深度测序 (图 3c, 3, 516个细胞已成功测序,图 3c) 相比, 浅测序估计预测为 3 480个细胞。
- 或者使用细胞恢复近似值 (从步骤 5.3), 或者使用制造商协议20中的恢复图表来规划车道分布, 以便进行更深入的排序。每个样本都应获得可比的覆盖范围, 因此, 如果浅层测序显示每个样本中的细胞数量不同 (通常是这种情况), 则应相应计算车道分布。一个 hiseq 流单元 (包括8条车道) 可以对多达24亿个自定义配对的端读取进行排序。图 3d显示了流单元设置的示例。
6. 处理读取文件
注: 使用此协议对单个单元 3 ' 库进行排序, 以二进制基本调用 (bcl) 格式生成原始数据。细胞游侠包用于从 bcl 文件生成基于文本的 fastq 文件, 执行基因组和转录对齐、基因计数、去多路复用和样本聚合。在本节中, 介绍了使用户能够从测序设施下载原始 bcl 数据并生成可用于下游生物信息学的过滤基因条形码矩阵的关键步骤。
- 使用集中式服务器运行程序。bcl 文件、fastq 文件和大部分下游生物信息学处理都需要强大的处理能力。
- 将所有原始读取文件下载到服务器 (或 fastq 文件 (如果可用))。
- 请咨询服务器管理员, 在集中式服务器或群集上设置帐户, 并熟悉 unix21。
- 使用适用于服务器操作系统的提取命令从排序工具的服务器下载所有文件。
- 大多数排序工具提供了从可从命令行运行的安全路径下载文件的命令 (请参见下面的示例)。
- 将命令行中的 "< 用户名 >" 和 "< 密码 >" 占位符替换为提供的凭据。
wget-o-"htps://your_sequencing_facilitys_server.com/path_to_raw_read_files/--无饼干--无检查证书--后数据" j _ 用户名 = 用户名 & j _ 密码 = 密码 'wget-无饼干-无检查证书-后数据 ' j _ 用户名 = 用户名 & j _ 密码 = 密码 '-ci-
- 如果只提供文件的绝对路径 (即http://your_sequencing_facilitys_server.com/path_to_raw_read_files/), 则将此路径插入到提取命令中。
- 解压缩文件: 如果下载的文件以 ". gz" 扩展名结尾, 则使用 "gzip" 命令对其进行压缩。若要解压缩, 请在命令行中运行解压缩命令 (请参见下面的示例)。
生的。 - 将最新版本的细胞游侠作为独立的. tar 22 下载到服务器上。
-
关键:在下载之前, 请确保 linux 系统满足最低要求23。确保至少有8核英特尔处理器具有 64 gb ram 和 1 tb 的可用磁盘空间。
注: 细胞游侠提供预先构建的人类和啮齿类动物参考转录。这些可以使用大提琴手 mkref 命令进行修改, 以检测 gfp24 等基因。
-
关键:在下载之前, 请确保 linux 系统满足最低要求23。确保至少有8核英特尔处理器具有 64 gb ram 和 1 tb 的可用磁盘空间。
- 使用设场询程序 mkfastq 命令从排序器的基本调用文件 (bcls) 生成 fastq 文件。
注: 程序将使原始读取 (从 fastq 文件) 对齐到参考基因组, 并生成基因细胞矩阵进行下游分析。它使用 star aligner 对参考基因组执行读数的拼接感知对齐。只有自信地映射的读数 (即与单个基因注释兼容的读取) 才用于 umi 计数。- 例如, 使用大提琴手 mkfastq 命令:
拉热格 mkfastq -id = 样品名称 \
--------------------------------
--csv = csv _ file _ 装有 _ lane _ t例 _ index. csv
- 例如, 使用大提琴手 mkfastq 命令:
- 运行选区管理员计数 fastq 文件产生的 mkfastq 生成单细胞基因计数。
- 例如, 使用牧者计数命令:
测距仪计数--id = 样本名称 \
--转录组 = 重新数据处理的细胞-mm10-1.2。0
-----------------------------------------------------------
--样本 = 同一个 _ 示例 _ name _ 拿给 _ cellranger _ mkfastq\
----本地 coresf 30
- 例如, 使用牧者计数命令:
- 多库聚合 (可选): 要组合样本, 使用电保护器格器将池游光器计数输出。这将生成一个包含来自多个库的数据的基因条形码矩阵。示例牧者格格命令:
拉热员报告--id = 样本 _ name \
--csv=csv_with_libraryID_&_path_to_molecule_h5.csv
--规范化 = 映射
注: 可以使用三种规范化模式 (映射、原始、无) 聚合库。建议映射, 因为它对更高的深度库进行子采样, 直到所有库具有相等的排序深度25。 - 为了立即可视化/分析数据, 请将. 最大的云输出文件 (使用电保护器计数或电镜格生成) 导入到10倍的放大单元格浏览器26中。
7. scrna-seq 数据集的高级分析
注: 一个完整的 scrna-seq 工具数据库可在 scrna 工具3,27中找到。下面是使用 urat2和使用单核细胞6的伪小波排序的无监督细胞聚类框架.尽管这些工作大多可以在本地计算机上完成, 但以下步骤假定将使用机构服务器完成计算。
- 使用 linux 平台28将最新版本的 miniconda 下载到服务器帐户上.
- 使用 conda29安装最新版本的 r。
- 使用提供的 seurat r 脚本作为模板 30绘制数据。
注: seurat 是一个基于 r 的工具包, 可实现质量控制检查、聚类分析、差异基因表达分析、标记基因识别、降维和 scrna-seq 数据的可视化。在 satija 实验室网站31上可以找到 seurat 编码和教程的全面描述。 - 使用提供的 monocle r 脚本作为模板 32绘制数据。
注: 单核细胞是另一个基于 r 的工具包, 它可以可视化表达在伪时间内的变化, 并确定细胞命运决定背后的基因。在 monocle 网站33上可以找到 monocle 编码和教程的全面描述。 - 由于池数据集 34, 可以使用 r 包 (如 kbet) 来测试和纠正批处理效果.
