
Method Article
利用人类诱导多能干细胞生成肿瘤抗原特异性T细胞
* 这些作者具有相同的贡献
摘要
本文介绍了一种利用OP9/DLL1共培养系统生成功能性肿瘤抗原特异性诱导多能干细胞衍生CD8++单阳性T细胞的方法。
摘要
功能性T细胞在体外产生和扩展,可带来广泛的临床应用。其中一种用途是治疗晚期癌症患者。高富化肿瘤抗原特异性T细胞的采用T细胞转移(ACT)已被证明在一些患者中引起转移性癌症的持久回归。然而,在扩张过程中,这些细胞可能会耗尽或衰老,从而限制其作用和体内的持久性。诱导多能干细胞(iPSC)技术可以克服这些障碍,导致在体外产生大量分化程度较低的肿瘤抗原特异性T细胞。人类 iPSC (hiPSC) 有能力分化成任何类型的体细胞,包括淋巴细胞,当 T 细胞用作起始细胞时,这些细胞保留原始 T 细胞受体 (TCR) 基因组重排。因此,将人类肿瘤抗原特异性T细胞重新编程为hiPSC,然后再分化为T细胞系具有产生恢复性肿瘤抗原特异性T细胞的潜力。本文介绍的是一种使用OP9/DLL1共培养系统从hiPSC生成肿瘤抗原特异性CD8++单阳性(SP)T细胞的方法。该方法是体外T细胞系生成的有力工具,将促进体外衍生T细胞的发展,用于再生医学和基于细胞的治疗。
引言
除了生理优势,T细胞还有许多潜在的治疗应用。体外T细胞的产生和扩展可用于疾病建模和治疗验证,以及遗传和后天免疫缺陷状态的治疗来源(即病毒免疫缺陷和淋巴性继发性化疗或移植),以及根除癌症。后一种质量已导致采用T细胞转移(ACT)用于治疗晚期癌症患者1的发展。
ACT包括切除病人的肿瘤,提取肿瘤渗透淋巴细胞(TLL),扩大TiLs前体内,然后重新注入扩大的细胞到患者2。它已被证明是一种有效的治疗方式,为一些转移性癌症患者。 不幸的是,并非所有患者都对这种疗法有反应。以前的报告已经表明,转移细胞的分化状态3,4,5,6,7,8,9,使用大数高富集的癌症抗原特异性T细胞10,和持续T细胞转移11,12所有与更持久的反应13,14相关。因此,当ACT未能引起抗肿瘤反应时,部分原因可能是癌症抗原特异性T细胞产量低,低效率的外体扩张导致反应性克隆的耗尽和丧失,或转移后缺乏持久性4.据推测,这些障碍可以通过在体外产生大量分化性较差的癌症抗原特异性T细胞来克服。
造血干细胞/祖细胞(HSPCs)是体外T细胞生成的常规来源,尽管这种方法受到可以从单个供体1中恢复的少量细胞的限制。胚胎干细胞(ESCs)也已被证明能产生T细胞,但产量低17,因此在临床应用中效率很低。此外,由于T系细胞在早期发育阶段经历其T细胞受体(TDR)的随机基因重组,如果不进一步基因组,就不可能使用HSPC或ESCs来生成纯抗原特异性T细胞群修改,如TCR基因转导。
克服这些警告的一种方法是重新编程TILs到人类诱导多能干细胞(hiPSCs),这可能为体外T细胞生成提供无限的来源。已经表明,癌症抗原特异性TILs可以重新编程到hiPSC,并重新分化为T细胞谱系,这保留了与原始T细胞18、19相同的T细胞受体(TCR)基因重新排列。 这个细节对ACT很重要,因为单个患者肿瘤具有独特的突变谱,并且很少癌症抗原被证明在患者之间共享20。因此,利用癌症抗原特异性TiLs作为体外产生hiPSC衍生T细胞的来源,可以为转移性癌症患者的个性化治疗提供一种新的策略。
此处详细介绍了使用 OP9/DLL1 共培养系统将 hiPSC 衍生的 T 系质细胞区分为功能抗原特异性 CD8++单阳性 (SP) T 细胞的协议。该方法是体外T细胞分化的hiPSC,造血祖体,胚胎干细胞,以及其进一步应用于再生医学和基于细胞的治疗的有力工具。
研究方案
1. 在小鼠胚胎纤维细胞(MEF)上培养人类iPSC(hiPSC)
注:也可以使用培养 hiPSC 的替代方法,包括但不限于:在 6 孔板上播种,预涂有明胶、胶状蛋白混合物、重组层蛋白 511,或用于 hiPSC 扩展的任何其他细胞外基质,以及使用专为人类多能干细胞培养而配制的定明介质培养。
-
栽培 MEF
- 涂上4 mL 0.1%明胶的10厘米细胞培养培养培养皿,在37°C下孵育30分钟。
- 将一小瓶4 x 106辐照MEF快速解冻至10 mL的37°C MEF介质(DMEM = 10% FBS = 1x青霉素-链霉素 = 1x L-谷氨酰胺补充剂)。在 300 x g下在 4°C 下离心 5 分钟。吸出上清液,在MEF介质的9 mL中重新悬浮细胞颗粒。
