
Method Article
RNA 下一代测序和生物信息学管道, 以识别点对位的表达 Line-1
摘要
在这里, 我们提出了一个生物信息学方法和分析, 以识别 LINE-1 表达在位点特定的水平。
摘要
长插入元素-1 (Lines/l1) 是重复的元素, 可以复制并随机插入基因组, 导致基因组不稳定和突变。了解 L1 位点在个体层面的表达模式, 将有助于了解这种诱变元素的生物学。这种自主元素占人类基因组的很大一部分, 有超过 50万份, 尽管99% 被截断和有缺陷。然而, 它们丰富和占主导地位的有缺陷的副本数量使得从作为其他基因一部分表达的 L1 相关序列中真正表达的 L1 变得具有挑战性。由于元素的重复性质, 确定表达哪些特定 L1 位点也具有挑战性。克服这些挑战, 我们提出了一个 RNA-Seq 生物信息方法来识别 L1 表达在位点特定的水平。总之, 我们收集细胞质 rna, 选择多腺化转录, 并利用特异性 RNA-Seq 分析, 将读取唯一映射到人类参考基因组中的 L1 位点。我们用独特的映射读数直观地管理每个 L1 位点, 以确认从其自身的启动子中进行转录, 并调整映射的转录读数, 以考虑每个 L1 位点的映射能力。这种方法被应用于前列腺肿瘤细胞系 DU145, 以证明该方案检测出少量全长 L1 元素表达的能力。
引言
逆转录酶是重复的 DNA 元素, 可以通过 RNA 中间体在复制和粘贴机制中 "跳" 在基因组中。后移子区的一个子集被称为 Long infos散元长-1 (LINEs/L1s), 占人类基因组的六分之一, 有超过 500, 000份副本1。尽管这些副本很多, 但它们大多是有缺陷的, 被截断的只有估计的 80-120 L1 元素被认为是活跃的 2。全长 l1 的长度约为 6 kb, 具有 5 ' 和 3 ' 未翻译的区域, 内部启动子和相关的反义启动子, 两个非重叠的开放阅读帧 (orf), 以及信号和多a 尾 3,4,5.在人类中, l1 是由因进化年龄而区分的亚系组成的, 与最年轻的亚科 L1s6,7 相比, 年龄较大的家庭随着时间的推移积累了更多独特的序列突变。L1 是唯一的自主的人的反转座子及其 Orf 编码逆转录酶, 内切酶, 和 RNPs 的 rna 结合和陪同活动所需的再转相和插入基因组在一个过程称为目标底漆逆转录酶8,9,10,11,12。
据报告, l1 的逆行通过各种机制导致人类生殖素疾病, 包括插入突变、靶点缺失和重新排列13、14、15、 16. 最近有人假设, l1 可能在肿瘤发生和肿瘤进展中发挥作用, 因为在各种上皮癌中观察到这种诱变元素的表达和插入事件增加 17,18.据估计, 每200个新生儿中就有一个新的 L1 插入率19。因此, 更好地理解主动表达 L1 的生物学是势在必行的。在其他基因的转录记录中发现的有缺陷的副本的重复性质和丰富, 使这一水平的分析具有挑战性。
幸运的是, 随着高通量测序技术的出现, 在特定于库的级别上解析和识别真实表达 L1 的方法取得了长足的进步。关于如何使用 RNA 下一代测序最好地识别表达的 L1, 有不同的理念。建议在特定于用户的层面绘制 L1 记录的方法只有两种。你只注重通过 L1 多腺苷酸信号读入侧翼序列20的潜在转录。我们的方法利用 L1 元素之间的微小序列差异, 只映射那些唯一映射到一个位置21的 RNA-Seq 读取。这两种方法在成绩单水平的定量方面都有局限性。通过为每个 L1 位点21的 "唯一映射性" 添加校正, 或者使用更复杂的算法来重新分配无法唯一映射到特定位点22的多映射读取, 可以潜在地改进量化。在这里, 我们将逐步详细介绍 RNA 提取和下一代测序和生物信息学协议, 以确定在特定于位置的级别上表达的 L1 元素。我们的方法最大限度地利用了我们对功能 L1 元素生物学的了解。这包括知道功能 L1 元素必须从 L1 启动子中生成, 在 L1 元素开始时启动, 必须在细胞质中转换, 并且它们的转录应与基因组呈线性关系。简单地说, 我们收集新鲜的细胞质 RNA, 选择多腺苷酸转录, 并利用特异性 RNA-Seq 分析, 将读取唯一映射到人类参考基因组中的 L1 位点。然后, 这些对齐的读取仍然需要广泛的手动策划, 以确定是否从 L1 启动子开始, 然后再指定一个轨迹作为一个真正表达的 L1。我们将此方法应用于 DU145 前列腺肿瘤细胞系样本, 以证明它如何从大量的非活性拷贝中识别相对较少的主动转录的 L1 成员。
研究方案
1. 细胞质 RNA 提取
- 通过以下方法获取单元格。
- 收集活细胞从2.75%–100% 融合, T-75 瓶。
- 在5毫升的冷 PBS 中清洗烧瓶 2次, 最后清洗刮掉细胞, 转移到15毫升的锥形管。在 1, 000 x g和4°c 下离心 2分钟, 并小心去除和丢弃上清液 (材料表)。
- 从组织标本中收集细胞。
- 准备组织, 以便在被解剖后一小时内提取细胞质 RNA, 并始终保持在冰上。对于长期储存, 请使用 RNA 抑制剂溶液在按照制造商的协议 (材料表) 进行解剖后最多72小时内储存组织。
- 在无菌弹跳均质机中, 用5毫升的冷 pbs 将新鲜样品共质, 转移到15毫升锥形管中, 离心机在4°c 时在 1, 000 x 克 g 处进行 2分钟, 并小心去除和丢弃上清液 (材料表 )。
- 收集活细胞从2.75%–100% 融合, T-75 瓶。
- 在细胞颗粒混合中加入2毫升裂解缓冲液, 在冰上孵育5分钟。
- 用 150 mM Ncl、50 mM HEPES (pH 7.4) 和25μgml 数联 (材料表) 制备新鲜的裂解缓冲液。
- 由于穿透质膜所需的裂解缓冲液中的数字蛋白的最低浓度可能因细胞类型而异, 显微镜下证实, 使用裂解缓冲液处理的细胞会失去质膜并保留完整的核膜。
- 使用前, 加入 1, 000 U/ml RNase 抑制剂 (材料表)。
- 在 1, 000 x g和4°c 下离心 1分钟, 并收集上清液。
- 在预冷7.5 毫升的三唑和1.5 毫升的氯仿中加入上清液。所有需要氯仿的步骤都必须在干净的化学罩内完成 (材料表)。
- 在 3, 220 x g 和4°c 下离心35分钟。
- 将水部分 (顶层) 转移到新鲜的预冷 15 mL 管中。
- 加入4.5 毫升氯仿和涡旋。
- 在 3, 220 x g和4°c 下离心10分钟。
- 将水的部分转移到新鲜的预冷管中。