8. ncbi 的全球环境展望和 sba 提交材料
注: 由于易于访问原始测序文件可确保重现性和重新分析, 因此建议或要求在提交文稿之前提交免费提交的文件。国家生物技术信息中心 (ncbi) 基因表达综合 (geo) 和序列读取存档 (sara) 是可公开访问的数据存储库, 用于高通量测序数据35,36。
- 注册 ncbi 的 geo 提交帐户37。
- 完成 geo 提交, 其中包括编译为目录文件夹的三个组件 (标题为 geo 提交者的用户名): 1) 元数据记录 (每个项目提交一个电子表格);2) 原始数据文件;3) 处理过的数据文件。
- 下载并完成元数据电子表格38。以下公开提交的全球环境展望可作为指南 (gse100320)39。将电子表格放在目录中。
- 将所有库的列表管理员计数脚本生成的原始数据文件放入目录中。
- 将从所有库的筛选器计数脚本生成的已处理数据文件 (筛选为份使用. tsv、gen. tsv 和矩阵. mtx 文件) 放入目录。
- 使用 geo 提交者的 ftp 服务器凭据传输包含所有三个组件的目录。对于 linux/unix 用户: 可以使用 ncftp、lftp、ftp、sftp 和 ncftp。
- 通知所有转让的 geo 38。
结果
用于分析 srna-seq 数据集的开源包的曲目已显著增加了 40个, 其中大多数包使用基于 r 的语言3。在这里, 介绍了使用其中两个包的代表性结果: 评估基于基因表达的单个细胞的无监督分组, 并沿轨迹排序单个细胞, 以解决细胞异质性和解构生物过程。
图 4显示了使用 seurat 进行预处理质量检查和下游生物信息学分析的情况。首先, 过滤和去除异常细胞的分析是必不可少的质量检查。这是用小提琴 (图 4a) 和散点图 (图 4a) 来显示线粒体基因的百分比、基因数量 (ngene) 和 umi (nGene) 的数量来识别细胞双位和异常值。任何具有明确异常数量的基因、umi 或线粒体基因百分比的细胞都被使用 seurat 的过滤细胞功能删除。由于 seurat 对集群细胞使用主成分 (pc) 分析分数, 因此确定要包括的具有统计意义的 pc 是一个关键步骤。用弯头图 (图 4c) 进行 pc 选择, 排除了超过 "pc 标准差" 轴高原的 pc。还对聚类的分辨率进行了操作, 表明集群的数量可以改变, 从 0.4 (低分辨率导致较少的细胞集群,图 4d) 到 4 (高分辨率导致更高的细胞集群,图4d).