- 从培养箱中取出明胶涂层的盘子。吸气明胶,并添加7 mL的MEF介质。将 MEF 悬架的 3 mL 板(从步骤 1.1.2)板到明胶涂层盘上。从侧面和前后摇动菜,确保 MEF 在盘子上均匀分布。在37°C孵育8-36小时。
-
在 MEF 上传递 hiPSC
注:数据是使用从长期培养的黑色素瘤TIL中提取的MART-1 iPSC生成的,该类型在HLA-A_02:01上下文中专门识别MART-1肽,如前所述18。- 当菌落直径在 0.8 - 1.2 mm 之间时,通过 hiPSC。在通过之前,在立体显微镜中检查hiPSC菌落,并使用200 μL尖端的塑料边缘去除培养体的任何分化区域。
- 吸气废介质,并添加10 mL hiPSC介质(人类ES培养基 [材料表]= 10纳克/mL 人类基本成纤维细胞生长因子[hbFGF])辅以10μM ROCK抑制剂。
- 一手握住细胞培养皿,将一次性细胞传递工具卷过整个盘子,朝一个方向。施加足够的压力,使整个滚轮叶片接触培养皿,并在滚动操作过程中保持均匀的压力。
- 旋转培养皿 90°并重复步骤 1.2.3。在显微镜中查看板,以目视确认对菌落的正确切割,这些菌落应呈方格。使用 200 μL 移液器通过温和的机械冲洗分离切割菌落。
注:用滚筒切割菌落后,必须立即通过机械冲洗分离切割菌落,因为 3 分钟后,切割菌落将开始重新连接到盘子,并且很难通过冲洗分离同质大小的菌落。 - 将 350 - 600 团切割菌落转移到新的 10 厘米的 MEF 盘中(在 hiPSC 传化前 8 - 36 小时)与 10 mL 的新鲜 hiPSC 介质辅以 10 μM ROCK 抑制剂。在37°C孵育。
注:600团表示约1.0 x 106 MART-1 iPSC,将在第35天产生0.5-1.0 x 106 DP细胞。但是,预期数量将因起始细胞系的效力和培养条件而异。 - 第二天,吸气废介质并添加 10 mL 的新鲜 hiPSC 介质。根据 hiPSC 增长率,每 1-2 天更换一次 hiPSC 介质。
2. 制备OP9/DLL1细胞,用于与hiPSC共培养
- OP9培养基中[最小必需介质(β-MEM)+20%胎儿牛血清(FBS)+1x青霉素-链霉素]中的培养蛋白在37°C下。当OP9/DLL1细胞达到汇合时,吸气培养基用5mL的1x镁、钙和无苯酚无红磷酸盐缓冲盐水(PBS)。洗涤一次。
- 吸气PBS,并添加2 mL 0.05%胰蛋白酶-EDTA。 在37°C下孵育5分钟。然后,加入4 mL的OP9介质,通过移液机械分离细胞层,使单细胞悬浮。
- 通过100μm细胞过滤器将细胞悬浮液转移到50 mL锥形管中,以避免细胞团块。在 300 x g下在 4°C 下离心 5 分钟。吸出上清液,并在 OP9 介质的 12 mL 中重新悬浮。
- 在 6 个新的 10 厘米细胞培养培养皿中,每增加 8 mL 的 OP9 介质。从步骤 2.3 到每个新的 10 厘米盘的 OP9/DLL1 细胞悬浮液的板 2 mL。从正面到背面摇动菜,以确保 OP9/DLL1 在盘上均匀分布。
- 在37°C孵育。当细胞到达汇合时,每2-3天重复一次。
注:制作足够的OP9/DLL1细胞冷冻库存,每4-6周解冻一次新股票,这一点很重要。
3. 在体外分化高PSC到CD8+++单正(SP)T细胞
- 在与 hiPSC 共同培养前一周准备明胶化 OP9/DLL1 菜肴。要制备0.1%明胶溶液,将5mL室温(RT)组织级库存明胶溶液加入500 mL的PBS中。
- 涂层 3 新的 10 厘米细胞培养培养培养皿,每道菜加入 4 mL 0.1% 明胶。 在37°C下孵育30分钟。
- 吸气明胶,并在每道菜中加入8 mL的OP9介质。将 OP9/DLL1 的一个汇盘中(如上文第 2 节所示)移至三道明胶预涂盘。
- 4天后,在明胶上每 10 厘米的 OP9/DLL1 盘中添加 10 mL 的 OP9 介质,每道菜共 20 mL 的介质。
- 7 - 8天后,开始对OP9/DLL1汇盘(分化日0)进行hiPSC共培养。
- 吸气花费媒体从融合 10 厘米盘的 hiPSC 在 MEF.添加 10 mL 的 OP9 介质。使用一次性细胞通过工具切割和分离 hiPSC 菌落,如步骤 1.2.3 和 1.2.4 中所做的那样。
- 使用 200 μL 移液器将 350 - 600 团切割菌落转移到 10 厘米预明胶化 OP9/DLL1 盘(步骤 3.1)上,使用 10 mL 的新鲜 OP9 介质。从正面到背面摇动文化菜,以确保菌落的均匀分布。