- 加入4.5 毫升异丙醇, 摇匀, 在-80°c 下隔夜 (材料表)。
- 离心机在 3, 220 x g和4°c 下进行45分钟。
- 去除异丙醇, 加入15% 毫升的100% 乙醇 (材料表)。
- 离心机为 3, 220 x g , 10分钟。
- 除去乙醇, 排水干燥约1小时。
- 使用无菌棉签将剩余的乙醇 (材料表) 抹掉。
- 根据颗粒尺寸 (材料表), 在100至200μl 的 RNase 无水中重新悬浮样品。
- 分位样品采用电泳技术, 根据制造商的介绍确定样品的质量和浓度23 (材料表)。
- 如果 RIN > 824,则样本符合 rna-seq 分析的条件。
2. 下一代测序
- 提交细胞质 rna 样本, 使用下一代测序平台进行测序, 旨在产生至少 5000万个 paired-end 100 bp 读数。
- 选择多腺苷酸 Rna 和特异性测序。
3. 创建批注 (如果有现有批注, 则可选择)
- 创建全长 L1 批注或下载全长 L1 批注 (补充文件 1a-b)。
- 使用表格浏览器工具 (https://genome.ucsc.edu/cgi-bin/hgTables) 从 UCSC 基因组浏览器下载 LINE-1 元素的重复标记。指定哺乳动物的克隆、人类基因组、hg19 组件 (或 hg19 用于更新的基因组), 并在 "类名" 下筛选 "LINE1"。下载为. gtf 文件和标签为 FL-L1-BASTT. gtf。
- 运行 L1.3 全长 L1 元素的前 300 bp 的本地 blast 搜索, 其中包括人类基因组中的启动子区域, 并在下游添加 6, 000 bp, 以便在注释文件中创建 L1 坐标的末端。保存在 gtf 文件中, 并将其标记为 fl-l1-rm. gtf。
- 使用床工具将重复扫描器注释和基于标记的 L1 注释相交, 并将其标记为 FL-1-blast _ Rm. txt (软件包)。
- 在 Linux 终端中使用此命令:床工具相交-fl1-blast-gtf-b fl1-rm > fl1-blast _ rm. txt.
- 将相交的 Fl-l1 批注分上、下链。
- 将 FL-L1-BLAST _ rm. Txt 复制到电子表格软件中, 按 "减号" 和 "加号" 链进行排序, 然后按染色体位置进行排序。
- 创建两个新的电子表格文档, 一个具有负链上全长 L1 的相交坐标, 另一个保存为 FL1-BLAST _ mem. Xls 和 FL1-BL1-BLAST _ Rm _ pluss. xls。
- 将这两个新文档另存为. txt 文件。
- 使用 mac2unix 程序将. txt 文件转换为正确的注释文件 (软件包)。
- 在终端中使用此命令: mac2unix. sh fl-1-bl1 _ rm _ minuse. gff.
- 在终端中使用此命令: mac2unix. sh FL-1-bl1 _ rm _ pluse. gff.
- 使用. gff 扩展名保存新文件。
- 或者, 使用 AWK 筛选与 + 和–链关联的行。
- 使用以下命令获取 + 链: awk '/+/"fl-l L1_BLAST_RM.gtf > fl-L1_BLAST_RM_plus.gtf。
- 使用以下命令行获取-链: awk '//' fl-fl L1_BLAST_RM.gtf >/-/。
4. 读取对齐管道, 以识别表示的 L1
| 选项 | 描述 |
| –p | 这将详细说明计算机应使用运行对齐方式的线程数。较大的计算机内存将允许更多的线程, 并且应该是经验 d。 |
| –m 1 | 这告诉程序只接受基因组中有一个比任何其他基因组匹配都要好的读数。 |
| –y | 这是 tryhard 开关, 它使映射搜索所有可能的匹配, 不允许它退出后达到固定数量的匹配。 |
| –v 3 | 这只允许程序利用内存映射读取3或更少的不匹配的基因组。 |
| –X 600 | 这只允许在600个基内对地图进行配对读取。这确保了读取对在基因组中是共线性的, 并针对涉及经过加工的 RNA 分子的。 |
| –chunkmbs 8184 | 此命令为处理每个与 L1 相关的读取可能的大量对齐分配额外的内存。 |
表 1: 鲍蒂的命令行选项。
- 使用 Bowtie 运行与 RNA-Seq 样本的对齐对齐配端测序快速 q 文件。
注: 必须使用 Bowtie1, 而不是 Bowtie2, 因为唯一对齐所需的参数仅在此版本的 bowtie (软件包) 中专门找到。鲍蒂是用来的花式感知的标志, 如 STAR, 以评估一致性, 连续读取更相关的 L1 生物学和表达。- 在 Linux 终端中使用此命令行: bowtie-p 10-m 1-S-y-v 3-x 600-chunkmbs 8184 hg _ y _ m _ index-1 hg_sample_1.fq-2 hg_sample_2.fq samtools 视图-hbus-samibs 排序–g _ 示例 _ sorted. bam.有关 Bowtie 的命令行选项的说明, 请参阅表 1 。
- Strand 使用 samtools (软件包) 和以下 linux 命令分隔输出 bam 文件。请注意, 如果不使用标准的下一代排序协议, 则实际标志值可能会有所不同。
- 使用此命令行可选择顶部链: samtools 视图-h hg _ sorted. bam ' awk ' substr($0,1,1) = = "@" 2 = 83 = 2 = 163 {打印} ' 采样器视图-bs-> hg _ 示例 _ top _ top无限制. bam. bam.
- 使用此命令行选择底部链: samtools 视图-h hg _ sorted. bam ' awk ' substr($0,1,1) = = "@" 2 = = 99 2 = 147 {打印} ' samtools 视图-> _ hg _ 示例 _ 第二部 strands. bam. bam.
- 使用床工具 (软件包) 根据 l1 位点的批注生成读取计数。
- 使用此命令行可以在顶部链的感觉方向生成 L1 的读取计数:床工具覆盖-abam fl-1-blast _ rm _ plus. gff-b hg _ 示例 _ top前来 > bam _ 示例 _ tryhard _ tplus _ topx. txt.
