Brain Pericyte Calcium and Hemodynamic Imaging in Transgenic Mice In Vivo
In This Article
Summary
This protocol presents steps to acquire and analyze fluorescent calcium images from brain ensheathing pericytes and blood flow data from nearby blood vessels in anesthetized mice. These techniques are useful for studies of mural cell physiology and can be adapted to investigate calcium transients in any cell type.
Abstract
Recent advances in protein biology and mouse genetics have made it possible to measure intracellular calcium fluctuations of brain cells in vivo and to correlate this with local hemodynamics. This protocol uses transgenic mice that have been prepared with a chronic cranial window and express the genetically encoded calcium indicator, RCaMP1.07, under the α-smooth muscle actin promoter to specifically label mural cells, such as vascular smooth muscle cells and ensheathing pericytes. Steps are outlined on how to prepare a tail vein catheter for intravenous injection of fluorescent dyes to trace blood flow, as well as how to measure brain pericyte calcium and local blood vessel hemodynamics (diameter, red blood cell velocity, etc.) by two photon microscopy in vivo through the cranial window in ketamine/xylazine anesthetized mice. Finally, details are provided for the analysis of calcium fluctuations and blood flow movies via the image processing algorithms developed by Barrett et al. 2018, with an emphasis on how these processes can be adapted to other cellular imaging data.
Introduction
The central nervous system vasculature consists of penetrating arterioles, capillaries, and ascending venules. Within this network, mural cells such as vascular smooth muscle cells encase arterioles and pericytes extend cellular processes along the first arteriole branches and capillaries1. Pericytes appear to have several roles within the brain including maintenance of the blood-brain-barrier1,2, migration and motility3, potential stem-cell properties, and the regulation of brain blood flow4,5,6. Many of the functional roles of pericytes have been linked to fluctuations in intracellular calcium that may regulate the dilation or contraction of these cells4,5,6.
Several recent studies have set criteria for identifying different types of brain pericytes7,8. Mural cells within the first 4 branches of penetrating arterioles are ensheathing pericytes based on their expression of the contractile protein α-smooth muscle actin (αSMA) and their protruding, ovoid somata with processes that wrap around vessels7,8,9. To visualize calcium fluctuations in ensheathing pericytes, this protocol uses a novel transgenic mouse line, Acta2-RCaMP1.07, also known as Tg(RP23-370F21-RCaMP1.07)B3-3Mik/J10. These mice express the red genetically encoded calcium indicator, RCaMP1.07, in αSMA expressing cells (vascular smooth muscle cells and ensheathing pericytes). Breeding colonies are maintained by crossing noncarrier animals with hemizygotes. RCaMP1.07 is a red fluorescent protein with a calmodulin binding domain, which increases fluorescence when binding to the intracellular calcium10,11. This protocol outlines the steps for combined calcium imaging of ensheathing pericytes and blood flow measurements by two photon microscopy including procedures for tail vein injection of fluorescent dyes, microscope image acquisition in anesthetized mice, and data analysis with programming platforms (Figure 1). These techniques are useful for addressing questions about mural cell physiology but can be adapted to study calcium transients in any cell type in the brain or other organ system.
A 10-month-old female Acta2-RCaMP1.07 mouse was used for the experiment presented in this article. The mouse underwent surgery for chronic cranial window and head post implantation two months prior. Details for the surgical protocol are discussed in previous studies12,13 and similar procedures have been performed in other previously published protocols14,15. The vasculature is labeled with green fluorescein-Dextran (70,000 MW, anionic solution, 2.5% w/v) injected intravenously. This dye is cost effective and readily available from commercial sources, but it has a wider emission spectrum that may overlap with RCaMP emission and bleed through during microscope image acquisition. Steps for spectral unmixing are outlined in Section 4 below to circumvent this, but other green dyes with narrower emission spectra, such as those based on EGFP, may also be used.
Protocol
All of the procedures involving experimental animals outlined below have been approved by the Animal Care Committee of the University of Manitoba, which is governed by the Canadian Council on Animal Care.
1. Procedure setup and preparation
NOTE: The following items are required for a tail vein catheter injection: insulin syringes, a 15 cm piece of PE10 tubing, 30 G needles, gauze, saline, forceps, green fluorescein dextran dye, pliers, and scissors. Also, have a needle ready with ketamine/xylazine anesthesia that will be injected before the imaging session.
CRITICAL: All materials and equipment in Steps 1 and 2 must be sterilized prior to the use by autoclaving or rinsing with 70% ethanol. If the pliers cannot be properly sterilized, the use of a pair of large needle holders is recommended. Catheter assembly must be done with pliers and forceps to avoid accidental needle punctures.
- Cut approximately 15-20 cm of Polyethylene Tubing, PE10 (I.D. 28 mm; O.D. 61 mm).
- Fill a 27 G insulin syringe with 0.9% saline and lace the needle of the syringe into the tip of the polyethylene tube. Push saline through the tube, making sure there are no leaks.
- Using pliers, bend a 30 G needle (0.3 mm x 25 mm) back and forth until it breaks from the hub. The needle must be clean with no bends.
- Holding the needle with forceps, carefully insert the needle into the end of the PE10 tubing that is attached to the saline filled syringe and remove air bubbles. This is the catheter for injection.
- Filter 30 µL of 2.5% (w/v) fluorescein dextran through a 13-25 µm filter prior to the injection.
- Fill another insulin syringe with the 30 µL aliquot of dextran and make sure that there are no bubbles in the filled syringe.
2. Tail vein injection
- Anesthetize the mouse with isoflurane (4% induction, 1.5% maintenance) or ketamine/xylazine (60 mg/kg; 10 mg/kg; i.p.) and apply eye lubricant gel. Ketamine/xylazine is recommended for more stable blood flow measurements during imaging and the dose can be increased to 90 mg/kg; 10 mg/kg ketamine/xylazine for longer imaging sessions.
- When the mouse is at the surgical plane of anesthesia, place a glove filled with warm water on the tail to dilate the lateral vein.
- Remove the glove after 30 s and clean the tail with ethanol.
- Place the tail between the thumb and the middle finger. Provide pressure with the index finger on the tail to dilate the vein. With the other hand, pick up the needle of the catheter with forceps orienting the bezel upwards towards the ceiling.
- After cleaning the tail with 70% ethanol, smoothly, insert the needle into the vein at a 0° angle and gently inject saline through the catheter to ensure the needle is placed correctly.
NOTE: If there is no resistance on the plunger and no swelling of the tail, then the needle is in the vein. If there is significant resistance or swelling, the needle should be removed. Steps 2.2-2.5 can be repeated up to 3 times on each side of the tail, replacing the 30G needle in the end of the catheter every second trial until placement is correct. - Once the needle is in the vein, switch the saline syringe at the end of the catheter with the syringe containing fluorescein dextran (Step 1.6). Slowly inject the dextran into the catheter tubing, ensuring no bubbles enter the tube. If an air bubble is apparent, cut the tubing containing the bubble to remove it and reattach the syringe.
- When all the dextran (30 µL aliquot) has been injected, remove the syringe, and replace it with the saline syringe. Inject the remaining dextran from the tubing into the mouse until no dye is left in the tube.
- Remove the needle from the tail and provide pressure with gauze for 10-30 s until the bleeding stops.
CRITICAL: If after 6 attempts the tail vein injection is not successful, the animal should be imaged in another session. Also, the total volume (saline and dextran) injected into the mouse should not exceed 100 µL.