注: 或者,可以使用预成型的 hiPSC 胚胎体 (IB) 或小团块悬浮液。 然而,使用一次性细胞传递工具或EB形成系统是首选产生统一大小的hiPSC团块。
- 在第 1 天,吸气废旧介质,并替换为 20 mL 的新鲜 OP9 介质。在OP9/DLL1上共同培养1天的hiPSC团群将显示为小圆单层菌落(图1)。
- 第 5 天,吸出 10 mL 的废介质,并添加 10 mL 的新鲜 OP9 介质。hiPSC菌落将开始分化成原始中皮,其特点是多层黑暗中心。
- 第 9 天,吸出 10 mL 的废介质,并添加 10 mL 的新鲜 OP9 介质。此时,多层中心结构将演化为圆顶状形状,外围网络状区域将开始变得明显。
- 第13天,收获造血祖细胞(HPCs)(图1)。第13天HiPSC衍生结构的特点是一个黑暗的中央器官,周围是圆顶状区域网络,代表先前报告包围人类胚胎干细胞衍生造血祖体的造血区(HZs)21.
注:在没有暗中心的情况下,圆顶状结构的存在表明过程是成功的。无法生产 HPC 可能是由于 OP9/DLL1 质量差、FBS 批次质量低、种子到 OP9/DLL1 上的 iPSC 团块的汇合(350-600 团群是最佳),以及/或 iPSC 管路效力的变化以产生造血前体。- 吸气废介质和洗涤1x与5mL的1x苯酚无红汉克斯的平衡盐溶液与钙和镁(HBSS)修饰。
- 吸气HBSS,在10 mL的HBSS中加入250μL的5000单位/mL胶原酶IV。在37°C孵育45分钟。用胶原酶IV吸脂HBSS,用5mL的PBS洗涤一次。
- 吸气PBS,并加入5 mL 0.25%胰蛋白酶-EDTA。在37°C孵育20分钟。然后,加入4 mL的OP9介质,通过移液分离细胞层,使单细胞悬浮。
- 通过100μm细胞过滤器将细胞悬浮液转移到50 mL锥形管中。 在 300 x g下在 4°C 下离心 5 分钟。吸出上清液,并在 OP9 介质的 10 mL 中重新悬浮。
- 板细胞悬浮到新的明胶10厘米细胞培养培养皿(见步骤3.1.1和3.1.2)。在37°C孵育45分钟。然后,通过温和的移液收集非粘附细胞。
- 通过100μm细胞过滤器将收集的细胞悬浮液转移到50mL锥形管中。 在 300 x g下在 4°C 下离心 5 分钟。在10 mL的分化介质中吸出上清液并重新悬浮[OP9介质,具有5纳克/mL人类干细胞因子(hSCF)、5纳克/mL人FLT3配体(hFLT3L)和5纳克/mL人类白细胞介素7(hIL-7)]。
- 将细胞悬架盘到新的 10 厘米 OP9/DLL1 汇盘中。
- 第16天,通过细胞。
- 通过温和的移液和过滤100μm细胞过滤器,机械地分离非粘附细胞。在 300 x g下在 4°C 下离心 5 分钟。吸出上清液,并在10 mL分化介质中重新悬浮。
- 将细胞悬架盘到新的 10 厘米 OP9/DLL1 汇盘中。
- 之后每5-7天通过重复步骤3.8继续传递非粘附细胞。
- 在第35天,丰富CD4+CD8+双阳性(DP)总体,刺激产生CD8+++SP T细胞(图2)。
- 通过温和的移液和过滤100μm细胞过滤器,机械地分离非粘附细胞,以去除细胞团块。根据制造商的协议,通过CD4磁珠隔离来丰富CD4+细胞群。
注:使用CD4磁珠的原理是从培养基中去除CD4-CD8-DN细胞,因为已经证明这些细胞在刺激后会导致CD4+CD8+DP细胞的直接杀伤。 - 使用 Neubauer 血细胞计和 Trypan 蓝色染料计数活 CD4 富集细胞。 以总浓度 0.5 x 106细胞/mL 悬浮在 OP9 介质中。将1 mL的细胞悬浮液(0.5 x 106细胞)放入每个孔的组织培养平底24孔板的汇合OP9/DLL1。
- 加入100 IU人白莱素2(hIL-2),5纳克/mL hIL-7,500纳克/mL抗人CD3抗体,2μg/mL抗人CD28抗体,然后在37°C培养。
- 在刺激后第4-7天,收集细胞进行分子分析(图3)或与肽脉冲抗原呈现细胞(APC)共培养。
- 通过温和的移液和过滤100μm细胞过滤器,机械地分离非粘附细胞,以去除细胞团块。根据制造商的协议,通过CD4磁珠隔离来丰富CD4+细胞群。
4. 测量hiPSC衍生CD8+++SP T细胞的抗原特异性