在低分辨率下, 每个集群很可能表示已定义的细胞类型, 而在高分辨率下, 这也可能表示细胞群的亚类型或过渡状态。在这种情况下, 低分辨率的集群设置用于进一步分析表达式热图 (使用 seurat 的 doheatmap 函数), 以识别给定集群中表达最强烈的基因 (图 4f)。在这种情况下, 通过评估给定星系团中的差异表达和所有其他集群的组合, 确定了表达最强烈的基因, 这表明每个集群都是由定义的基因唯一地表示的。此外, 可以使用 seurat 的特点点函数在 tssee 图上可视化单个候选基因 (图 4g)。这就可以破译是否有代表巨噬细胞的星团。利用特征图, 我们发现第2组和第4组都表达了 cd68-一个泛巨噬细胞标记。
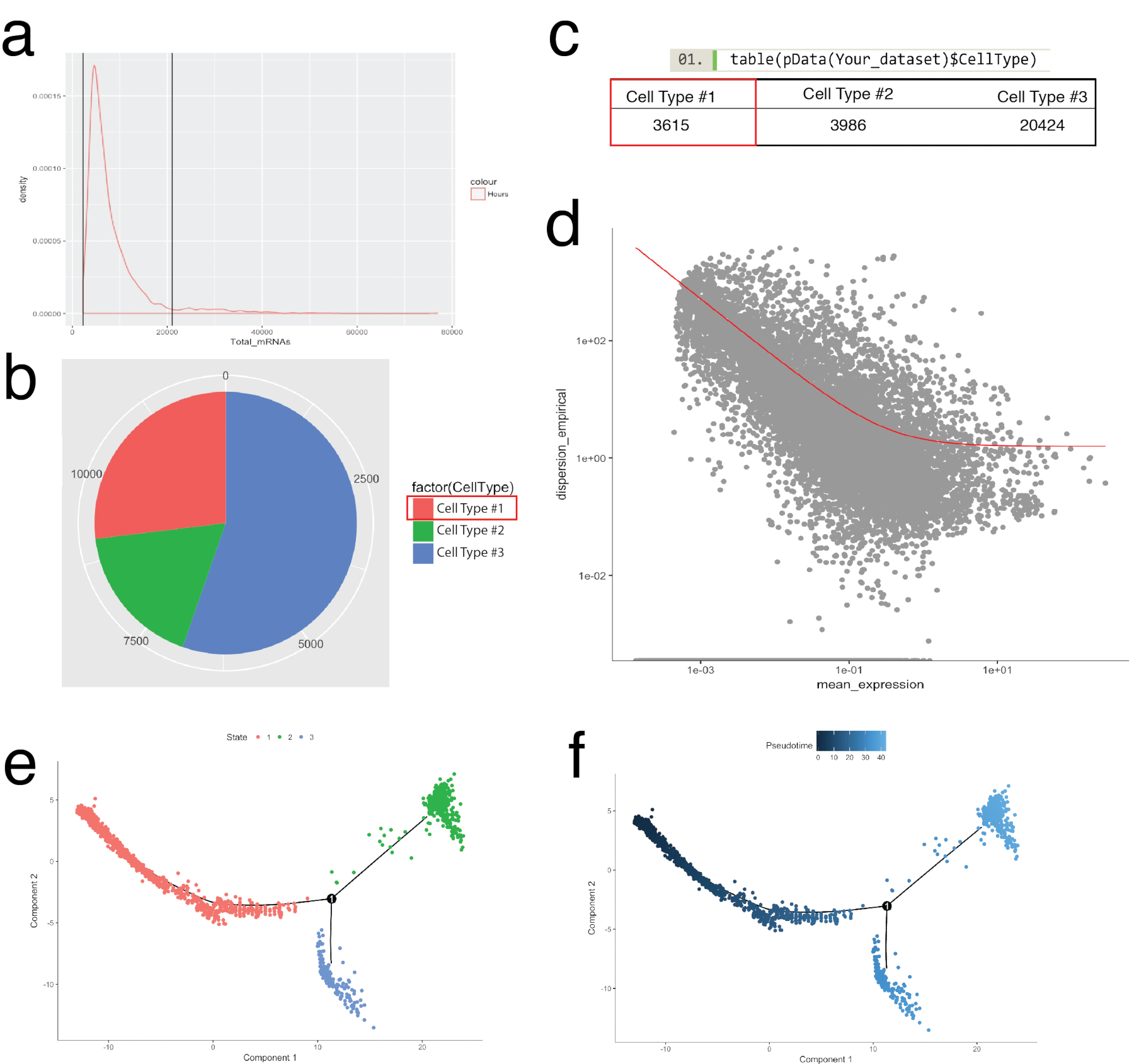
monocle 包用于证实在 seurat 中确定的细胞群, 并用于构建细胞轨迹, 或伪分子排序, 以重述生物过程 (图 5)。在单细胞表达谱应遵循生物时间过程的样品中, 可以使用伪分子排序。细胞可以沿着一个伪分子连续体排序, 以解决两个替代细胞命运的中间状态、分叉点, 并识别每个命运获取的潜在基因特征。首先, 与 seurat 的过滤类似, 劣质细胞被去除, 因此所有细胞的 mrna 分布都是正常的, 并在图5a 中确定的上限和下限之间。然后, 利用 monocle 的新细胞类型层次函数, 使用已知的沿袭标记基因对单个细胞进行分类和计数 (图 5b, 5b)。例如, 表达 pdgf 受体α或成纤维细胞特异性蛋白1的细胞被分配到细胞类型 #1, 以创建定义成纤维细胞的标准。接下来, 对这一群体 (细胞型 #1) 进行了评估, 以破译成纤维细胞的轨迹。为了做到这一点, 使用了 monocle 的差异基因测试功能, 该功能比较了代表群体中极端状态的细胞, 并发现了用于订购群体中剩余细胞的差异基因 (图 5d)。通过在所有细胞中应用流形学习方法 (一种非线性降维方法), 给出了沿伪电流路径的坐标。然后通过单元格状态 (图 5e) 和伪时间 (图 5e) 对这一轨迹进行可视化。

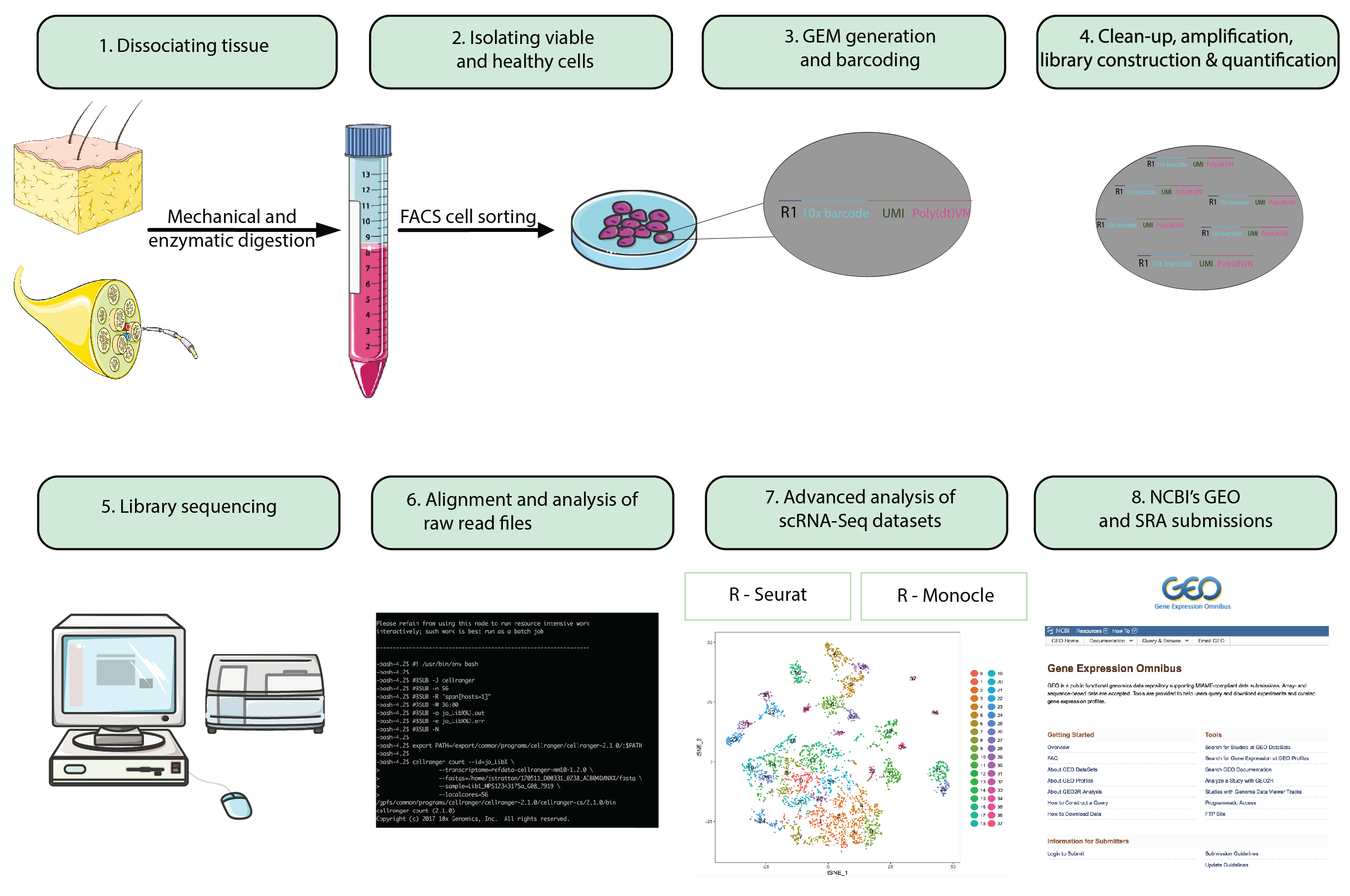
图 1: 流程图.从整个动物准备到分析单个单元 rna-seq 数据集, 再到将最终数据集提交到公开的存储库的步骤。乳化液 (gm) 中的凝胶微珠是指含有含条形码寡核苷酸的微珠, 它封装了数千个单个细胞。请点击这里查看此图的较大版本.