3. Two photon microscopy
- Focusing on the cranial window
NOTE: Use ketamine/xylazine anesthesia during data acquisition because it has less vascular effects (vasodilation) than isoflurane. If using isoflurane in the steps above, inject the mouse with ketamine/xylazine i.p. (recommended dose described above) before imaging.- Fix the mouse with a screw through its head post to a platform with a heating pad under the microscope.
- Apply eye lubricant to the eyes of the mouse.
- Clean the cranial window with damp dental applicators. Make sure that there are no particles left that could interfere with the imaging process.
- Apply ultrasound gel on the window.
- Focus through the two-photon microscope objective until the pial blood vessels can be seen under the window.
- Check the breathing of the mouse and ensure that the heating pad is providing sufficient temperature support.
- Image acquisition
NOTE: The two-photon microscope used in this experiment has a tunable Ti-Sapphire laser for fluorescence excitation with a Pockel cell that controls the amount of laser that reaches the sample. Emitted light is split by a 565 long pass dichroic to two GaAsP photomultiplier tubes (PMTs) with 595/50 band pass filter (red) and 525/70 band pass filter (green) for detection.
The procedures described in Steps 3.2 to 3.4 are performed using specific software of the two-photon microscope from this protocol (see Table of Materials). These steps can be adapted to other microscope software and equipment.- With the room lights off, set the desired wavelength in the microscope software to 990 nm to excite both RCaMP and fluorescein-dextran by clicking on the 2-P Laser box.
- Set the laser power by clicking on the Power/Gain box/Lasers section and adjusting the Pockels 1 cell voltage at 30% or a value of 300 on a scale of 1000. The laser power that reaches the sample at this setting was previously determined to be ~30 mW.
- Set the PMT detector sensitivity by clicking on the Power/Gain box/PMTs section and adjusting the value to 700-800.
NOTE: These values can be adjusted relative to the intensity of the fluorescent sample and should be set to zero before turning on the lights in the room. - Go to Image Resolution section and click on the 512 x 512 resolution for a larger image size.
- Click on 2-P Laser/Open to open the 2-P Laser shutter.
- Go to Scanning Section and click on the Live Scan button.
NOTE: Live scanning with these parameters and higher resolution, RCaMP positive mural cells and the fluorescently labelled blood plasma can be seen. If the signal is weak, the pockel value can be increased until the picture is clear.
CRITICAL: In the superficial tissue layers, the laser power should not exceed 50 mW, which is about 600 in the Pockels cells settings in this example.
- Acquiring a depth stack of the mural cells and the vascular network
NOTE: Acquisition of a depth stack is recommended to properly locate pericytes in the vascular network. Ensheathing pericytes are located on the first to fourth branches from the penetrating arteriole7,8,9. The microscope software utilized in this protocol refers to depth stacks as "Z-series".- Moving the microscope objective in the X, Y and Z plane, localize a large artery on the surface of the brain based on the smooth muscle cell labelling of RCaMP.
- Click on the Z-Series box.
- Focus at the top of the tissue near the pial vessels, set this as the zero point and top of the Z-series stack by clicking on the Current Z-series/ Start position [µm] section. Click on the box with four black stripes and one red stripe on the top.
- Focus down in the tissue to the desired depth and set this as the bottom of the stack by clicking on the Current Z-series/Stop position [µm] section. Click on the box with four black stripes and one red stripe on the bottom.
- Set the thickness of each image plane (step size) to 1-2 µm by typing the desired value in the box below the "Step Size" button (Step Size button is localized in Z-series/Current Z-series section). This will define the number of pictures that are acquired in the stack.
- Set the laser power to increase exponentially as the microscope moves deeper through the stack by clicking on the Laser/PMT Compensation box and selecting Relative (Exponential Gradient).
- Name the file, chose a folder to save it and click on Start Z-series.
- After acquisition, open the Z-series in the imaging processing software.
- Merge the two channels as colored images and scan through the stack looking for pericytes and blood vessels of interest by clicking in the box Image | Color | Split Channels; Image | Color | Merge Channels.
- Select Regions of interest (ROIs) that contain pericytes and save the positions to help locate these spots again in future imaging sessions.
- T-series calcium imaging movie acquisition (time)
- Using the depth stack and ROIs from above as a reference, move the microscope objective in the X, Y and Z axis during live scan mode until a pericyte of interest is found.
- To collect a movie of pericyte calcium events, increase the acquisition frame rate (>10 frames per second) by going to Image Resolution section, and clicking on the 128x128 box.
- Set the imaging duration to 60 s by clicking on the T-series box and entering the time in the duration box.
- Beside the Save Path box, click on the button with three dots to update the save path with a unique file name.
- Zoom optically on the vessel to account for the lower resolution and to gain a closer view of the pericyte by adjusting the value in the Optical Zoom [mag] section.
- Acquire the T-series by clicking on Start T-series.
- Hemodynamics measurements with kymographs (line scans)
- Focus on the vessel of interest at 512 x 512-pixel resolution in Live Scan mode.
- To measure blood vessel diameter and red blood cell velocity, click on the Line Scan to start a one-dimensional scan with the microscope.
- Set the duration of the scan (30-60 s) in milliseconds.
- Draw a line that bisects the vessel of interest and moves parallel along the vessel. This will generate a kymograph of the vessel diameter on the left and streaks of the red blood cells moving through the vessel on the right.
NOTE: Multiple blood vessels can be measured with the same line as long as they are in the same imaging plane. - Name the file and click on the Start Linescan(s) to acquire the data.
4. Image Analysis
- Calcium movie analysis.
NOTE: This protocol outlines the steps for spectral unmixing (Figure 2) and two different methods to analyze ensheathing pericyte calcium events using manual, hand-selected ROIs (Figure 3) and automated, activity-based ROI selection (Figure 4)16,17. In order to detect and classify the signal peaks with the normalized calcium trace from each ROI, the data is long-pass and band-pass filtered which helps to smooth the data for amplitude and width estimations and also to identify peaks of different shapes: single peaks, multi-peaks, and plateaus (Figure 3B). The parameters for this analysis can be optimized to detect different types of dynamic cellular signals. The steps below will require the use of image processing software and programming software with image processing packages which contain different codes to analyze calcium movies as mentioned above. Please see the materials table for a complete list of programs and packages used in this protocol. Imaging data from different types of microscopes can be imported with these packages maintaining the metadata from the images.
NOTE: Steps 4.1.1-4.1.7 describe how to select ROIs by hand in image processing software for subsequent use in the manual calcium analysis method (Step 4.1.16)- Load the calcium imaging T series in the image processing software, by dragging the .xml file to the software tool bar. Click on the OK box.
- Take the average of the stack (the average of the stack is labeled as "Z projection" by the image processing software). This can be done by clicking on Image | Stacks | Z projection in the tool bar.
- Make a colored image from both channels as in step 3.3.9.
- Open the ROI manager window by clicking in the box Analyze | Tools | ROI Manager, or simply pressing the letter "T" in the keyboard.
- Select the polygon tool by clicking on the polygon shape on the image processing software tool bar and outline the visible ensheathing pericyte structures, such as soma and processes.
- Click on the Add button located in ROI manager window to add selected ROIs in the ROI manager.
- Give each region of interest a unique name by clicking the Rename button and save them as a zip folder that can be loaded later in the programming software by clicking More>> | Save.
NOTE: Steps 4.1.8-4.1.14 describe how to import the calcium T-series into the programming platform and how to unmix the different fluorophores detected by the microscope PMTs into different channels (Figure 2). - Open the programming software and make sure the folders for the image processing packages are on the path (see Table of Materials).
- Import the calcium T-series into the programming software by calling the BioFormats function in the programming platform command window, which automatically opens the file selection window.