注:用于此测试的 APC 类型取决于 hiPSC 衍生 T 细胞的 MHC 限制。在这里,使用T2细胞系,这是T和B淋巴细胞系的杂交。T2细胞表达HLA-A_02:0123,这是由JKF6细胞识别,其中MART1-iPSC派生18。此 T2 细胞系可在 RPMI 1640 + 20% FBS + 1x 青霉素-链霉素中扩展,当细胞密度达到 5 x 105细胞/mL 时,可通过。
- 使用 Neubauer 血细胞测定仪和锥形蓝色染料计数实时 HLA-A_02:01= T-B 混合淋巴细胞 T2 细胞。在24孔组织培养板中孵育APC,在37°C下用1μg/mL MART-1肽孵育2小时。
注:最佳肽浓度是可变的,取决于细胞系和抗原特异性。 - 收集APC,用10 mL的PBS洗涤2倍,以去除任何额外的肽。
- 在 OP9 介质中计数 APC 并悬浮在 2 × 5 x 105个单元/mL,介质中具有 100 IU IL-2 和 5 纳克/mL IL-7。将100 μL的细胞悬浮液(2-5 x 104细胞)放入超低附件U底部96孔板的每个孔中,或直接放入预涂的ELISpot板中。
- 使用细胞分拣机对HIPSC衍生的CD8++++SP T细胞(抗人CD3/CD28抗体刺激后1周)进行分类,并在100 IU IL-2和5 ng/mL IL-7的OP9介质中悬浮在1 x 106细胞/mL。将100μL的细胞悬浮液(1 x 105细胞)到APC的每口井中,在37°C下培养16-20小时。
- 16-20小时后,根据制造商的协议(图4)通过ELISpot测定分析细胞因子分泌配置文件。
结果
13天后hiPSCs与OP9/DLL1共同培养,CD34+CD43+造血祖细胞出现(图1)。在hSCF、hFLT3L和hIL-7的存在下,在非明胶化OP9/DLL1上再培养22天后,造血原代细胞分化成CD3+CD7+CD4+CD4+CD8+双阳性(DP)T系系细胞,其中大多数表示特定于 MART-1 表位 (四联体) 的 TCR (图 2)。
此前已经表明,CD8+ SP T细胞可以通过激动剂肽或抗体驱动的TCR刺激24、25从CD4+CD8+DPT细胞诱导。因此,在培养的第35天,hiPSC衍生的CD4+CD8+DP T细胞在hIL-7和hIL-2的存在下受到抗人CD3和反人类CD28抗体的刺激。 刺激四天后,CD3+CD8+ +SP细胞的数量急剧增加,并且对 MART-1 表位保持特异性,确认其遗传抗原特异性的保存(图3)。
为了确定hiPSC衍生CD8+++SP T细胞的功能特性,分析了干扰素伽马(IFN-+)的抗原依赖性活化和分泌。在用抗人CD3和抗人类CD28抗体刺激1周后,使用细胞分拣机分离出HIPSC衍生CD8++SP T细胞,并与T2细胞系共同培养,表达HLA-A_02:01,带或不带共理MART1肽16-20小时。ELISpot测定表明,当在MART-1肽存在的情况下培养时,hiPSC衍生的CD8+++-SP T细胞分泌的IFN-*与CD8+++和SP T细胞相比,分泌量更高。IFN-α的表达对于T细胞和APC来说是无效的,表明人类T-iPSC衍生的T细胞具有抗原特异性和功能性(图4)。

图1:产生hiPSC衍生造血原细胞。(A) 使用 OP9/DLL1 共文化将 hiPSC 分化为造血系的原理概述。(B) 第 1 天(左上)、3(右上)、7(左下)和 13 日(右下角)出现 hiPSC 派生结构。刻度条 = 100 μm. (C) 在 13 日对 hiPSC 派生 CD34+CD43+造血祖细胞的流量细胞学分析。数据代表六个独立实验(n = 1 到 2)。请点击此处查看此图的较大版本。
{kind=link}

图2:hiPSC分化为Mart1+ CD4+CD8++DPT细胞。 (A) 使用OP9/DLL1共培养的hiPSC衍生造血系与未成熟的T细胞分化的原理概述。(B) 第35天,CD4与CD8+、CD3与CD8+和MART-1四元体表达在hiPSC衍生T细胞中的流动细胞学分析。门在淋巴细胞,单细胞,PI阴性。数据代表三个独立实验(n = 3 - 8)。请点击此处查看此图的较大版本。
{kind=link}

图3:CD8+++SP T细胞表型诱导。在人抗CD3和人体抗CD28抗体驱动刺激后4天对CD4- hiPSC衍生T细胞进行流细胞分析。门在淋巴细胞,单个细胞,PI负数(n = 4)。 请点击此处查看此图的较大版本。
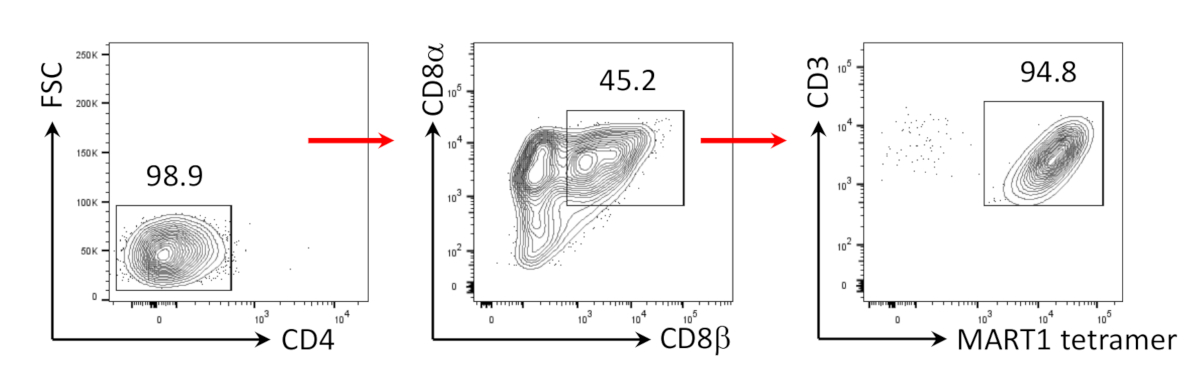{kind=link}