{kind=link}

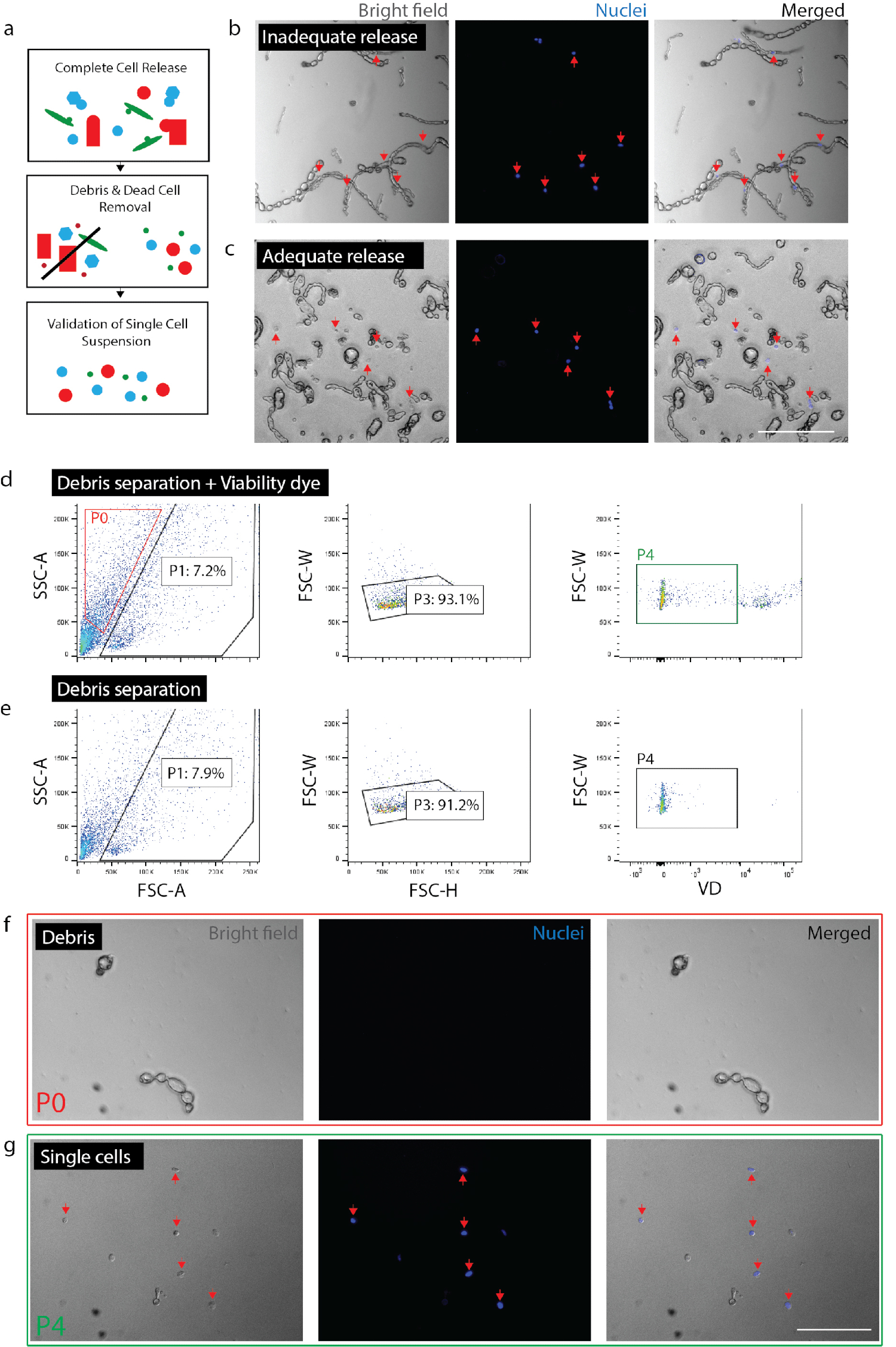
图 2: 从神经组织中创建活的单细胞悬浮液.(a) 质量控制检查的卡通概述。(b) 仍被纳入碎片的细胞和碎片 (红色箭头)。(c) 碎片释放的细胞 (红色箭头)。(d) 外地资产管制系统的细胞分离。p0: 碎片分数;p1: 细胞样分数;p3: 排除重复;p4: 活力染料 (sytox orange) 负分数。(e) 不控制活力染料。(f) 代表孤立碎片的 p0 分数图像。(g) 代表分离的活细胞 (红箭) 的 p4 分数图像。(b)(c)(f) 和 (g) 在成像前20分钟添加了核染料。刻度棒: 80μm. 请点击这里查看这个数字的更大版本.