- 使用此命令行可以在底部链的感觉方向生成 L1 的读取计数:床工具覆盖-abam fl-1-blast _ rm _ minus. gff-b _ 样本 _ 刻字) _ 底应变. bam > bam _样本 _ tryhard _ tryhard _ 值 _ 底价. txt。
- 索引 bam 文件从步骤 5.1.1, 使其在集成基因组学查看器 (IGV) 25 (软件包)中可见。
- 使用此命令行: samtools index hg _ samtication _ sortete. bam
- 若要使用批处理模式来增加一次通过管道传输的 RNA-Seq 样本的数量, 请使用超级计算机脚本完成称为人手 _ bowti. sh 的步骤4.1、已创建了一个脚本来完成步骤 4.2-4.3, 称为 human_L1_pipeline.sh, 以及一个脚本来完成步骤4.4 已被创建为 bam _ index. sh。这些脚本可以在补充文件 2中找到, 其中包含运行脚本的相关超级计算机命令。
5. 手动策划
- 创建映射到每个带注释的 L1 位点的读取的电子表格。
- 复制 hg _ 示例 _ b文蒂 _ tryhard _ 减去 _ 收底. txt, 在步骤中创建4.3.2 并将页面标记为 "最小底部"。
- 根据列 j 中找到的最高到最低读取数对所有列进行排序。
- 复制 hg _ 示例 _ b文蒂 _ b文蒂 _ tryhard _ plus _ topt. txt, 在步骤中创建4.3.1 并在另一个电子表格中标记为 "顶加"。
- 根据列 j 中找到的最高到最低读取数对所有列进行排序。
- 创建标记为 "组合" 的第三页, 并添加所有位点, 其中包含 "最小底部" 和 "加顶部" 页面中的10个或更多读取。
- 根据列 j 中找到的最高到最低读取数对所有列进行排序。
- 将以下文件加载到 IGV25 (软件包) 中: 1) 感兴趣的参考基因组, 2) Fl1-blast _ rm. gff 可视化 l1 注释, 3) hg _ 样品 _ sorted. bam 可视化映射的成绩单感兴趣的样本, 和 4) hg _ 基因组分解. bam, 以评估基因组区域的可映射性。
- 删除与每个 bam 文件关联的覆盖范围和联接行。
- 压缩 hg _ samit _ sorted. bam 和 hg _ genomicDNA _ sorted. BAM, 以便所有的 IGV 轨道都适合在一个屏幕上。
- 复制 hg _ 示例 _ b文蒂 _ tryhard _ 减去 _ 收底. txt, 在步骤中创建4.3.2 并将页面标记为 "最小底部"。
- 手动策划。
- 使用电子表格 "组合" 页上列出的位点中的坐标, 查看 IGV25 (软件包) 中称为位点的位点。
- 如果在 L1 方向上没有上游的读取, 则将一个轨迹进行真实的表达, 最高可达5kb。
- 用颜色标记行绿色, 并注意为什么它是真实表达的 L1。
注: 如果 L1 上游的区域未映射, 则存在此规则的异常。如果是这种情况, 请用颜色标记行红色, 并注意不能计算 L1 启动子上游区域的表达式, 因此无法自信地确定 L1 的表达式。
- 用颜色标记行绿色, 并注意为什么它是真实表达的 L1。
- 如果上游有高达 5 kb 的读取, 则将一个轨迹进行说明, 使其无法真实地表达其自身的启动子。
- 用颜色标记行红色, 并注意为什么它不是真实表达的 L1。
- 将位点指示为假, 如果它在表达的基因的内含物中与 L1 的上游读取相同的方向, 如果它是在与 L1 上游读取相同方向的表达基因的下游, 或为未注释的表达模式与重新的L1 上游的广告。
注: 当 L1 启动子启动站点的最小读取直接重叠, 但 L1 的上游稍有上游时, 则应用此规则的例外。如果这样的 L1 案例在上游没有其他读取, 请考虑将此 L1 真实表达。标记行绿色, 并注意为什么它是一个真实表达的 L1。
- 如果映射到位点的模式与特定 L1 的映射区域不相关, 则将 L1 位点的定位定为 false。
注: 例如, 如果 L1 具有高度可映射性, 但在 L1 内的压缩区域中只有大量读取, 则它不太可能与其自身启动子的 L1 表达式相关, 更有可能来自未注释的源 (如外联或 Ltr)。在这样的情况下, 把位点假装橙色, 并注意为什么位点可疑。通过检查 UCSC 基因组浏览器中的 L1 位置来验证可疑堆积物的来源。 - 如果一个位点是在零星表达的未注释区域的基因组环境中, 则确定它不被真实地表达
注: 例如, 读取可以表示 L1 上游 10 kb, 但每 10 kb 左右就有映射读取, 其中一些读取与 L1 对齐。这些 L1 不太可能从自己的启动子上表达, 更有可能由于没有注释的基因组表达模式而绘制读数。在这样的情况下, 把位点假装橙色, 并注意为什么位点可疑。
6. 读取对齐策略, 以评估参考基因组中的可映射性 (如果有现有的对齐基因组 DNA 数据集, 则可选择)
- 下载整个基因组 DNA 序列文件并转换为. fq 文件
- 转到 NCBI 网站在这里找到: https://www.ncbi.nlm.nih.gov/sra
- 键入Wgs HeLa 配对端。
- 在 "分类结果" 下选择智人。
- 选择一个已配对结束并具有100个或更多 bp 读取的示例, 如下面的示例: https://www.ncbi.nlm.nih.gov/sra/ERX457838[accn]
- 通过选择"运行",然后选择"元数据" 确认读取长度, 如下所示: https://trace.ncbi.nlm.nih.gov/Traces/sra/?run=ERR492384
- 要下载整个基因组 DNA 序列数据, 请在 Linux 终端中输入此命令: Sratoolkit.2.9.2-mac64/bin/prefetch-10G ERR492384
注: SRA 工具包预取功能下载 ncbi 网站 (软件包) 中的加入号 "ERR492384"。"100G" 将下载的数据量限制为100G 兆字节。 - 在 Linux 终端中输入此命令: fastq-转储--拆分文件 ERR492384
注: 这会将下载的基因组 DNA 数据集拆分为两个快速数据集。
- 使用 Bowtie 运行对齐。
- 使用此命令在 Linux 中对齐: bowtie-p 10-m 1-s-y-v 3-x 600-chunkmbs 8184 hg _ x _ index-1 Hg_genomicDNA_1.fq-2 hg_genomicDNA_2.fq samtools视图-hbus-samibs 排序–g _ genomicdna _ sortedd. bam.
- 请参阅步骤 4.1, 以了解 Bowtie 对齐 (软件包) 中使用的参数。
- 下载基因组对齐的 bam 文件, 以评估可根据作者的要求进行映射。
- 使用此命令在 Linux 中对齐: bowtie-p 10-m 1-s-y-v 3-x 600-chunkmbs 8184 hg _ x _ index-1 Hg_genomicDNA_1.fq-2 hg_genomicDNA_2.fq samtools视图-hbus-samibs 排序–g _ genomicdna _ sortedd. bam.