图4:hiPSC衍生CD8++SP T细胞的抗原特异性。 IFN-α 分泌通过ELISpot检测的hiPSC衍生CD8++ + SP,CD8+++SP,和散装T细胞后20小时共培养与或不T2细胞脉冲(或不)与MART-1肽。 请点击此处查看此图的较大版本。
{kind=link}
讨论
OP9鼠位细胞的共同培养是一个完善的系统,用于从HSPC和多能干细胞体外生成淋巴细胞(即NK、B和T细胞)。Notch信号是诱导T系承承诺的,可以通过Notch配体DLL1或DLL4的异位表达来完成,后者对T细胞生成1具有可比的疗效。因此,OP9/DLL1共培养系统已成为在体外产生T细胞的广泛使用方法。 此外,该方法适用于多种类型和来源的人体细胞,包括脐带血、骨髓HSPC和ESCs。 然而,从这些源中产生T细胞受到来源细胞检索不足或对T细胞1的低效分化的限制。此外,具有单个 TCR 重组的 T 单元产品不能从这些开放式剧目源生成。通过使用再生医学技术,即诱导多能干细胞(iPSC)技术,有可能产生大量抗原特异性T细胞,用于基于细胞的治疗15。
hiPSC 与多能 ESC 类似,其自我更新、无限扩张和能够区分体内任何类型的体细胞的能力;然而,他们缺乏关于使用胚胎源产品用于临床应用的道德问题。此外,hiPSC可以从任何体细胞生产,允许开发个性化药物的细胞产品。在以前的报告中,hiPSC是从人类T细胞中产生的,使用全外周单核细胞、CD3+细胞或分离的细胞毒性T淋巴细胞(CTLs)作为来源18,19,22, 26.当HiPSC从T细胞源(T-iPSCs)生成时,原始TCR基因重新排列被继承。因此,患者T-iPSC衍生T细胞可以通过靶向患者独特的癌症抗原,为个性化的ACT治疗提供模型。
人类多能干细胞分化为T系细胞分为两个步骤:造血祖细胞(HPCs)27及其进一步分化为T系细胞21。这两个步骤都可以使用 OP9/DLL1 共区域性系统完成。重要的是,OP9/DLL1进纸细胞的质量对T细胞分化的成功至关重要。由于OP9/DLL1细胞不是不朽的同质细胞系,FBS质量和培养条件对于保持其扩张而不丧失支持hiPSC分化的能力至关重要。因此,建议在细胞与细胞质接触开始发生时,一致地预先评估FBS的批次和通道,以防止细胞分化和衰老。需要考虑的一点是,根据显微镜的相位对比度和放大倍率,细胞与细胞接触可能与背景无法区分。根据我们的经验,大多数 OP9/DLL1 菜肴在准备通过时看起来有 80% 的康收。
研究表明,OP9/DLL1共培养从T-iPSC产生的重新分化的T系系细胞在刺激18、19时可产生CD8+SP T细胞。然而,再生CD8+SP T细胞获得先天状CD8+homodimer22,28,这是TCR信号29的无效共受体。 此外,这些再生的CD8+SP T细胞表现出强烈的TCR无关细胞毒性,使这些细胞不利于临床使用30。该协议描述了最近一种方法,涉及刺激纯化CD4+ CD8+ DP细胞,以产生CD8++ SP T细胞与更传统的表型和改进抗原特异性细胞毒性22。虽然由于继发性TR®等位重排引起的抗原特异性损失是在长期培养后的DP阶段,但可以通过T-iPSCs31中的基因组编辑来克服。 根据我们的经验,hiPSC衍生的DP细胞开始出现在培养的第30-35天,这些新生成的DP细胞尚未经历二次TCR+重排。因此,第35天的大多数DP细胞保留抗原特异性,可用于生成抗原特异性CD8+++SP T细胞。
在人类抗CD3和反CD28刺激之前,CD4-CD8-DN细胞必须从培养基中取出,因为这些细胞已被证明在刺激后导致CD4+CD8+DP细胞直接杀伤。使用CD4磁珠富集(步骤3.10)将丰富DP和CD4+CD8-中间单阳性(ISP)细胞1,我们已经证明没有负面影响22。或者,通过流式细胞测量对荧光激活细胞进行分类,以分离DP细胞。然而,磁珠分离是首选,因为它避免了流动细胞学引起的机械应力。
CD8+++SP T细胞从人类多能干细胞生成,无需活化介导的激动剂选择,随后通过使用3D鼠位细胞培养32来证明。然而,生理阳性选择取决于TCR与自肽-MHC复合物的相互作用,这些复合物由胸腺皮质上皮细胞33进行独特的处理和呈现。此外,TCR对选择肽的亲和力已被证明确定成熟CD8+++SP T细胞34的后续功能。 目前,没有证据表明,基于Notch基质细胞的共培养系统可以提供生理阳性选择所需的定义选择肽和MHC复合物。