- Define what is on each channel by entering the desired number. In this example data, Channel 1 answer=6 (cellular_signal), Channel 2 answer=1 (blood_plasma).
- Plot the data as a movie within the programming software to facilitate visualization by calling the plot function.
- For removing green fluorescence from fluorescein-dextran that bleeds through into the red RCaMP channel, unmix the channels in the image processing package by calling the unmix_chs function in the programming platform command window.
- Select a region that only contains fluorescence from this fluorophore in Channel 1, such as RCaMP in this case.
- Select a region that only contains fluorescence from fluorophore 2, such as fluorescein in the blood plasma in this example.
- Select a background area that does not have fluorescence from either fluorophore. This generates a spectral contribution matrix which is applied to each pixel in each channel. It significantly improves the localization of the RCaMP signal which will enhance the detection of calcium events in these structures.
NOTE: As mentioned above, there are multiple ways that the calcium imaging data can be analyzed within the image processing packages. Steps 4.1.16-4.1.23 describe the method to analyze ensheathing pericyte calcium events using manual, hand-selected ROIs. - Run the cellular signaling analysis on the unmixed calcium movie by calling the CellScan function in the programming platform command window.
- The code will ask "Which ROI detection method would you like to use?". Type the number 2 to load the hand-selected ROIs to the programming platform.
- Load the regions of interest from the zip folder that were selected from the pericytes by hand earlier (Step 4.1.6).
- The code will ask "What's the scale factor?". Determine the scale factor for the hand-selected ROIs relative to the image series being analyzed and type the number of the scale. In this example, the scale factor is 1 because the ROIs do not need to be resized since they were selected on images with 128x128 pixels, the same resolution as the original calcium movie.
- Generate plots of each ROI and the normalized calcium traces in different colors (Figure 3A) by calling the process and plot functions in the command window.
- If the code does not detect majority of calcium events in the individual traces, modify the built-in parameters within the configuration optimization box by calling the opt_config function and adjusting the values, such as decreasing the threshold for the short-pass filtered data to three times the standard deviation of the baseline period, which is the first 30 frames of the T-series.
- Select the Process button in the optimization box to apply the new parameters.
NOTE: In order to detect and classify the signals, the normalized calcium trace is long pass and band pass filtered, which helps to smooth the data for amplitude and width estimations, but also to determine if signals are single peaks, multi-peaks or plateaus (Figure 3B). - Output the data as a .csv file that contains spatial information about the regions of interest and the peaks that were identified by calling the output_data function in the command window. Give the file a unique name for further analysis in a statistics program.
NOTE: Steps 4.1.24-4.1.31 describe the method to analyze ensheathing pericyte calcium events using analysis of activity-based ROIs. - Repeat steps 4.1.8.-4.1.16 to import the calcium movie, unmix the channels, and call the CellScan function in the programming platform.
- The code will ask "Which ROI detection method would you like to use?". Type the number 6 to select the automated region of interest identification based on the activity and change in fluorescence in 3 dimensions (x, y, and time; "3D FLIKA algorithm").
- Plot the processed results to view the identified regions of interest as different colors by calling the process and plot functions in the command window. Each ROI is distinguished in time and space and represented as an overlayed mask (Figure 4).
- If the algorithm does not detect ROIs that are clearly visible by eye, modify the built-in parameters within the optimization box by calling the opt_config function and adjusting the values, such as increasing the gaussian filter that smooths the data in time (by 2 s) and decreasing the threshold for finding ROIs to 3 times the standard deviation of the baseline.
- Select the process button in the optimization box to apply the new parameters. With the optimization process more ROIs should be identified (Figure 4B).
- Plot the ROIs as a movie to clearly identify the areas of activity (outlined in rainbow colors) by changing the mode of the default box to movie within the optimization window for further visualization.
- Output the data as a csv file by calling the output_data function in the command window. This file can be analyzed further in a statistics program.
NOTE: The analysis parameters can be adjusted to fit any type of dynamic cellular signal (calcium, FRET ratios, etc.). All of the steps above can be automated with simple programming code in order to batch process many calcium movies with the same settings.
- Line scan blood flow analysis.
- Import the line scan kymograph data file acquired in section 3.5 into the programming software.
- The code will ask "What is shown on channel 1 and 2?". Define what is on each channel when prompted. In this example, Channel 1 is blank (type 0) and Channel 2 is blood_plasma (type 1).
- Run the diameter analysis function on the line scan by calling the LineScanDiam function, which opens a box to select the area that corresponds to the diameter in the kymograph (Figure 5B, left).
- Draw a box outside the kymograph fluorescence boundaries that corresponds to the vessel diameter.
- Process this data class by calling the process function in order to measure the full width at half maxima for vessel diameter and generate a plot (Figure 5C) with the plot function.
- Output the data as a csv file by calling the output_data function in the command window. This file can be analyzed further in a statistics program.
- Run the velocity radon transformation analysis by calling the LineScanVel function, which opens a box to select the area that corresponds to the RBC velocity in the kymograph (Figure 5B, right).
- Draw a box inside the border of the kymograph fluorescence that corresponds to the vessel velocity.
- Process this data class by calling the process function in order to calculate the velocity, flux, and linear density of red blood cells from the angle of the streaks in the fluorescence. Generate a plot (Figure 5D) with the plot function.
- Output the data as a csv file by calling the output_data function in the command window. This file can be analyzed further in a statistics program.
NOTE: The kymographs must have clear fluorescence with well-defined edges between the black spaces in order for the diameter and velocity analysis to be accurate (Figure 5A, B). It is very important to draw the orthogonal and parallel lines in a precise manner, otherwise reliable analysis of the kymographs will not be possible. Similar to calcium analysis with the image processing algorithms, the parameters for diameter and velocity calculations can be optimized.