{kind=link}

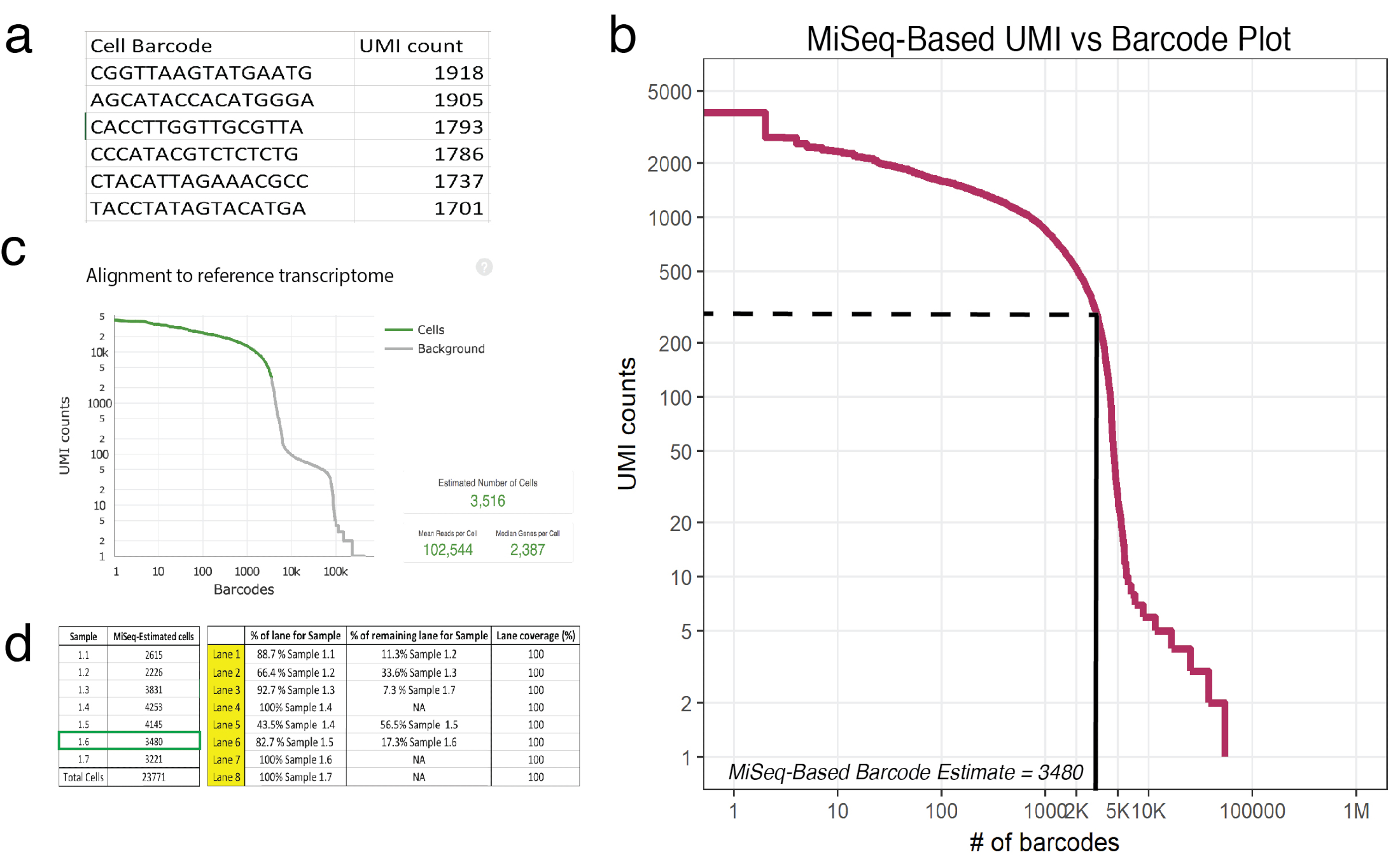
图 3: 浅层测序预测10倍处理样品中恢复的细胞数量.(a) miseq 生成的 csv 列出单元条形码的一个例子 (样本 1.6) 及其相应的 umi 计数, 这些计数由自信映射的读数确定。(b) 示例1.6 的条形码等级图显示, umi 计数作为细胞条形码的函数有一个显著下降。虚线和实线表示通过目视检查确定的单元格和背景之间的截断。(c) 使用 hiseq 后的细胞游侠管道观察到的细胞条形码显示, 浅层测序准确地接近样本1.6 的细胞数量。(d) 根据浅测序衍生细胞估计建立流动细胞的一个例子。对于样本 1.6, 由于浅测可预测3480细胞, 1.17条车道被分配, 以确保每个细胞测序覆盖率达到 gt;100,000。注意: 所有车道必须增加到100%。请点击这里查看此图的较大版本.