此前,在一个小鼠模型中,仅使用OP9/DLL1就从肿瘤抗原特异性T细胞衍生的HiPSCs产生的T系质细胞未能经历常规成熟。然而,由OP9/DLL1系统产生的iPSC衍生的不成熟T细胞可以通过在3D培养系统中进一步进行生理胸腺教育,成熟成天真的T细胞。因此,这里提出的生产由OP9/DLL1系统产生的iPSC衍生的不成熟T细胞的协议对于进一步尝试产生真正的人类肿瘤抗原特异性后胸肌T细胞至关重要,这些细胞能够在体内长期持久。治疗已建立的血管化肿瘤的效率。
披露声明
作者没有披露。
致谢
我们感谢艾伦·胡弗林和埃里娜·他寻求图形援助。这项研究得到了国家癌症研究所(ZIA BC010763)的内学研究计划(ZIA BC010763)和基于细胞的癌症免疫治疗的内NCI癌症月射计划的支持。
材料
| Name | Company | Catalog Number | Comments |
| 10 cm dish | Corning, Inc. | 353003 | |
| Anti-CD3, human | BD Biosciences | Cat# 561812, RRID:AB_1089628 | |
| Anti-CD34, human | BD Biosciences | Cat# 348791, RRID:AB_400381 | |
| Anti-CD4, human | Biolegend | Cat# 344612, RRID:AB_2028479 | |
| Anti-CD43, human | BD Biosciences | Cat# 560198, RRID:AB_1645460 | |
| Anti-CD7, human | BD Biosciences | Cat# 555361, RRID:AB_395764 | |
| Anti-CD8a, human | BD Biosciences | Cat# 555369, RRID:AB_398595 | |
| Anti-CD8b, human | BD Biosciences | Cat# 641057, RRID:AB_1645747 | |
| Anti-TCRb, human | BD Biosciences | Cat# 555548, RRID:AB_395932 | |
| CD28 human monoclonal antibody (15E8), pure functional grade | Miltenyl Biotec | 130-093-375 | |
| CD3 human monoclonal antibody (OKT3), pure functional grade | Miltenyl Biotec | 130-093-387 | |
| CD4 Microbeads, human | Miltenyl Biotec | 130-045-101 | |
| Cell strainer 100 um | Fisher Scientific | 22-363-549 | |
| Fetal Bovine Serum (FBS) | Gemini | 100-500 | |
| Flt-3 ligand | R&D Systems | 427-FL | |
| Gelatin Solution 2% | SIGMA-Aldritch | G1393-100ML | |
| GlutaMAX (100X) | Thermo Fisher Scientific | 35050-061 | L-Glutamine supplement |
| HBSS Mg+Ca+ Phenol-Red Free | Gibco | 14025-092 | |
| Interleukin-2 | R&D Systems | 202-IL | |
| Interleukin-7 | R&D Systems | 407-ML | |
| iTAG MHC Tetramer HLA-A*0201 Mart1 Tetramer -ELAGIGILTV | MBL | Cat#TB-0009-2 | |
| Mart1-hiPSC | Vizcardo et al., Cell Stem Cell 2013 | RIKEN-IMS | |
| Melan-A, MART 1 (26-35) | InnoPep | 3146-0100 | |
| MEM Non-Essential Amino Acids Solution | Gibco | 11140050 | |
| αMEM powder | Gibco | 61100061 | |