Representative Results
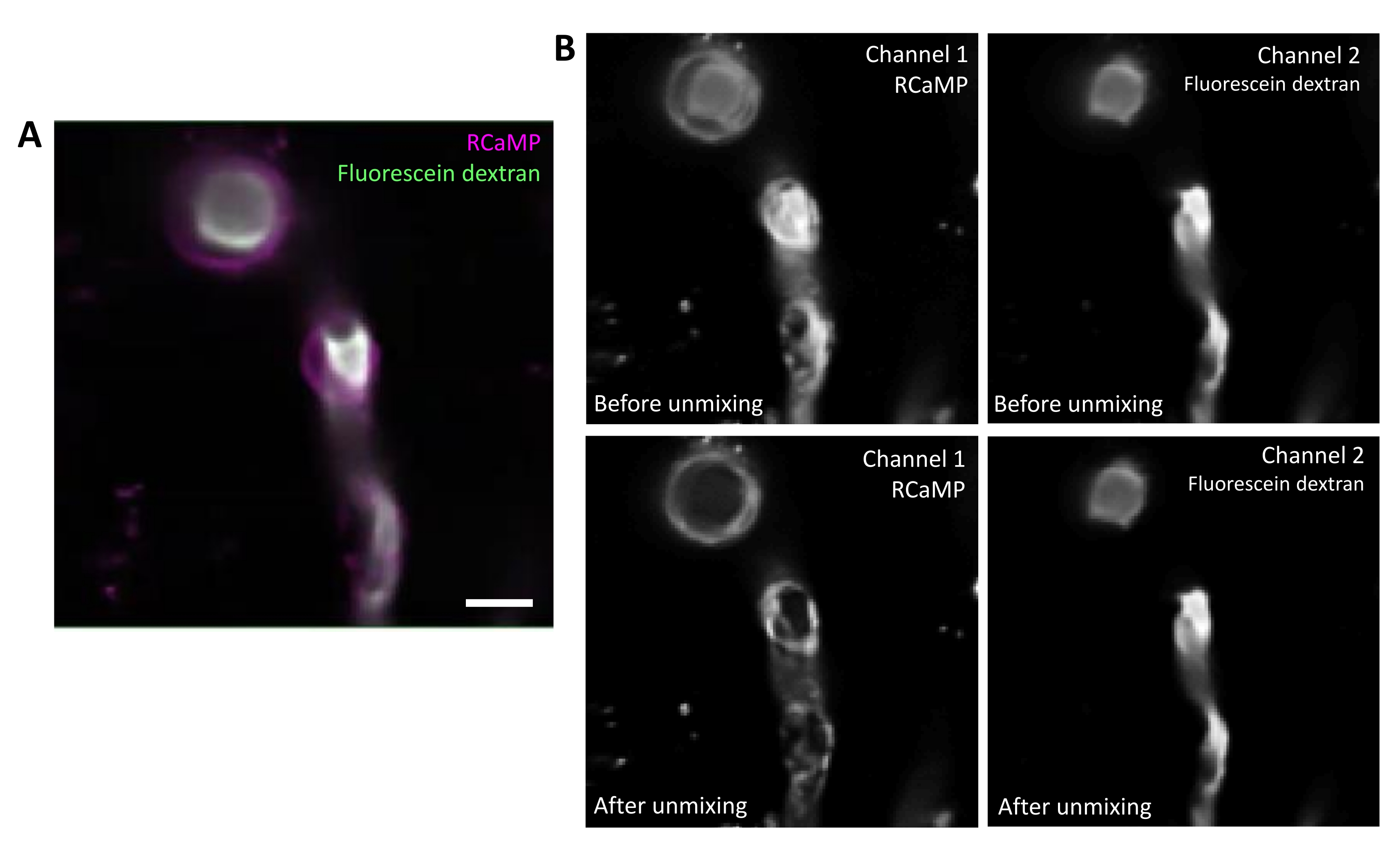
Fluorescein-dextran has a broad emission spectrum that can bleed through into the red channel, impacting RCaMP detection in ensheathing pericytes (Figure 2A). Spectral unmixing after data acquisition in the software program reduces the fluorescein bleeding through (Figure 2B, lower), enhancing calcium signal detection in subsequent analysis steps.
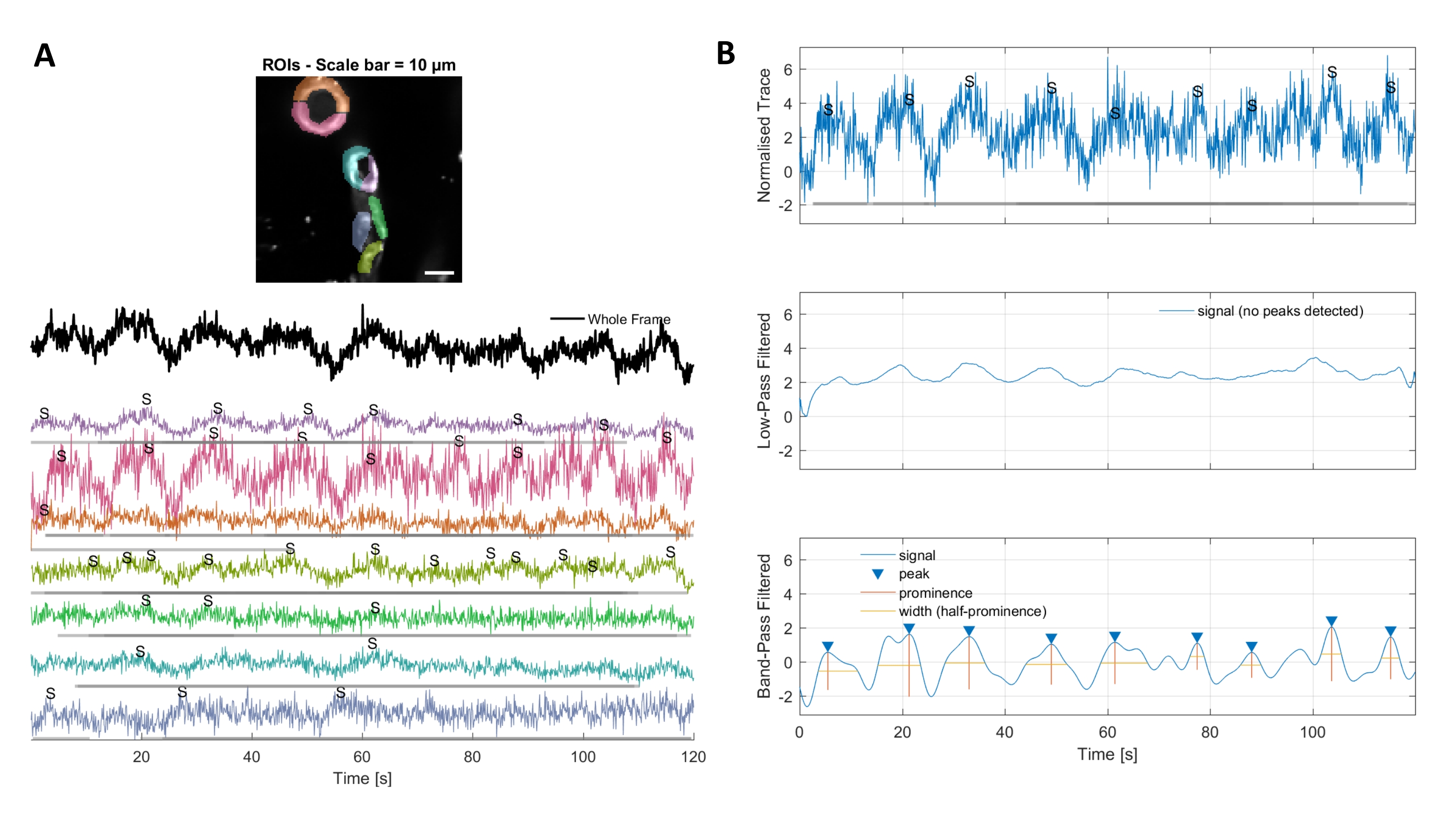
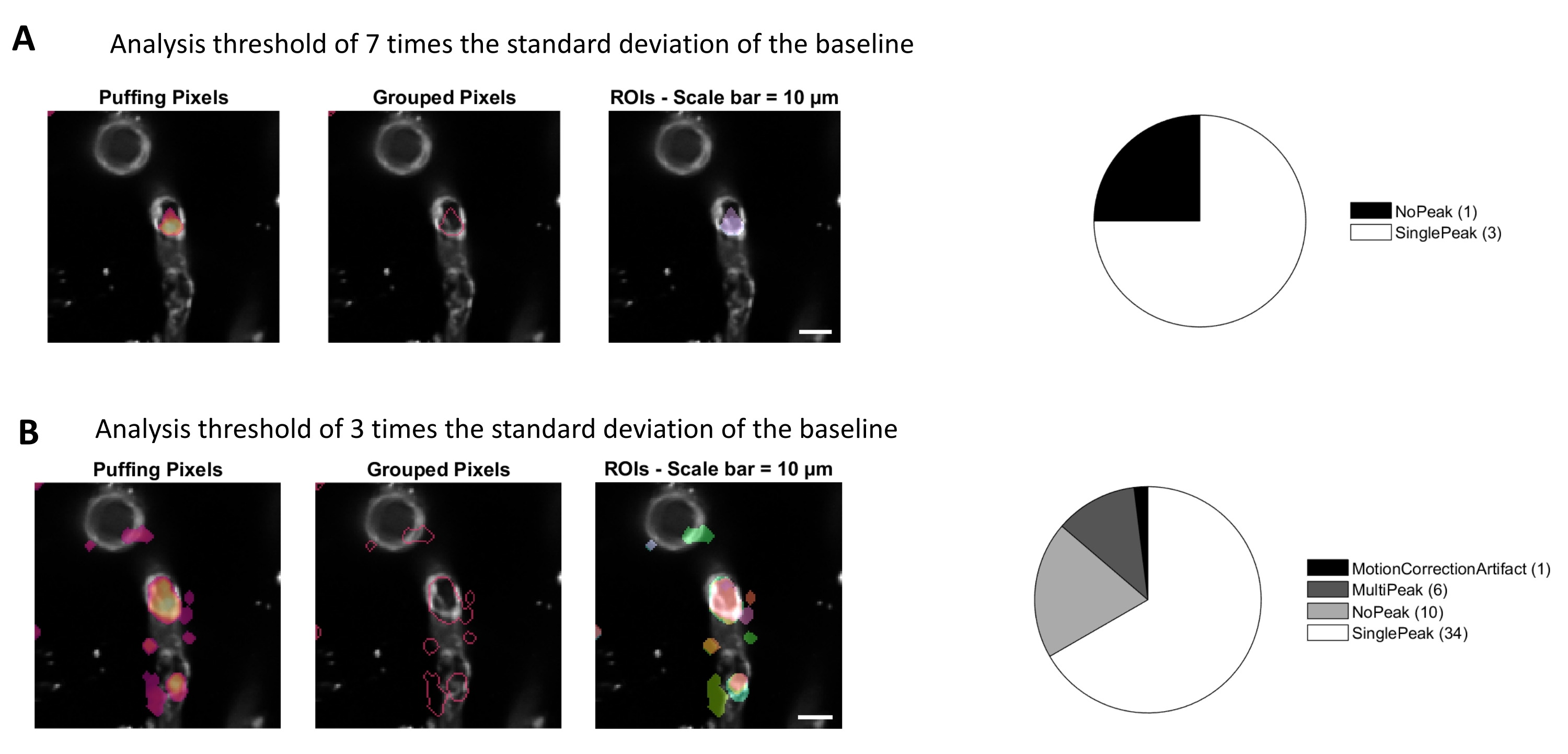
Calcium analysis with the image processing algorithms used in this protocol allows several different approaches to identify ROIs and intracellular calcium fluctuations (i.e., calcium signals). Selecting cellular structures by hand permits detection of calcium fluctuations within these regions (Figure 3A), including different types of signal peaks, such as single peaks and multi-peaks, after the normalized calcium traces are low-pass and band-pass filtered (Figure 