- 索引 bam 文件从步骤4.2.1 使用采样工具, 使其在 IGV 25 (软件包)中可见, 以进一步通知手动策划。
- 在 Linux 中使用此命令行: samtools 索引 hg _ genomicDNA _ sorted. bam
- 评估每个 L1 位点的可映射性
- 使用床工具程序、Fl-l1 注释和对齐的基因组序列数据 (软件包) 确定唯一映射到 l1 位点的读取数。
- 使用 Linux 中的此命令行:床上用品覆盖-abam fl-1-blast _ rm. gtf-b hg _ genomicDNA _ sorted.bam >。
- 当400个唯一读取与 l1 对齐时, 指定一个 L1 位点具有完全覆盖映射。
- 确定每个 L1 的基因组 DNA 对齐读数向上或向下排列为400所需的因素。
- 要根据单个 L1 位点的映射性进行缩放的表达测量, 请将步骤中确定的因子 6.4.3 RNA 转录读数乘以与第4-5 节中确定的真实表达的 L1 对齐的 Rna 转录数。
- 使用床工具程序、Fl-l1 注释和对齐的基因组序列数据 (软件包) 确定唯一映射到 l1 位点的读取数。
结果
上述步骤和图 1中的图形描述已应用于人类前列腺肿瘤细胞系 du145。RNA 样本是细胞质体准备的, 并在多 a 选择的、特异性的、配体的协议中进行了下一代测序。使用 Bowtie, 对配对端测序文件进行了对齐, 只允许唯一的匹配, 在这种匹配中, 与任何其他基因组位置相比, 配对端读取更适合一个基因组位置。DU145 序列文件与人类参考基因组对齐, 创建了一个 bam 文件, 可根据作者的请求使用。使用床工具, 从用于映射到全长 L1 的读取数的 DU145 带分隔的 bam 文件中提取数据。这些读数在电子表格中进行了从最大到最小的排序, 并通过检查 IGV 中每个 L1 位点周围的基因组环境进行手动管理, 以确认其真实性 (补充表 1)。如果一个样本被组织为真实地表示, 它被颜色编码绿色与解释为它的采纳在最右边的专栏。图 2a-b显示了建议按照方法部分中描述的准则真实表达的 l1 位点的示例。如果一个样本被拒绝, 以真实地表示, 它被颜色编码为红色与拒绝的原因在最右边的列。图 2c-e详细介绍了由于方法部分中描述的启动子以外的启动子的表达而被拒绝的 l1 位点的示例。
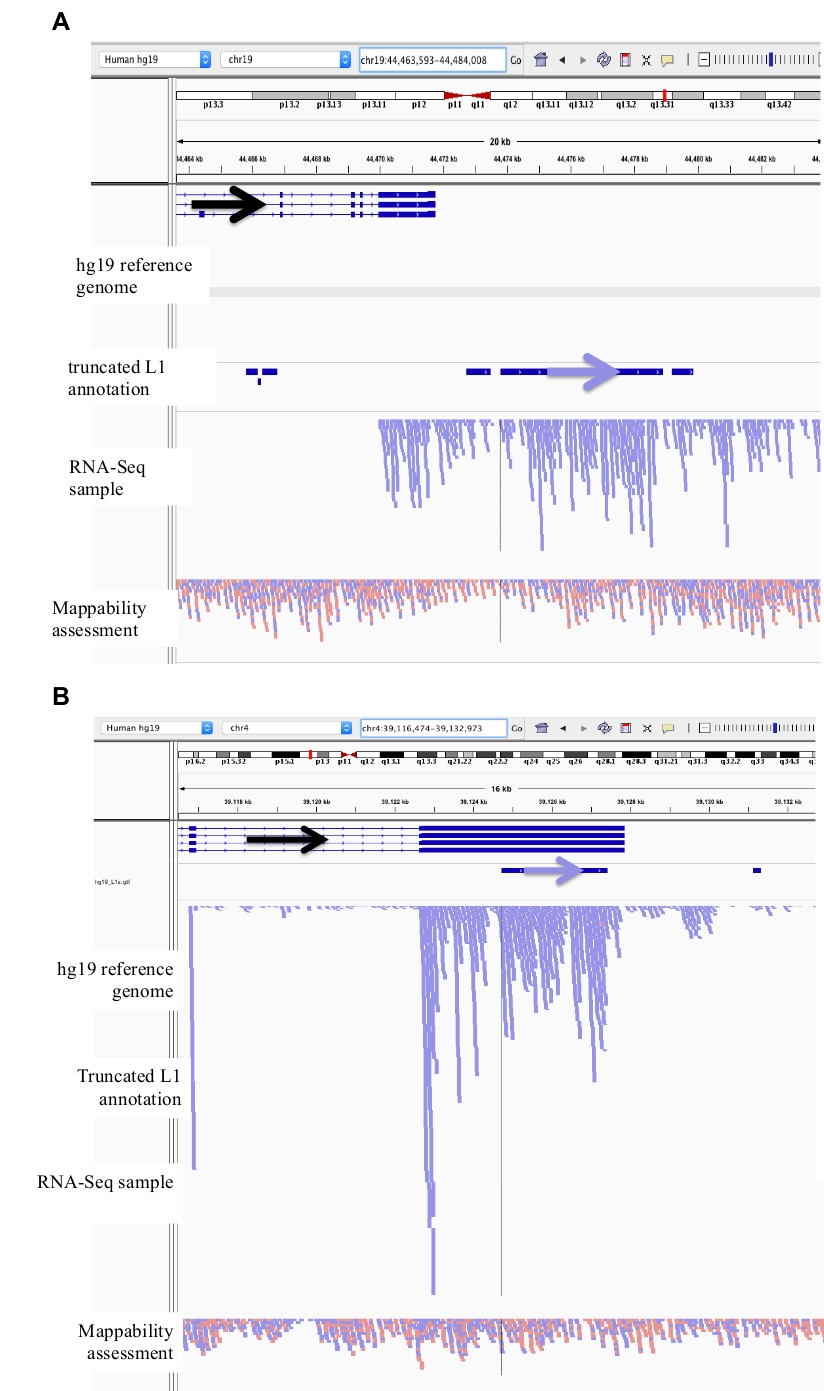
在这里, 只研究了具有完整启动子区域的全长 L1。如果不进行这种区分, 则引入了来自截断 L1 的大转录噪声源。在 DU145 中截断 L1 的示例如图 3a-b所示, 其中它们被标识为具有唯一映射的 RNA-Seq 读取。然而, 在 IGV 中, 这些记录显然不是从截断的 L1 开始的, 而是从基因中或从表达的基因下游纳入 L1 序列开始的。