| Mouse Embryonic Fibroblasts (MEF) | Thermo Fisher Scientific | C57BL/6 MEF MITC-TREATED 4M EACH; A34962 | |
| OP9/N-DLL1 | Riken Bioresource center | Cat# RCB2927; RRID:CVCL_B220 | OP9/DLL1 |
| Penicillin/streptomycin | Thermo Fisher Scientific | 15140-122 | |
| Phosphate buffered saline pH 7.4 (1x) | Thermo Fisher Scientific | 10010-023 | |
| Primate ES Cell Medium | Reprocell | RCHEMD001 | Human ESC Culture Media |
| Rhok inhibitor (Y-27632 dihydrochloride) | Tocris | 1254 | |
| RPMI 1640 | Gibco | 11875093 | |
| Stem Cell Factor (SCF) | R&D Systems | 455-MC | |
| StemPro | EZPassage | 23181-010 | |
| T2-tumor | ATCC | T2 (174 x CEM.T2) (ATCC® CRL-1992™) | |
| Trypsin-EDTA (0.05%), phenol red | Thermo Fisher Scientific | 25300-062 | |
| Trypsin-EDTA (0.25%), phenol red | Thermo Fisher Scientific | 25200-072 | |
| U Bottom 96 well plate | Corning, Inc. | 3799 |
参考文献
- Brauer, P. M., Singh, J., Xhiku, S., Zuniga-Pflucker, J. C. T Cell Genesis: In Vitro Veritas Est. Trends in Immunology. 37 (12), 889-901 (2016).
- Rosenberg, S. A., Restifo, N. P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 348 (6230), 62-68 (2015).
- Gattinoni, L., et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. Journal of Clinical Investigation. 115 (6), 1616-1626 (2005).
- Rosenberg, S. A., et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clinical Cancer Research. 17 (13), 4550-4557 (2011).
- Crompton, J. G., et al. Lineage relationship of CD8(+) T cell subsets is revealed by progressive changes in the epigenetic landscape. Cellular and Molecular Immunology. 13 (4), 502-513 (2016).
- Henning, A. N., Klebanoff, C. A., Restifo, N. P. Silencing stemness in T cell differentiation. Science. 359 (6372), 163-164 (2018).
- Henning, A. N., Roychoudhuri, R., Restifo, N. P. Epigenetic control of CD8(+) T cell differentiation. Nature Reviews Immunology. 18 (5), 340-356 (2018).