3B). Additionally, ROIs are identified by grouping active pixels together where the fluorescence intensity changes over time using image processing algorithms developed by Ellefsen et al. 201416 and Barrett et al. 201817 (Figure 4). This can be applied to any dynamic cellular signal by adjusting the time, threshold and spatial parameters to encompass the expected size and shape of the signal. Decreasing the threshold for signal identification finds more regions of interest (Figure 4B).
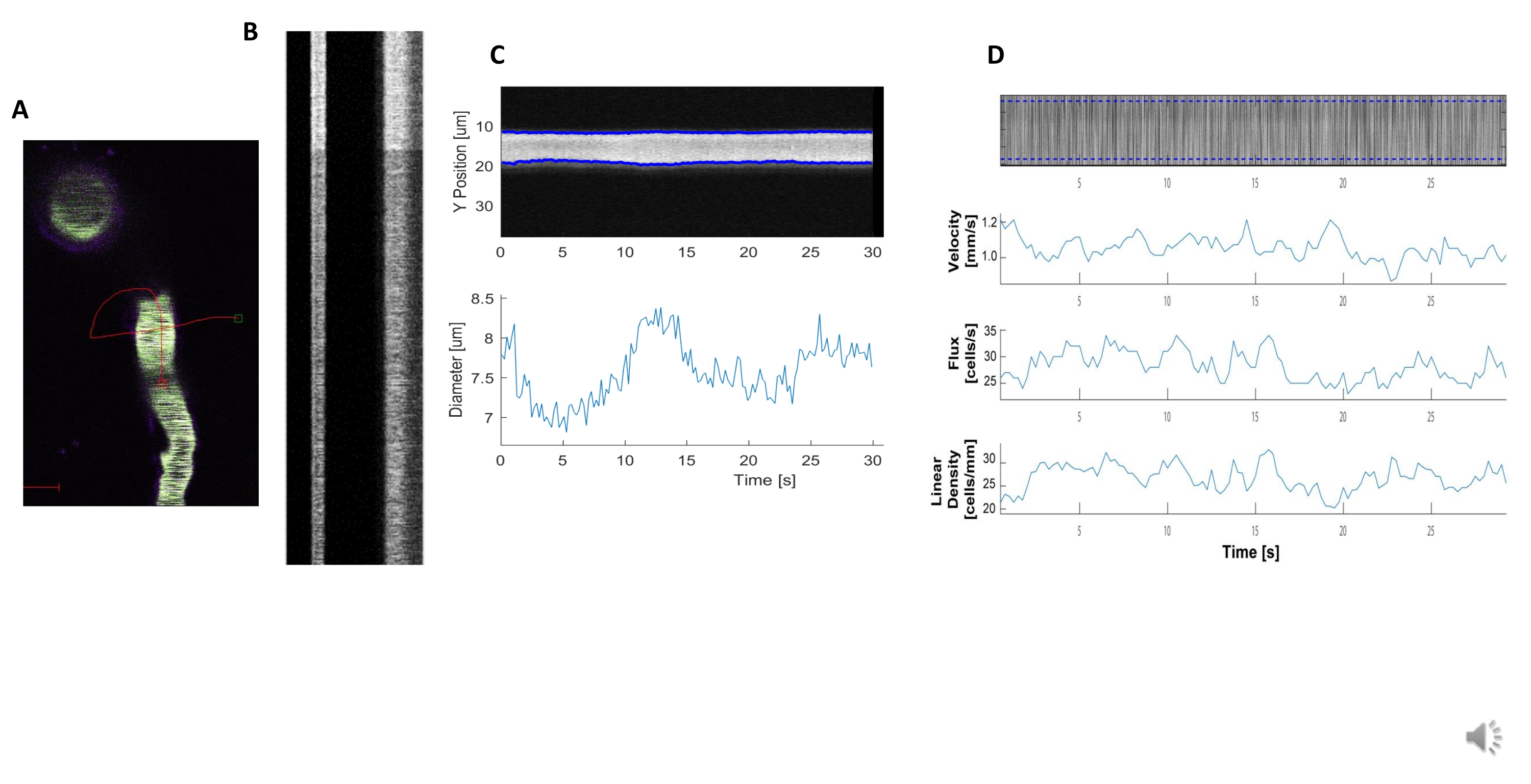
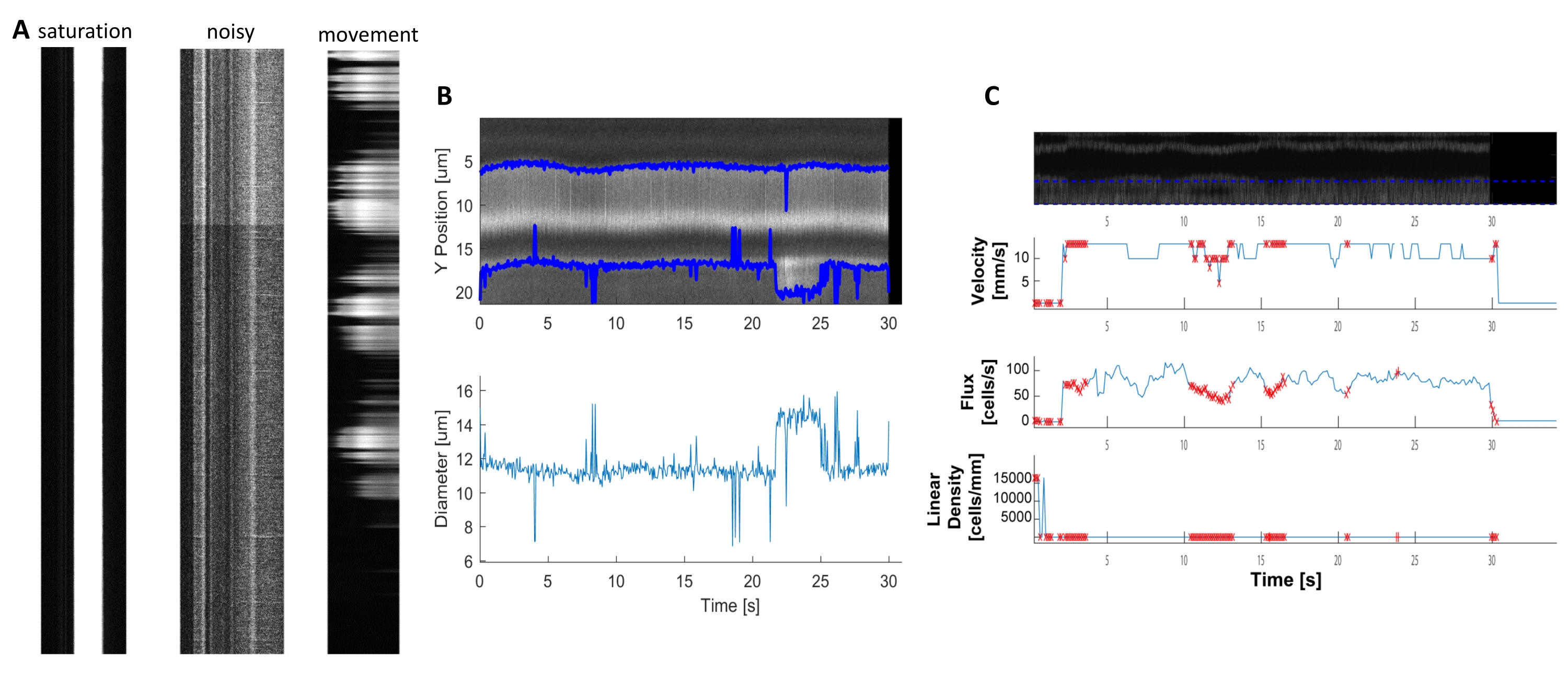
Bright and clear hemodynamic kymographs can be analyzed to measure diameter and RBC velocity in blood vessels near ensheathing pericytes (Figure 5A, B). The diameter is calculated from the full width at half maximum of the fluorescence (Figure 5C). The RBC velocity is approximated from the streaks made from unlabeled RBCs, where the angle is input into a Radon transformation to calculate the velocity, flux (cells/s) and linear density (cells/mm; Figure 5D). Poor-quality kymographs where there is fluorescence saturation, poor signal to noise ratio or movement of the imaging field (Figure 6A) creates unreliable plots with error points (red crosses) where data cannot be determined (Figure 6B, C). The quality of the acquired data is critical for a good outcome and following the steps described in this protocol ensures good results.

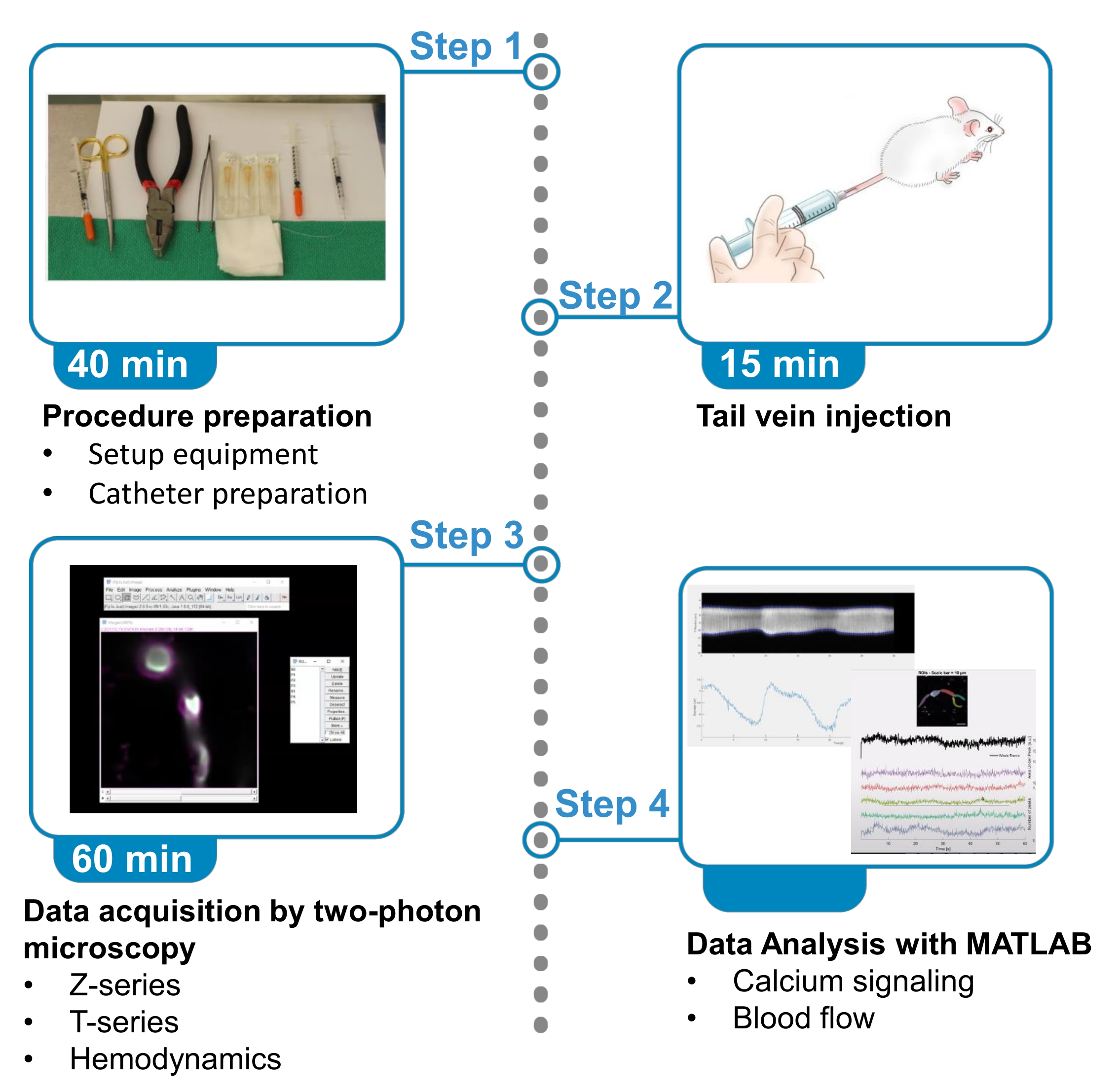
Figure 1. Summary of protocol. The protocol presents the steps to acquire and analyze fluorescent calcium images from brain ensheathing pericytes and blood flow data from nearby blood vessels in anesthetized mice. The protocol is divided in 4 steps. 1) Procedure preparation: set up of equipment and catheter preparation; 2) Tail vein injection; 3) Data acquisition by two-photon microscopy; 4) Data analysis with image processing algorithms. Please click here to view a larger version of this figure.