总体而言, 在 DU145 中, 在人工管理后被拒绝为真实表达的 L1 位点的全长 L1 位点和读数的百分比约为 50% (补充表 2), 显示了高水平的 l1 映射记录, 这将否则, 无需人工策划, 则被记录为误报。具体而言, 在 DU145 有114个总全长 L1 位点有独特地映射读在感觉方向与总 3 152个读, 但仅有60个位点被确定被表达在他们自己的促进器在手工策展 1, 879 读以后 (补充表 1)。即使采取步骤通过选择细胞质 mRNA 来减少与 L1 生物学无关的表达时, 情况也是如此。请注意, 在 DU145 中映射的记录级别最高的位点被拒绝, 因为它不是真实表示的 L1 (图 4)。总体而言, 映射的记录数与特定 L1 位点的数量在被接受和被拒绝的 L1 位点之间相似地作为真实地表达在手工策展以后 (图 4)。
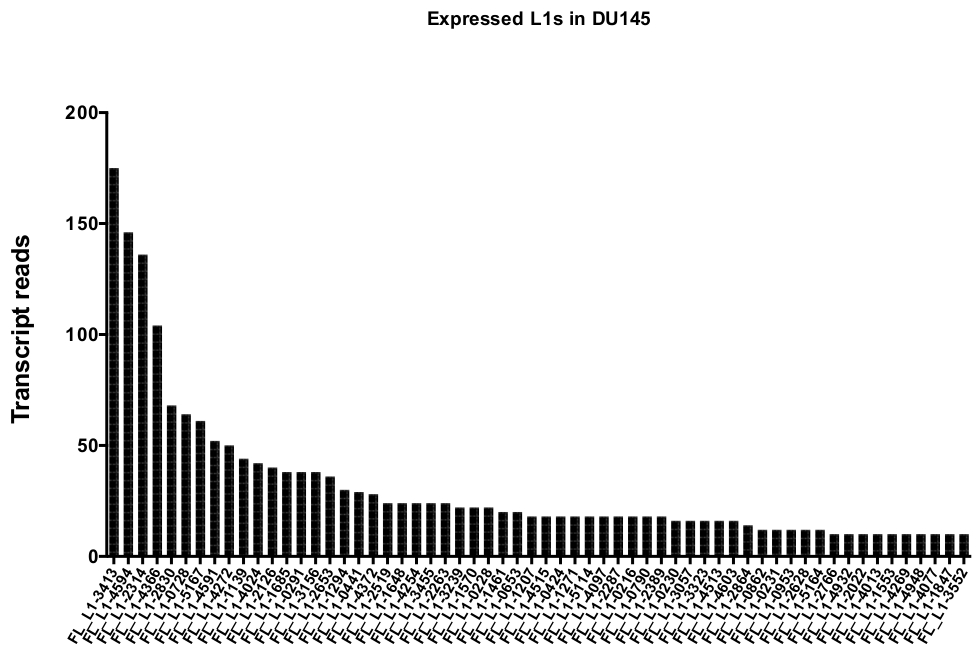
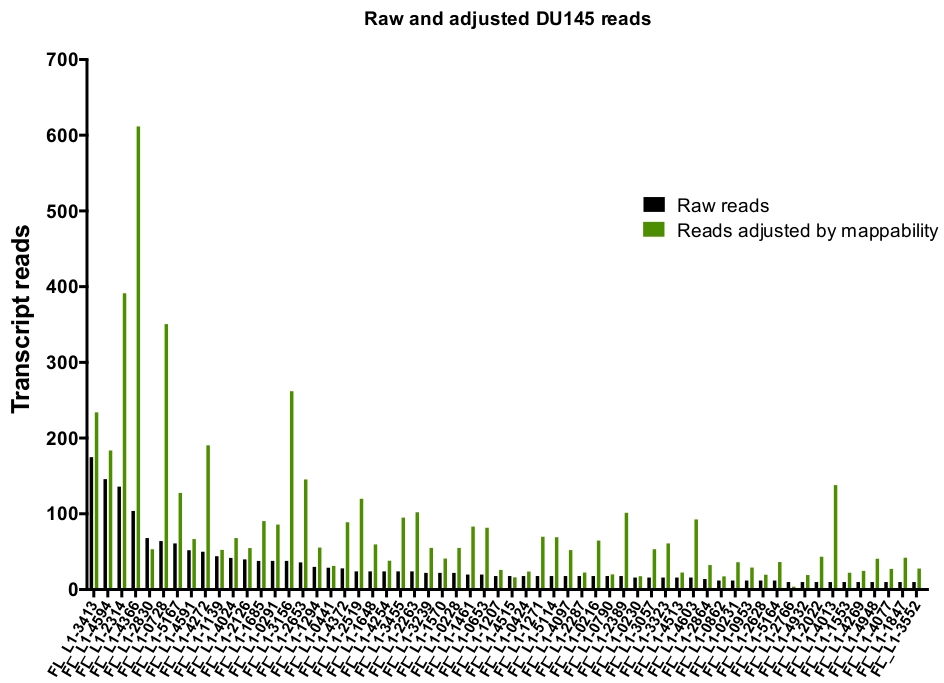
手动策划后, 地图唯一表示 du145 范围内的特定 L1 位点的读取次数从175个读取到任意选择的10个读取的最小切数 (图 5)。这种识别唯一映射到 L1 的记录读取的方法限制了准确量化表达的能力。为了说明这一点, 根据每个位点的映射能力创建了一个校正因子。为了创建此校正因子, 使用了第一个床工具从 HeLa 基因组 bam 文件中提取唯一映射的读取数, 这些读取与所有全长 L1 位点对齐, 并绘制了从最高到最低映射的记录读取的这些位点 (补充) 图 1)。它被任意地选定 L1 与400个读有充分的覆盖映射。能够映射到 HeLa 基因组测序样本中 L1 位点的读取数相对于400次读取进行了缩放, 然后将该缩放数量乘以映射到 DU145 中每个真实表达的 L1 位点的读取数 (补充表 2).不出所料, 具有较大的映射校正分数的 L1 元素来自像 L1PA2 这样的年轻亚科 (补充表 2)。一旦根据每个位点的映射分数调整了读取, 大多数位点的表达量的定量就会增加 (图 6)。在 DU145 中唯一映射到具有映射更正的真实表达的特定 L1 位点的读取数从612到4个读取不等, 并且有最高到最低表达位点的重新排序 (图 6)。

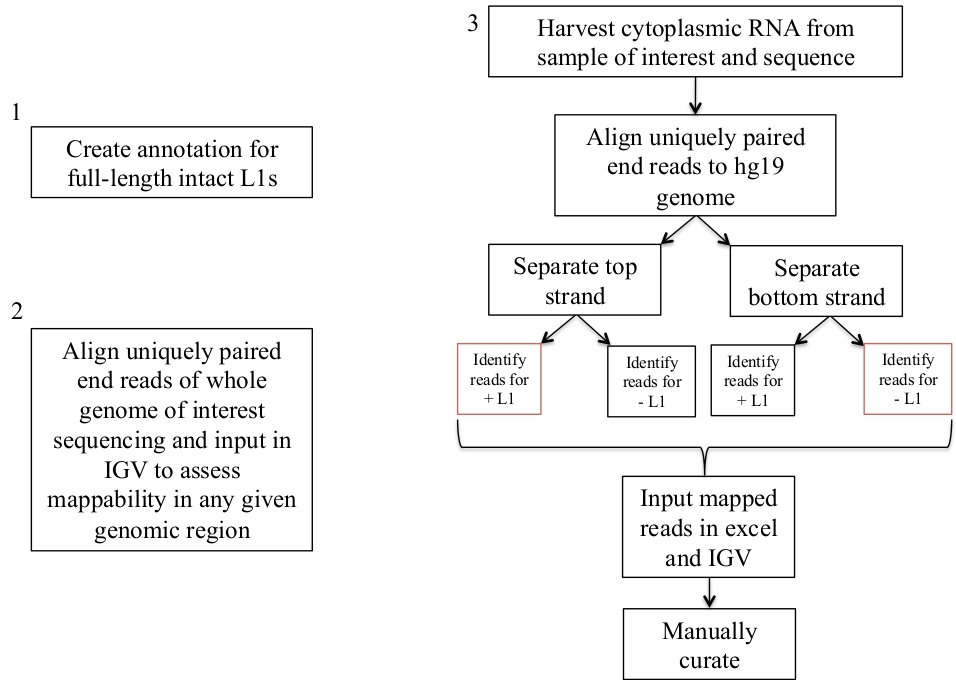
图 1: 工作流原理图。
图形描述的是识别人类样本中表达的 L1 的步骤。请注意, 如果相应的文件已经可用, 则不需要重复步骤1和2。这些适当的文件可以从补充文件1a-b 和补充文件 2下载。红色的框表示使用床工具覆盖程序在同一意义上计算映射到 L1 的读取数的步骤。这些具有面向感官的映射读取的位点是应该手动管理的 L1。请点击这里查看此图的较大版本.
{kind=link}

图 2: DU145 中的计算 L1 位点的示例。
加载到 IGV 是参考基因组, 全长 L1 gff 注释文件匹配的参考基因组版本 (补充文件 1), du145 文件, 最后是基因组 heabam 文件, 以评估映射性, 这都是可在作者请求。添加了箭头以帮助显示带注释的 L1 的方向。红色的箭头和读取按顺序从右到左。蓝色的箭头和读取按从左到右的顺序定向。a) 在 igv 中, 这个 l1 位点似乎是由它自己的启动子表示出来的, 因为在 l1 的上游没有超过5kb 的感觉方向上的读取。这种 L1 具有较低的映射能力, 它不在基因中, 并有预期反义启动子活性的证据 26。