{kind=link}

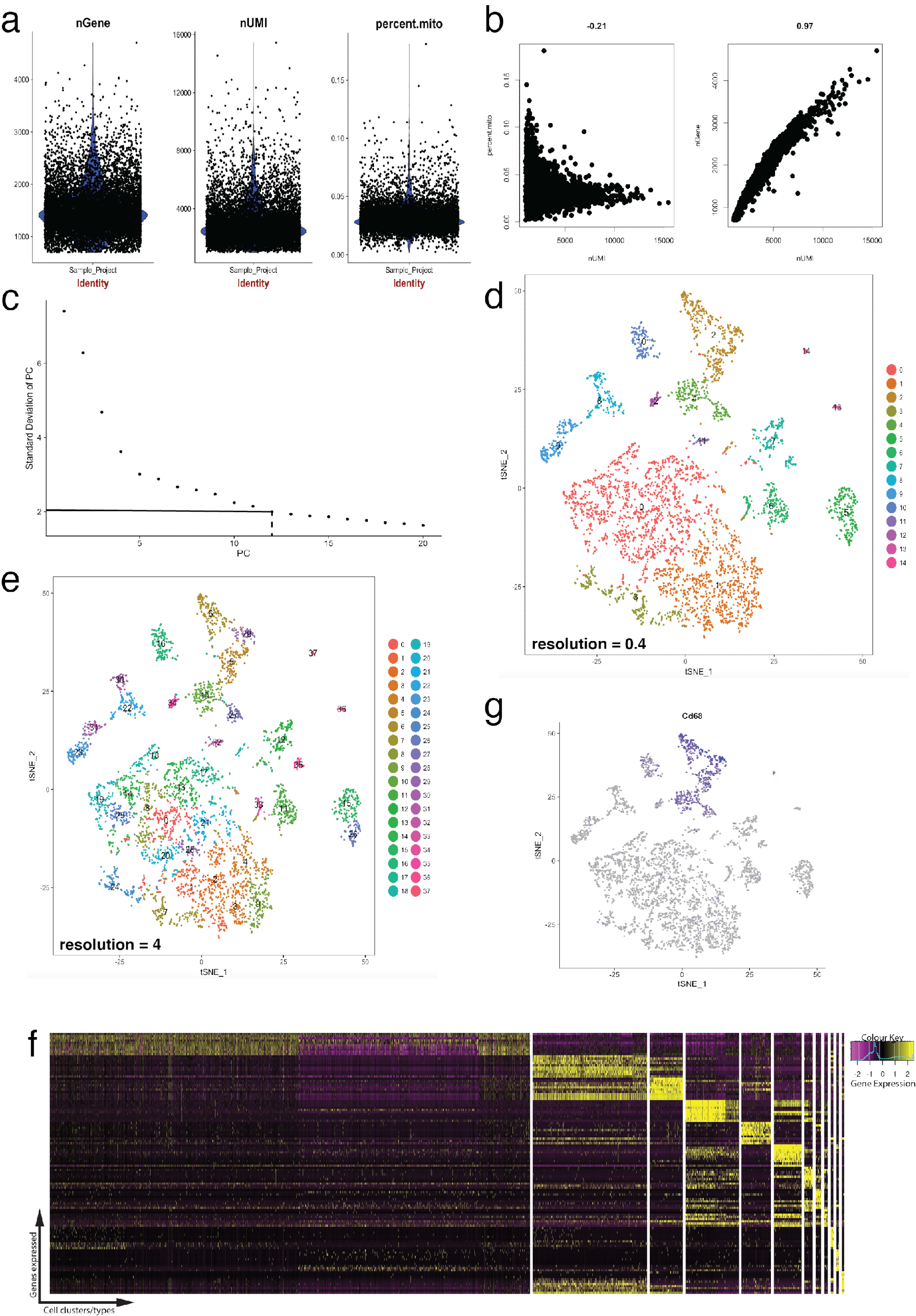
图 4: 使用 seurat r 封装的单细胞 rna-seq 数据集的质量控制和生物信息学.(a) 制定质量控制指标, 其中包括基因数量、唯一分子标识符的数量以及与线粒体基因组映射的记录的百分比。(b) 检测线粒体转录和 umi 异常水平的细胞的样本基因图。(c) 用于临时确定具有统计意义的个人电脑的弯头图样本。虚线和点虚线表示在图形中明显出现清晰 "弯头" 的截止线。此弯头之前的 pc 尺寸包括在下游分析中。(d, e)使用 tSNE 图形, 在低维空间中以两种不同分辨率可视化基于图形的单元格集群。(f) 使用 seurat 的 do暖气贴图函数在表达热图上可视化的每个星系的顶部标记基因 (黄色)。(g) 可视化标记表达, 例如, 使用 seurat 的特征点功能, 表示巨噬细胞 (紫色) 的 cd68 基因。这表明此数据集的第2和第4组 (在面板 d 中) 表示巨噬细胞。请点击这里查看此图的较大版本.
{kind=link}

图 5: 使用 monocle 工具包沿 pudotepoctocp 轨迹进行单元分类和排序.(a) 检查样品中所有细胞的 mrna 分布 (根据 umi 计数推断)。仅使用具有 0 ~ 20, 000 之间 mrna 的细胞进行下游分析。(b, c)根据已知的沿袭单元标记分配和计数单元格类型。例如, 表示 pdgf 受体α或成纤维细胞特异性蛋白1的细胞被分配到细胞类型 #1 代表泛成纤维细胞使用单核细胞的新细胞类型。不同单元格类型的数量可以可视化为饼图 (b) 和表 (c)。(d) 以细胞型 #1 (成纤维细胞) 为例, 可以使用显示基因分散与平均表达的散点图来可视化用于订购细胞的基因。红色曲线显示了用于排序的基因的截止时间, 该模型使用单核细胞的估计色散函数由均值方差模型计算。满足这种截止日期的基因用于下游伪时间排序。(e, f)在减少的二维空间中可视化细胞轨迹, 由单元格的 "状态" (e) 和由单点分配的 "伪时间" (f) 着色。请点击这里查看此图的较大版本.