{kind=link}

Figure 2. Spectral unmixing of fluorophores. A) Representative average image of RCaMP ensheathing pericyte and fluorescein-dextran labelled blood vessel from a T-series acquisition. Scale bar= 10 µm. B) Upper: When considering individual channels, bleed-through from Channel 2 is apparent in Channel 1 (left). Lower: After spectral unmixing, the bleed-through is reduced and signal from RCaMP is more prominent in the pericyte structure. Please click here to view a larger version of this figure.
{kind=link}

Figure 3. Hand selected ROIs and optimized calcium traces. A) Regions of interest selected in the used image processing software (rainbow shapes) can be used to identify calcium signal traces. B) Signal peaks from normalized traces are identified by low pass and band pass filtering the data. We defined the signal threshold as 3 times the standard deviation of the baseline period (first 30 frames) and any peaks over this threshold were considered a signal (lower trace). Please click here to view a larger version of this figure.
{kind=link}

Figure 4. Automated, activity-based ROIs for calcium analysis. The same data was analyzed with a threshold of 7 times the standard deviation of the baseline (A) and 3 times the standard deviation of the baseline (B). Decreasing the threshold for identifying active pixels finds more ROIs (B) and signal peaks (pie chart) within the pericytes. Please click here to view a larger version of this figure.
{kind=link}

Figure 5. Kymograph hemodynamic measurements. A) Example line scan through the vessel. B) Example of well-defined kymographs for diameter (left) and velocity (right). The black streaks within the right band of fluorescence correspond to RBCs. C) Diameter analysis with clear vasomotion fluctuations. D) Velocity analysis with plots for Y axis= RBC flux(cells/s), line density (cells/mm), velocity (mm/s), and streak angle (degree), signal to noise ratio (arbitrary units, a.u.), X axis=time (sec). Please click here to view a larger version of this figure.
{kind=link}

Figure 6. Representation of Poor-Quality Hemodynamic measurements. A) Examples of poor-quality kymographs with fluorescence saturation, poor signal to noise ratio, or movement of the imaging field during acquisition. B and C) Plots similar to Figure 7 of diameter and velocity data that have error points (red dots) because of the poor-quality of the kymographs. (image E, Y axis=Diameter (µm), X axis=time (sec); image F, Y axis= RBC flux(cells/s), line density (cells/mm), velocity (mm/s), and streak angle (degree), signal to noise ratio (a.u.), X axis=time (sec). Please click here to view a larger version of this figure.
{kind=link}
Discussion
The present method provides details on mouse tail vein injection with a catheter, two-photon microscope image acquisition for depth stacks, cell calcium signaling movies, creation of hemodynamic kymographs, and calcium and hemodynamic analysis with our image processing algorithms17 (Figure 1). There are several advantages to these techniques that improve the in vivo imaging outcome and reduce time, resources, and animal stress during the session. First, the use of a catheter for tail vein injection provides more control over the needle, the syringe and the amount of substance injected into the circulation of the mouse. Additionally, it prevents dye injection into the tail tissue, saving expensive reagents. Second, we use transgenic mice which express genetically encoded calcium sensors in ensheathing pericytes and demonstrate how to localize them within the brain vascular network with a depth z-stack, which facilitates cell identification and relocation in subsequent imaging sessions long-term. This is an important factor in pericyte studies and ensures proper cell classification6,7. Third, we provide our parameters for collecting calcium movies and hemodynamic line scan data which are a good starting point for measuring dynamic cellular signals. Finally, we present our image processing algorithms17, a comprehensive image processing toolbox which contains multiple approaches for image pre-processing (such as spectral unmixing), calcium image analysis, and hemodynamic analysis (diameter, velocity, etc.). These algorithms can generate plots for a quick and easy visualization of the data, while minimizing the level of user expertise required to analyze results. Furthermore, it can be automated with a few lines of code to quickly batch process multiple datasets with the same parameters. This can potentially improve data visualization and the time investment of the researcher.
The key to collecting good calcium imaging data is to adjust the laser power and PMT settings for clear fluorescence signal acquisition, but to also collect data at a sufficient frame rate to capture the entire calcium event. The data in this protocol was acquired at 10-11 frames per second, which captures the slower calcium oscillations in ensheathing pericytes. There are also several steps during analysis that can improve the analysis outcome. First, spectral unmixing is beneficial if there is significant overlap between the emission spectra of fluorophores (Figure 2). Fluorescein-dextran was used in this protocol because it is a cost-effective and commercially available dextran conjugate that is commonly used for hemodynamic measurements5. Spectral unmixing helps to clean up the data for enhanced detection of calcium signals, but alternative fluorophores with narrower emission spectra could also be used. Second, hand selecting cellular structures as ROIs (Figure 3) is useful for classifying calcium events in different sub-cellular regions such as the soma or process branches. Activity-based ROI selection (Figure 4)16 provides more spatial and temporal information about individual calcium events. This can be helpful when determining the frequency of calcium events in a given area or the propagation of events to other cellular areas. The use of programming software to analyze imaging data can save researchers hours of time when data is batch-processed, but it requires some initial time investment to adjust the parameters for optimal results. The most important factors are the expected size (in µm2) of the active region as well as the duration of the signal (minimum signal time and maximum signal time must be defined). Researchers must examine some example T-series movies first to best determine which parameters fit their data. Finally, poor quality data acquired on the microscope can greatly hinder the analysis of calcium and hemodynamics (Figure 6). Therefore, care should be taken to optimize the microscope acquisition settings in the beginning. With these factors in mind, this protocol that can be adapted to fit calcium imaging or analysis of other dynamic cellular signals (e.g., fluorescent sodium, potassium, metabolite, or voltage fluctuations) in other tissues or cell types.
There are several limitations to this protocol. First, the data is collected under anesthesia, which affects brain activity and could impact blood flow. Similar imaging can be done in awake mice that are trained to accept head fixation for more physiological results. Additionally, it is important to remember that we collect 2-dimensional images of a 3-dimensional cell and blood vessel in vivo. Therefore, we can only capture a faction of the calcium events within these cells or the blood flow in a single section of blood vessel at a time.
Another limitation to note is that two-photon calcium imaging is sensitive to motion artifacts, where movement in and out of the focal plane can be mistaken for calcium fluctuations. This protocol was performed under anesthesia, which limits movement of the animal; however, motion artifacts can be introduced by the breathing rate of the mouse, heart rate, possible tissue swelling, and in the case of ensheathing pericytes, vessel contraction or vasomotion 4,6,18,19. Motion artifacts can be mitigated by several strategies. The image processing packages used in this protocol include an optional motion correction step, which utilizes a 2D convolution engine to align the images within the T-series based on the visible vasculature13,17. Frames with significant changes in the focal plane are identified by this algorithm and can be excluded from analysis. Additionally, it is possible to use statistical strategies within the imaging processing packages, such as a Z-score when generating the fluorescence traces to normalize the movement induced calcium fluctuations20. The most robust approach to account for motion artifacts in two-photon imaging is to combine expression of two fluorescent indicators within the same cell, such as a calcium indicator (e.g., GCaMP) and a fluorescent reporter (e.g., mCherry) that is calcium-independent. Fluctuations in the fluorescent reporter can then be attributed to movement and are subtracted from the calcium indicator signal to normalize motion artifacts.