b) 在 igv 中, 这个 l1 位点似乎是由它自己的启动子表示出来的, 因为在感觉方向超过5kb 的 l1 上游没有读取。这种 L1 具有较低的映射能力, 并且在相反方向的基因内。c) 在 igv 中, 此 l1 位点被拒绝为表示的 l1, 因为在5kb 范围内存在相同方向的上游读取。这个 L1 是在同一方向的基因内, 所以文字记录的阅读很可能源于表达的基因的启动子。(d) 在 igv 中, 此 l1 位点被拒绝为表示的 l1, 因为在5kb 范围内存在相同方向的上游读取。这个 L1 是一个高度表达的基因在同一方向的下游, 所以文字阅读很可能来自该表达的基因的启动子, 并延伸到正常的基因终止符之外。e) 在 igv 中, 此 l1 位点被拒绝为表示的 l1, 因为在5kb 范围内存在相同方向的上游读取。这个 L1 不在参考基因中的一个附加注释的基因之内或附近, 因此这些转录的起源在 L1 元素的内部和上游表明一个未附加注释的启动子。请点击这里查看此图的较大版本.
{kind=link}

图 3: 背景噪声也来自截断的 L1。
我们的 L1 注释不包括截断的 L1, 因为它们是背景噪声的主要来源。添加了箭头以帮助显示带注释的 L1 的方向。蓝色的箭头和读取按从左到右的顺序定向。a) 演示的是 L1MB5 系列中的一个截断 l1 的例子, 该序列是 2706 bps。在 IGV 中, 读数显然源于表达的基因的下游延伸。b) 显示的是截断 l1 的另一个示例。此 L1 是一个 L1PA11, 长度为 4767 bps。在 IGV 中, 可以明显看出, 唯一映射到 L1 的读取映射来自于表示的外显子, L1 在其中。请点击这里查看此图的较大版本.
{kind=link}

图 4: 在 DU145 前列腺肿瘤细胞系中表达的人类基因组中唯一完整的 L1 的读写器读取。
在黑色是具体的位点被确定为真实地表示在手工策展以后和在红色是具体位点将被拒绝作为真实地表示读在手工策展以后。在灰色是位点与少于十个读取映射到每个。由于这些位点代表了文字记录读取的一小部分, 因此它们不是手动策划的。X 轴刻度线表示每100个全长、完整的 L1. 大约 4, 500个位点没有以图形方式显示, 因为它们的映射读数为零。请点击这里查看此图的较大版本.
{kind=link}

图 5: 在 DU145 前列腺肿瘤细胞系中, 独特地将地图映射到完整的 L1。
显示的是在人工策划后阅读到 DU145 单元中特定位点的文字记录的数量。请点击这里查看此图的较大版本.
{kind=link}

图 6: 在按映射性调整时, 读取映射到真实表示的 L1。
显示的是记录读取的数量, 这些读数由特定于 l因为子的可映射分数调整, 这些分数映射到在 DU145 单元中手动计算 L1 位点。请点击这里查看此图的较大版本.
{kind=link}
补充文件 1: 根据方向的全长完整的人类 L1 注释.Fl-l1-blast _ rm _ mins. gff。b) FL-L1-BLAST _ rm _ gff。请点击此处下载此文件.
补充文件 2: 用于自动化第4节详细介绍的生物信息学管道的超级计算机脚本.请点击此处下载此文件.