{kind=link}
讨论
该协议演示了单个细胞的适当制备如何揭示数千个单个细胞的转录异质性, 并区分组织内的功能状态或独特的细胞身份。该协议不需要荧光记者蛋白或转基因工具, 可应用于从各种感兴趣的组织 (包括人类组织) 中分离单个细胞;请记住每个组织是独特的, 这个协议将需要一定程度的调整/修改。
细胞内多样化和高度动态的转录程序强调了单细胞基因组学的价值。除了分离高质量的 rna 外, 高质量数据集所必需的一个关键样品制备步骤是确保细胞完全从组织中释放, 并确保细胞健康和完整。这对于收集容易释放的细胞来说是相对直接的, 例如循环细胞或细胞松散保留的组织中, 如淋巴组织中。但这对其他成人组织来说可能是具有挑战性的, 因为高度发达的细胞结构跨越很远的距离, 周围的细胞外基质和经常涉及维持细胞结构的刚性细胞骨架蛋白。即使有适当的分离技术来完全释放细胞, 严格的和往往需要的处理也有可能改变 mrna 的质量和细胞的完整性。此外, 用于酶辅助解离的高温也会影响转录特征29,30。该协议的目的是提出质量控制检查, 使用组织, 如髓鞘成人神经和细胞外基质丰富的成人皮肤, 以证明如何优化可以帮助克服这些障碍。
设计任何 scrna-seq 实验时的一个主要考虑因素是测序深度的选择。测序可以高度多路复用, 读取深度可以从使用 drop-seq2非常低到使用全长 rna-seq 方法 (如 smat-seq) 的多达500万个读数单元14不等。大多数 srna-seq 实验都能检测到中高表达记录, 测序低至 10, 000个读数/细胞, 这通常足以进行细胞类型分类41,42。在试图检测复杂组织中的罕见细胞群时, 浅层测序深度对于节省测序成本很有价值, 在这些组织中, 可能需要数千个细胞来自信地归因稀有人群。但是, 当需要关于基因表达和与微妙转录特征相关的过程的详细信息时, 浅层测序是不够的。目前, 据估计, 细胞中的绝大多数基因是用50万个读基因检测的, 但这可能因协议和组织类型43,44的不同而不同。虽然全长记录测序绕过了组装的需要, 因此可以检测到新的或罕见的拼接变种, 测序成本往往限制了缩放这种方法, 以检查由复杂的组织系统组成的数千个细胞。相反, 3 ' 标记的单细胞库 (如本协议中描述的库) 通常具有较低的复杂性, 需要较浅的排序。需要注意的是, 使用所述协议生成的库可以在五个受支持的序列器之一上进行排序: 1) novaseq, 2) hiseq 000千瓦, 3) hiseq 2500 快速运行和高输出, 4) nextseq 500/, 和 5) miseq。
单细胞 rna-seq 的另一种方法是分析单核45中的 rna, 它减少了对精细组织和细胞处理的需求, 同时又保持了单细胞 rna-seq 的一些好处。这种方法可以更快速地处理, 减少 rna 降解, 并采取更极端的措施, 以确保充分释放细胞核, 从而有可能更有信心地捕获代表特定组织内所有细胞的转录剖面。当然, 这只能提供特定细胞内存在的转录活性的一部分, 因此取决于这种方法可能是适当的, 也可能不是适当的实验目标。
除了对给定组织内的细胞身份进行完整的表征外, 对 scrna-seq 数据集最有价值的分析之一是评估 "定义" 细胞群的中间转录状态。这些中介状态可以深入了解已确定群体中细胞之间的谱系关系, 这在传统的批量 rna-seq 方法中是不可能的。目前已经开发了几种 scrna-seq 生物信息工具来阐明这一点。这些工具可以评估癌细胞过渡到癌基因转移状态、干细胞成熟到不同的终命运或免疫细胞在活动状态和静止状态之间穿梭所涉及的过程。细胞中微妙的转录组差异也可能表明血统偏见, 最近开发的生物信息工具, 如 fateid, 可以推断47。由于过渡细胞之间的区别可能很难确定, 因为转录差异可能是微妙的, 更深入的测序可能是必要的46。幸运的是, 如果有兴趣通过在另一个流单元格上重新运行库来进一步探测数据集, 则可以增加浅排序库的覆盖范围。
总之, 该协议提供了一个易于适应的工作流程, 使用户能够在一个实验中对数百到数千个单细胞进行转录。scrna-seq 数据集的最终质量取决于优化的细胞分离、流式细胞仪、cdna 文库生成和原始基因条形码矩阵的解释。为此, 该协议全面概述了所有可以轻松修改的关键步骤, 以便能够对不同的组织类型进行研究。
披露声明
无披露
致谢
我们感谢 ucsna 服务设施的支持人员以及卡尔加里大学的动物护理设施工作人员。我们感谢马特·沃伦廷的生物信息学支持和詹斯·杜鲁西的技术支持。这项工作的资金来自 cihr 赠款 (r. m. 和 j. b.)、cihr j. b. 新研究员奖和艾伯塔省儿童健康研究所研究金 (j. s.)。
材料
| Name | Company | Catalog Number | Comments |
| Products | |||
| RNAse out | Biosciences | 786-70 | |
| Pentobarbital sodium | Euthanyl | 50mg/kg | |
| HBSS | Gibco | 14175-095 | |
| Dispase 5U/ml | StemCell Technologies | 7913 | 5 mg/ml |
| Collagenase-4 125 CDU/mg | Sigma-Aldrich | C5138 | 2 mg/ml |
| DNAse | Sigma-Aldrich | DN25 | 10mg/ml |
| BSA | Sigma-Aldrich | A7906 | |
| 15 ml Narrow bottom tube VWR® High-Performance Centrifuge Tubes | VWR | 89039-666 | |
| Sytox Orange Viability Dye | Molecular Probes | 11320972 | 1.3 nM/µl |
| Nuc Blue Live ReadyProbes | Invitrogen | R37605 | |
| Agilent 2100 Bioanalyzer High senitivity DNA Reagents | Agilent | 5067-4626 | |
| Kapa DNA Quantification Kit | Kapa Biosystems | KK4844 | |
| Chromium Single Cell 3' reagents | 10x Genomics | ||
| Equipment | |||
| BD FACSAria III | BD Biosciences | ||
| Agilent 2100 Bioanalyzer Platform | Agilent | ||
| Illumina® HiSeq 4000 | Illumina | ||
| Illumina® MiSeq SR50 | Illumina | ||
| 10X Controller + accessories | 10x Genomics | ||
| Software | |||
| The Cell Ranger | 10x GENOMICS | support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome | |
| Loupe Cell Browser | 10x GENOMICS | support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest | |
| R | https://anaconda.org/r/r |
参考文献
- Shalek, A. K., et al. Single-cell RNA-seq reveals dynamic paracrine control for cellular variation. Nature. 510, 363-369 (2014).