- Vodnala, S. K., et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science. 363 (6434), (2019).
- Restifo, N. P., Gattinoni, L. Lineage relationship of effector and memory T cells. Current Opinion in Immunology. 25 (5), 556-563 (2013).
- Tran, E., et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 344 (6184), 641-645 (2014).
- Gattinoni, L., et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nature Medicine. 15 (7), 808-813 (2009).
- Gautam, S., et al. The transcription factor c-Myb regulates CD8(+) T cell stemness and antitumor immunity. Nature Immunology. 20 (3), 337-349 (2019).
- Klebanoff, C. A., et al. Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clinical Cancer Research. 17 (16), 5343-5352 (2011).
- Klebanoff, C. A., Gattinoni, L., Restifo, N. P. Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy. Journal of Immunotherapy. 35 (9), 651-660 (2012).
- Crompton, J. G., Clever, D., Vizcardo, R., Rao, M., Restifo, N. P. Reprogramming antitumor immunity. Trends in Immunology. 35 (4), 178-185 (2014).
- Crompton, J. G., Rao, M., Restifo, N. P. Memoirs of a reincarnated T cell. Cell Stem Cell. 12 (1), 6-8 (2013).
- Kennedy, M., et al. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Reports. 2 (6), 1722-1735 (2012).
- Vizcardo, R., et al. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell. 12 (1), 31-36 (2013).
- Nishimura, T., et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 12 (1), 114-126 (2013).
- Lo, W., et al. Immunologic recognition of a shared p53 mutated neoantigen in a patient with metastatic colorectal cancer. Cancer Immunology Research. , (2019).
- Timmermans, F., et al. Generation of T cells from human embryonic stem cell-derived hematopoietic zones. Journal of Immunology. 182 (11), 6879-6888 (2009).
- Maeda, T., et al. Regeneration of CD8alphabeta T Cells from T-cell-Derived iPSC Imparts Potent Tumor Antigen-Specific Cytotoxicity. Cancer Research. 76 (23), 6839-6850 (2016).
- Salter, R. D., Howell, D. N., Cresswell, P. Genes regulating HLA class I antigen expression in T-B lymphoblast hybrids. Immunogenetics. 21 (3), 235-246 (1985).
- Snauwaert, S., et al. In vitro generation of mature, naive antigen-specific CD8(+) T cells with a single T-cell receptor by agonist selection. Leukemia. 28 (4), 830-841 (2014).
- Takahama, Y., Suzuki, H., Katz, K. S., Grusby, M. J., Singer, A. Positive selection of CD4+ T cells by TCR ligation without aggregation even in the absence of MHC. Nature. 371 (6492), 67-70 (1994).
- Seki, T., et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell. 7 (1), 11-14 (2010).
- Vodyanik, M. A., Slukvin, I. I. Hematoendothelial differentiation of human embryonic stem cells. Current Protocols in Cell Biology. , (2007).
- Vizcardo, R., et al. Generation of Tumor Antigen-Specific iPSC-Derived Thymic Emigrants Using a 3D Thymic Culture System. Cell Reports. 22 (12), 3175-3190 (2018).
- McNicol, A. M., et al. CD8alpha/alpha homodimers fail to function as co-receptor for a CD8-dependent TCR. European Journal of Immunology. 37 (6), 1634-1641 (2007).
- Themeli, M., Riviere, I., Sadelain, M. New cell sources for T cell engineering and adoptive immunotherapy. Cell Stem Cell. 16 (4), 357-366 (2015).
- Minagawa, A., et al. Enhancing T Cell Receptor Stability in Rejuvenated iPSC-Derived T Cells Improves Their Use in Cancer Immunotherapy. Cell Stem Cell. 23 (6), 850-858 (2018).
- Montel-Hagen, A., et al. Organoid-Induced Differentiation of Conventional T Cells from Human Pluripotent Stem Cells. Cell Stem Cell. 24 (3), 376-389 (2019).
- Takada, K., Kondo, K., Takahama, Y. Generation of Peptides That Promote Positive Selection in the Thymus. Journal of Immunology. 198 (6), 2215-2222 (2017).
- Takada, K., et al. TCR affinity for thymoproteasome-dependent positively selecting peptides conditions antigen responsiveness in CD8(+) T cells. Nature Immunology. 16 (10), 1069-1076 (2015).
- Vizcardo, R., et al. A Three-dimensional Thymic Culture System to Generate Murine Induced Pluripotent Stem Cell-derived Tumor Antigen-specific Thymic Emigrants. JoVE. , e58672 (2019).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。