补充图 1: 用于确定 L1 映射能力的基因组 DNA 样本。
显示的是从 HeLa 细胞系样本中读取的基因组转录数, 这些样本独特地映射到基因组中的所有 5, 000个全长 L1 位点。当400次读取映射到 L1 时, 它被指定为具有完全覆盖映射。请点击此处下载此图.
补充表 1: DU145 中 L1 的手动饱和度.请点击此处下载此表格.
补充表 2: 在 DU145 中的计划 L1 与映射调整.请点击此处下载此表格.
讨论
L1 活动已被证明会造成遗传损害和不稳定, 导致疾病 27,28,29。在大约 5, 000份全长 L1 副本中, 只有几十份进化上年轻的 L1 占了逆转录数活动2的大部分。然而, 有证据表明, 即使是一些较老的、经后转移的 L1 仍然能够产生 DNA 破坏蛋白 30.为了充分理解 L1 在基因组不稳定和疾病中的作用, 必须了解 L1 在局部特定水平上的表达。然而, L1 相关序列的高背景包含在与 L1 反转换无关的其他 Rna 中, 这对解释真实的 L1 表达性提出了重大挑战。另一个挑战是识别并因此理解单个 L1 位点的表达模式, 这是因为它们的重复性质, 不允许许多简短的读取序列映射到一个唯一的位点。为了克服这些挑战, 我们开发了上述方法, 使用 RNA-Seq 数据识别单个 L1 位点的表达。
我们的方法过滤高水平 (超过 99%)通过采取一些步骤, 产生的与 L1 逆行无关的 L1 序列产生的转录噪声。第一步是制备细胞质 RNA。通过选择细胞质 RNA, 在细胞核中表达的内界 mRNA 中发现的 L1 相关读数被显著耗尽。在测序库准备中, 为减少与 L1 无关的转录噪声而采取的另一个步骤包括选择多腺苷酸化转录。这消除了在非 mrna 物种中发现的与 L1 相关的转录噪声。另一个步骤包括特定于结构的排序, 以识别和消除反义 L1 相关的记录。在识别映射到 L1 的 RNA-Seq 文字记录的数量时, 使用具有功能启动子区域的全长 L1 注释也消除了源自截断 L1 的背景噪声。最后, 消除与 L1 反转位无关的 L1 序列转录噪声的最后一个关键步骤是手动管理被确定为映射了 RNA-Seq 记录的全长 L1。手动策划涉及在其周围的基因组环境中对每个生物信息识别到表达的 L1 位点进行可视化, 以确认这种表达源自 L1 启动子。这种方法适用于 DU145, 前列腺肿瘤细胞系。即使采取了与准备有关的步骤来减少背景噪音, 在 DU145 中确定的生物信息 L1 位点中, 约有50% 被拒绝, 因为 L1 背景噪声来自其他转录源 (图 4),强调产生可靠结果所需的严谨性。这种使用人工策展的方法是劳动密集型的, 但在开发此管道时, 对于评估和了解全长 L1 周围的基因组环境是必要的。接下来的步骤包括通过自动化一些策展规则来减少必要的人工策展量, 不过由于基因组表达的性质尚不完全清楚, 参考基因组中没有注释的表达来源, 低区域映射性, 甚至是与参考基因组的构建相关的复杂因素, 在这个时候是不可能完全自动化的 L1 策展。
在识别具有排序的单个 L1 位点表达方面的第二个挑战涉及重复 L1 转录的映射。在此对齐策略中, 需要记录与参考基因组进行唯一一致的一致, 以便进行映射。通过选择一致绘制一致地图的配对端序列, 与参考基因组中的 L1 位点唯一对齐的转录量增加。这种唯一映射策略提供了对特定于单个 L1 位点的读取映射调用的信心, 尽管它可能低估了每个识别到真实表达的重复 L1 的表达量。为了大致纠正这种低估, 开发了基于每个 L1 位点的 "映射" 分数, 并将其应用于唯一映射的记录读取数 (图 6)。值得注意的是, 理想情况下, 映射性应根据匹配的 WGS 样本在全长 L1 中进行全覆盖读取。在这里, 我们使用 H但细胞的 WGS 来确定每个 L1 位点的映射分数, 以膨胀或降低读取映射到 DU145 前列腺肿瘤细胞系中的 L1 位点。这种映射计算是一个粗糙的校正评分, 但选择的 ' 完全覆盖映射 ' 的400次读数是在考虑肿瘤细胞系动态性质的情况下确定的。在补充图 1中可以看到, 有几个 l1 位点与 H过拉 wgs 具有非常高的映射读取数。这些可能来自在 HeLa 内的重复染色体序列, 这些序列不在参考基因组内, 这就是为什么这些位点没有被选择代表完全的映射覆盖。相反, 根据补充图 1 , 100% 阅读覆盖率的平均值发生在400次左右, 然后假定这一平均值也适用于 du145 肿瘤细胞系。
这种对齐策略与 100-200 bp 读取从 RNA-Seq 技术也优先选择在参考基因组中的进化上更老的 L1, 因为年龄较大的 L1 已经积累了独特的突变随着时间的推移, 使他们更可映射。因此, 这种方法在识别 L1 中最年轻的 L1 以及非引用的多态 L1 时的灵敏度有限。为了确定 L1 中最年轻的, 我们建议使用 5 ' RACE 选择 L1 文字记录和测序技术, 如 PacBio, 利用更长的读数21。这允许更独特的映射, 从而有信心地识别表达的年轻 L1. 使用 RNA-Seq 和 PacBio 方法可以共同产生更全面的真实表达 L1 的列表。为了识别真实表达的多态 L1, 接下来的第一步包括构建多态序列并将其插入参考基因组。
研究重复序列的生物和技术挑战是巨大的, 但通过上述严格的程序, 消除 L1 序列的转录噪声与重复转换无关, 我们开始筛选通过大水平的转录背景噪声, 并在单个位点水平上自信和严格地识别 L1 表达模式和数量。
披露声明
作者没有什么可透露的。
致谢
我们要感谢严东博士的 DU145 前列腺肿瘤细胞。我们要感谢 Nathan Ungerleider 博士在创建超级计算机脚本方面的指导和建议。其中一些工作是由国家卫生研究院资助的, 向 PD 提供了 R01 gm121812, 将 R01 AG057597 授予 VPB, 将5TL1TR001418 授予了传统知识。我们还要感谢癌症十字军和图兰癌症中心生物信息学核心的支持。
材料
| Name | Company | Catalog Number | Comments |
| 1 M HEPES | Affymetrix | AAJ16924AE | |
| 5 M NaCl | Invitrogen | AM9760G | |
| Agilent bioanalyzer 2100 | Agilent technologies | ||
| Agilent RNA 6000 Nano Kit | Agilent technologies | 5067-1511 | |
| bedtools.26.0 | https://bedtools.readthedocs.io/en/latest/content/installation.html | ||
| bowtie-0.12.8 | https://sourceforge.net/projects/bowtie-bio/files/bowtie/0.12.8/ | ||
| Cell scraper | Olympus plastics | 25-270 | |
| Chloroform | Fisher | C298-500 | |
| Digitonin | Research Products International Corp | 50-488-644 | |
| Ethanol | Fisher | A4094 | |
| Gibco (Phosphate Buffered Saline) | Invitrogen | 10-010-049 | |
| Homogenizer | Thomas Scientific | BBI-8541906 | |
| IGV 2.4 | https://software.broadinstitute.org/software/igv/download | ||
| Isopropanol | Fisher | A416-500 | |
| mac2unix | https://sourceforge.net/projects/cs-cmdtools/files/mac2unix/ | ||
| Q-tips | Fisher | 23-400-122 | |
| RNAse later solution | Invitrogen | AM7022 | |
| RNaseZap RNase Decontamination Solution | Invitrogen | AM9780 | |
| samtools-1.3 | https://sourceforge.net/projects/samtools/files/ | ||
| sratoolkit.2.9.2 | https://github.com/ncbi/sra-tools/wiki/Downloads | ||
| SUPERase·In RNase Inhibitor | Invitrogen | AM2694 | |
| Trizol | Invitrogen | 15-596-018 | |
| Water (DNASE, RNASE free) | Fisher | BP2484100 |
参考文献
- International Human Genome Sequencing. Initial sequencing and analysis of the human genome. Nature. 409, 860 (2001).