- Macosko, E. Z., et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 161, 1202-1214 (2015).
- Zappia, L., Phipson, B., Oshlack, A. Exploring the single-cell RNA-seq analysis landscape with the scRNA-tools database. bioRxiv:206573. , (2018).
- Dulken, B. W., Leeman, D. S., Boutet, S. C., Hebestreit, K., Brunet, A. Single cell transcriptomic analysis defines heterogeneity and transcriptional dynamics in the adult neural stem cell lineage. Cell Reports. 18 (3), 777-790 (2017).
- Llorens-Bobadilla, E., et al. Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Activated upon Brain Injury. Cell Stem Cell. 17 (3), 329-340 (2015).
- Trapnell, C., et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature Biotechnology. 32, 381-386 (2014).
- Aibar, S., et al. SCENIC: single-cell regulatory network inference and clustering. Nature Methods. 14, 1083-1086 (2017).
- Mayer, C., et al. Developmental diversification of cortical inhibitory interneurons. Nature. 555 (7697), 457-462 (2018).
- Stratton, J. A., et al. Purification and Characterization of Schwann Cells from Adult Human Skin and Nerve. eNeuro. 4 (3), (2017).
- Biernaskie, J. A., McKenzie, I. A., Toma, J. G., Miller, F. D. Isolation of skin-derived precursors (SKPs) and differentiation and enrichment of their Schwann cell progeny. Nature Protocols. 1 (6), 2803-2812 (2007).
- 10X Genomics. User Guides. , Available from: https://www.10xgenomics.com/resources/user-guides/ (2018).
- 10X Genomics. Chromium Single Cell 3' Training Module. , Available from: http://go.10xgenomics.com/training-modules/single-cell-gene-expression (2018).
- Agilent. , Available from: https://www.agilent.com/en-us/library/usermanuals?N=135 (2018).
- Kolodziejczyk, A. A. Single Cell RNA-Sequencing of Pluripotent States Unlocks Modular Transcriptional Variation. Cell Stem Cell. 17, 471-485 (2015).
- Jaitin, D. A., et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 343, 776-779 (2014).
- Pollen, A. A., et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nature Biotechnology. 32, 1053-1058 (2014).
- 10X Genomics. Sequencing Requirements for Single Cell 3'. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/sequencing/doc/specifications-sequencing-requirements-for-single-cell-3 (2018).
- Datacamp. Introduction to R. , Available from: https://www.datacamp.com/courses/free-introduction-to-r (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics#39; Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- 10X Genomics. User Guides. , Available from: https://www.10xgenomics.com/resources/user-guides/ (2018).
- UNIX Tutorial for Beginners. , Available from: http://www.ee.surrey.ac.uk/Teaching/Unix/ (2018).
- 10X Genomics. Creating a Reference Package with cellranger mkref. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/advanced/references (2018).
- 10X Genomics. System Requirements. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/overview/system-requirements (2018).
- 10X Genomics. Software Downloads. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest (2018).
- 10X Genomics. Aggregating Multiple Libraries with cellranger aggr. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/aggregate#depth_normalization (2018).
- 10X Genomics. Loupe Cell Browser Gene Expression Tutorial. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/visualization/latest/tutorial (2018).
- scRNA-tools. A table of tools for the analysis of single-cell RNA-seq data. , Available from: https://www.scrna-tools.org/ (2018).
- Conda. Downloading conda. , Available from: https://conda.io/docs/user-guide/install/download.html (2018).
- Anaconda. r / packages / r 3.5.1. , Available from: https://anaconda.org/r/r (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics' Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- Satija Lab. Seurat - Guided Clustering Tutorial. , https://satijalab.org/seurat/pbmc3k_tutorial.html (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics' Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- Monocle. , Available from: http://cole-trapnell-lab.github.io/monocle-release/docs/#constructing-single-cell-trajectories (2018).
- Github. An R package to test for batch effects in high-dimensional single-cell RNA sequencing data. , (2018).
- Edgar, R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Research. 30, 207-210 (2002).
- Leinonen, R., Sugawara, H., Shumway, M. The sequence read archive. Nucleic Acids Research. 39, D19-D21 (2011).
- NIH. GenBank Submission Portal Wizards. , Available from: https://www.ncbi.nlm.nih.gov/account/register/?back_url=/geo/submitter/ (2018).
- NIH. Submitting data. , Available from: https://submit.ncbi.nlm.nih.gov/geo/submission/ (2018).
- Shah, P. T., et al. Single-Cell Transcriptomics and Fate Mapping of Ependymal Cells Reveals an Absence of Neural Stem Cell Function. Cell. 173, 1045-1057 (2018).
- Anon, Method of the Year 2013. Nature Methods. 11, 1(2013).
- Adam, M., Potter, A. S., Potter, S. S. Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: a molecular atlas of kidney development. Development. 144, 3625-3632 (2017).
- Wu, Y. E., Pan, L., Zuo, Y., Li, X., Hong, W. Detecting activated cell populations using single-cell RNA-seq. Neuron. 96, 313-329 (2017).
- Zeigenhain, C., et al. Comparative Analysis of Single-Cell RNA Sequencing Methods. Molecular Cell. 65 (4), 631-643 (2017).
- Wu, A. R., et al. Quantitative assessment of single-cell RNA-sequencing methods. Nature Methods. 11 (1), 41-46 (2014).
- Habib, N., et al. Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science. 353 (6302), 925-928 (2016).
- Janes, K. A. Single-cell states versus single-cell atlases - two classes of heterogeneity that differ in meaning and method. Current Opinions in Biotechnology. 39, 120-125 (2016).
- Herman, J. S., Sagar,, Grün, D. FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data. Nature Methods. 15 (5), 379-386 (2018).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。