The purpose of this protocol is to provide a clear understanding of how to collect optimal calcium imaging and blood flow data in vivo and to present new methods and analysis tools that researchers can implement in order to improve their results. These techniques can be applied to study the role of different pericytes populations in blood flow control or in different brain disease states. These imaging parameters can also be used to study calcium and blood flow in other cell types and organ systems and similar principles apply to other dynamic imaging techniques that are made possible by other genetically encoded sensors, beyond calcium.
Acknowledgements
J. Meza is supported by fellowships from Mitacs and Research Manitoba. Funding for this work was provided by Canadian Institutes for Health Research, Research Manitoba, Manitoba Medical Service Foundation, start-up funding from the University of Manitoba and Brain Canada through the Canada Brain Research Fund, with the financial support of Health Canada and the Azrieli Foundation. The views expressed herein do not necessarily represent the views of the Minister of Health or the Government of Canada.
Materials
| Name | Company | Catalog Number | Comments |
| Acta2-RCaMP1.07 | The Jackson Laboratory | 28345 | In the video protocol the animal model used is a female mouse of 10 months, 1 day old. |
| Applicators (Regular) | Bisco | X-80250P | |
| BioFormats package for MATLAB | NA | NA | Denominated in this protocol as "image processing packages". Available in: https://docs.openmicroscopy.org/bio-formats/ |
| CHIPS MATLAB toolbox | NA | NA | Denomitaded in this protocol as "image processing algorithms". Barrett MJP, Ferrari KD, Stobart JL, Holub M, Weber B. CHIPS: an Extensible Toolbox for Cellular and Hemodynamic Two-Photon Image Analysis. Neuroinformatics. 2018;16(1):145-147. doi:10.1007/s12021-017-9344-y. Available in: https://github.com/EIN-lab/CHIPS |
| Clear Ultrasound Gel, Medium viscosity | HealthCare Plus | UGC250 | |
| Dextran, fluorescein, 70,000 MW, anionic | Thermo Fisher Scientific | D1823 | |
| Dextran, Texas Red, 70,000 MW, neutral | Thermo Fisher Scientific | D1830 | |
| Eye Lube Plus | Optixcare | NA | |
| FIJI | Image J | NA | Denominated in this protocol as "image processor software". Available in: https://imagej.net/Fiji/Downloads |
| GCaMP6sfl/fl | The Jackson Laboratory | ||
| Head Post fixing platform | University of Zurich | NA | |
| Ketamine (Narketan 100 mg/mL) | Vetoquinol | 440893 | |
| MATLAB R2020b | NA | Denominated in this protocol as "programming platform ". Available in: https://www.mathworks.com/downloads/ | |
| Needle 0.3mmx25mm | BD PrecisionGlide | 305128 | |
| Objective XLUMPLFLN20XW | Olympus | NA | https://www.olympus-lifescience.com/en/objectives/lumplfln-w/ |
| PDGFRβ-CreERT2 | The Jackson Laboratory | 30201 | |
| Polyethylene Tubing, PE10 I.D. 28mm (0.11”) O.D. 61mm (.024”) | BD Intramedic | 427401 | |
| Prairie View | Bruker Fluorescence Microscopy | NA | https://www.bruker.com/en/products-and-solutions/fluorescence-microscopy/multiphoton-microscopes/ultima-in-vitro.html |
| Ultima In Vitro Multiphoton Microscope | Bruker Fluorescence Microscopy | NA | https://www.bruker.com/en/products-and-solutions/fluorescence-microscopy/multiphoton-microscopes/ultima-in-vitro.html |
| Under Tank Heater | Reptitherm U.T.H | E169064 | |
| Xylazine (Rompun 20 mg/mL) | Bayer HealthCare | 2169592 |
References
- Armulik, A., Genové, G., Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Developmental Cell. 21 (2), 193-215 (2011).
- Armulik, A., et al. Pericytes regulate the blood-brain barrier. Nature. 468 (7323), 557-561 (2010).
- Berthiaume, A. -. A., et al. Dynamic remodeling of pericytes in vivo maintains capillary coverage in the adult mouse brain. Cell Reports. 22 (1), 8-16 (2018).
- Rungta, R. L., Chaigneau, E., Osmanski, B. -. F. F., Charpak, S. Vascular compartmentalization of functional hyperemia from the synapse to the pia. Neuron. 99 (2), 362-375 (2018).
- Shen, Z., Lu, Z., Chhatbar, P. Y., O'Herron, P., Kara, P. An artery-specific fluorescent dye for studying neurovascular coupling. Nature Methods. 9 (3), 273-276 (2012).
- Gonzales, A. L., et al. Contractile pericytes determine the direction of blood flow at capillary junctions. Proceedings of the National Academy of Sciences of the United States of America. 117 (43), 27022-27033 (2020).
- Hartmann, D. a., et al. Pericyte structure and distribution in the cerebral cortex revealed by high-resolution imaging of transgenic mice. Neurophotonics. 2 (4), 041402 (2015).
- Grant, R. I., et al. Organizational hierarchy and structural diversity of microvascular pericytes in adult mouse cortex. Journal of Cerebral Blood Flow & Metabolism. 39 (3), 411-425 (2017).
- Hill, R. A., Tong, L., Yuan, P., Murikinati, S., Gupta, S., Grutzendler, J. Regional blood flow in the normal and ischemic brain is controlled by arteriolar smooth muscle cell contractility and not by capillary pericytes. Neuron. 87 (1), 95-110 (2015).
- 28345 - STOCK Tg(RP23-370F21-RCaMP1.07)B3-3Mik/J. Jackson Laboratory Available from: https://www.jax.org/strain/028345 (2021)
- Ohkura, M., Sasaki, T., Kobayashi, C., Ikegaya, Y., Nakai, J. An improved genetically encoded red fluorescent Ca2+ indicator for detecting optically evoked action potentials. PLoS ONE. 7 (7), (2012).
- Stobart, J. L., et al. Long-term in vivo calcium imaging of astrocytes reveals distinct cellular compartment responses to sensory stimulation. Cerebral Cortex. 28 (1), 184-198 (2018).
- Stobart, J. L., et al. Cortical circuit activity evokes rapid astrocyte calcium signals on a similar timescale to neurons. Neuron. 98 (4), 726-735 (2018).
- Mostany, R., Portera-Cailliau, C. A craniotomy surgery procedure for chronic brain imaging. JoVE. (12), e680 (2008).
- Lin, X., et al. Imaging neural activity in the primary somatosensory cortex using Thy1-GCaMP6s transgenic mice. JoVE. (143), e56297 (2019).
- Ellefsen, K. L., Settle, B., Parker, I., Smith, I. F. An algorithm for automated detection, localization and measurement of local calcium signals from camera-based imaging. Cell Calcium. 56 (3), 147-156 (2014).
- Barrett, M. J. P., Ferrari, K. D., Stobart, J. L., Holub, M., Weber, B. CHIPS: an extensible toolbox for cellular and hemodynamic two-photon image analysis. Neuroinformatics. 16, 145-147 (2018).
- Hall, C. N., et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 508 (1), 55-60 (2014).
- Nilsson, H., Aalkjaer, C. Vasomotion: mechanisms and physiological importance. Molecular interventions. 3 (2), 79-89 (2003).
- Rungta, R. L., et al. Vascular arbors in layer II / III somatosensory cortex. Communications Biology. , (2021).
This article has been published
Video Coming Soon
关于 JoVE
版权所属 © 2024 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。