- Brouha, B., et al. Hot L1s account for the bulk of retrotransposition in the human population. Proceedings of the National Academy of Sciences of the United States of America. 100 (9), 5280-5285 (2003).
- Dombroski, B. A., Mathias, S. L., Nanthakumar, E., Scott, A. F., Kazazian, H. H. Isolation of an active human transposable element. Science. 254 (5039), 1805 (1991).
- Swergold, G. D. Identification, characterization, and cell specificity of a human LINE-1 promoter. Molecular and Cellular Biology. 10 (12), 6718-6729 (1990).
- Speek, M. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Molecular and Cellular Biology. 21 (6), 1973-1985 (2001).
- Deininger, L., Batzer, M. A., Hutchison, C. A., Edgell, M. H. Master genes in mammalian repetitive DNA amplification. Trends in Genetics. 8 (9), 307-311 (1992).
- Boissinot, S., Chevret, P., Furano, A. L1 (LINE-1) Retrotransposon Evolution and Amplification in Recent Human History. Molecular Biology and Evolution. 17 (6), 915-918 (2000).
- Khazina, E., Weichenrieder, O. Non-LTR retrotransposons encode noncanonical RRM domains in their first open reading frame. Proceedings of the National Academy of Sciences of the United States of America. 106 (3), 731-736 (2009).
- Martin, S. L., Bushman, F. D. Nucleic acid chaperone activity of the ORF1 protein from the mouse LINE-1 retrotransposon. Molecular and Cellular Biology. 21 (2), 467-475 (2001).
- Feng, Q., Moran, M. H., Kazazian, H. H., Boeke, J. D. Human L1 Retrotransposon Encodes a Conserved Endonuclease Required for Retrotransposition. Cell. 87 (5), 905-916 (1996).
- Mathias, S. L., Scott, A. F., Kazazian, H. H., Boeke, J. D., Gabriel, A. Reverse transcriptase encoded by a human transposable element. Science. 254 (5039), 1808 (1991).
- Luan, D. D., Korman, M. H., Jakubczak, J. L., Eickbush, T. H. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: A mechanism for non-LTR retrotransposition. Cell. 72 (4), 595-605 (1993).
- van den Hurk, J. A. J. M., et al. Novel types of mutation in the choroideremia (CHM) gene: a full-length L1 insertion and an intronic mutation activating a cryptic exon. Human Genetics. 113 (3), 268-275 (2003).
- Miné, M., et al. A large genomic deletion in the PDHX gene caused by the retrotranspositional insertion of a full-length LINE-1 element. Human Mutation. 28 (2), 137-142 (2007).
- Solyom, S., et al. Pathogenic orphan transduction created by a nonreference LINE-1 retrotransposon. Human Mutation. 33 (2), 369-371 (2012).
- Hancks, D. C., Kazazian, H. H. Roles for retrotransposon insertions in human disease. Mobile DNA. Mobile DNA. 7, 9-9 (2016).
- Tubio, J. M. C., et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 345 (6196), 1251343-1251343 (2014).
- Ewing, A. D., et al. Widespread somatic L1 retrotransposition occurs early during gastrointestinal cancer evolution. Genome Research. 25 (10), 1536-1545 (2015).
- Beck, C. R., Garcia-Perez, J. L., Badge, R. M., Moran, J. V. LINE-1 elements in structural variation and disease. Annual Review of Genomics and Human Genetics. 12, 187-215 (2011).
- Philippe, C., et al. Activation of individual L1 retrotransposon instances is restricted to cell-type dependent permissive loci. eLife. 5, e13926 (2016).
- Deininger, P., et al. A comprehensive approach to expression of L1 loci. Nucleic Acids Research. 45 (5), e31-e31 (2017).
- Jin, Y., Tam, O. H., Paniagua, E., Hammell, M. TEtranscripts: a package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics. 31 (22), 3593-3599 (2015).
- . . Agilent RNA 6000 Nano Kit Guide. , (2017).
- Mueller, O. L., Schroeder, A. . RNA Integrity Number (RIN) –Standardization of RNA Quality Control. , (2016).
- Robinson, J. T., et al. Integrative genomics viewer. Nature Biotechnology. 29, 24 (2011).
- Speek, M. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Molecular Cellular Biology. 21 (6), 1973-1985 (2001).
- Belancio, V. P., Deininger, L., Roy-Engel, A. M. LINE dancing in the human genome: transposable elements and disease. Genome Medicine. 1 (10), 97-97 (2009).
- Iskow, R. C., et al. Natural Mutagenesis of Human Genomes by Endogenous Retrotransposons. Cell. 141 (7), 1253-1261 (2010).
- Scott, E. C., et al. A hot L1 retrotransposon evades somatic repression and initiates human colorectal cancer. Genome Research. 26 (6), 745-755 (2016).
- Kines, K. J., Sokolowski, M., deHaro, D. L., Christian, C. M., Belancio, V. P. Potential for genomic instability associated with retrotranspositionally-incompetent L1 loci. Nucleic Acids Research. 42 (16), 10488-10502 (2014).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。