
需要订阅 JoVE 才能查看此. 登录或开始免费试用。
Method Article
参与蛋白质-蛋白质相互作用的潜在多特异性肽结合结构域的氨基酸偏好的计算预测
摘要
我们描述了一种基于序列多样化的方法,用于估计蛋白质-蛋白质相互作用 (PPI) 中多特异性结合位点的氨基酸偏好。在这种策略中,生成了数千个潜在的肽配体,并在 计算机中进行了筛选,从而克服了可用实验方法的一些限制。
摘要
许多蛋白质-蛋白质相互作用涉及短蛋白质片段与肽结合结构域的结合。通常,这种交互需要识别具有变量守恒的线性基序。同一配体中高度保守和可变区域的组合通常有助于结合的多特异性,这是酶和细胞信号转导蛋白的共同特性。肽结合结构域的氨基酸偏好表征对于蛋白质-蛋白质相互作用 (PPI) 介质的设计非常重要。计算方法是通常昂贵且繁琐的实验技术的有效替代方案,能够设计出潜在的介质,这些介质可以在以后的下游实验中得到验证。在这里,我们描述了一种使用 Rosetta 分子建模包的 Pepspec 应用程序来预测肽结合域的氨基酸偏好的方法。当受体蛋白的结构和肽配体的性质都已知或可以推断时,这种方法很有用。该方法从配体中一个特征明确的锚点开始,通过随机添加氨基酸残基进行延伸。然后通过柔性骨架肽对接来评估以这种方式生成的肽的结合亲和力,以选择具有最佳预测结合分数的肽。然后使用这些肽来计算氨基酸偏好,并选择性地计算可用于进一步研究的位置-权重矩阵 (PWM)。为了说明这种方法的应用,我们使用了人干扰素调节因子 5 (IRF5) 亚基之间的相互作用,以前已知是多特异性的,但在全球范围内由称为 pLxIS 的短保守基序引导。估计的氨基酸偏好与先前关于 IRF5 结合表面的知识一致。被磷酸化丝氨酸残基占据的位置表现出高频率的天冬氨酸和谷氨酸,这可能是因为它们带负电荷的侧链与磷酸丝氨酸相似。
引言
两种蛋白质之间的相互作用通常涉及氨基酸的短片段与肽结合结构域的结合,类似于蛋白质-肽界面。参与这种蛋白质-蛋白质相互作用 (PPI) 的受体蛋白通常具有识别一组重叠但不同的配体序列的能力,这种特性称为多特异性 1,2。多特异性识别是许多细胞蛋白的一个特征,但在酶和细胞信号转导蛋白中尤为显著3。与多特异性结合位点相互作用的蛋白质在其序列中通常具有更多和更不保守区域的组合 4,5,6。在这种情况下,更保守的序列基序参与严格的分子相互作用。相反,更多的可变序列以某种方式与受体结合位点中允许的表面相互作用。通常,这些不太保守但仍然具有功能相关性的片段是缺乏明确二级结构模式的环,或者具有更多的动态构象,例如那些典型的固有无序蛋白7。
鉴定结合位点的潜在肽配体通常是设计能够干扰相应 PPI 的介质的第一步8。然而,在多特异性结合位点的配体的大多数序列位置通常不太可能找到单个最常见的氨基酸残基。相反,这些位点可能根据其化学性质对特定类别的氨基酸有特定的偏好,例如,酸性和带负电荷的氨基酸,如天冬氨酸或谷氨酸,大体积芳香族氨基酸,如苯丙氨酸或更多的疏水残基,如脂肪族氨基酸丙氨酸、缬氨酸、亮氨酸或异亮氨酸3。几种实验方法可以提供有关蛋白质结合位点氨基酸偏好的见解,包括定向进化9 、多密码子扫描诱变10 和深度突变扫描11。所有这些方法都遵循序列多样化的方法,该方法基于将突变引入原始配体并进一步分析它们对受体蛋白功能的影响(参见 Bratulic 和 Badran12 的全面综述)。然而,这些方法通常需要对大型序列库进行调查,这使得它们更加繁琐、昂贵和耗时。
推断多特异性结合位点氨基酸偏好的计算方法有可能规避湿实验室方法的局限性。其中,计算机序列多样化方法评估了配体序列中各种氨基酸替换的能量影响,以此来表征 PPI 的结构可塑性13。该方法从与受体结合位点结合的肽配体的结构或模型开始,随后将突变引入配体序列。然后使用统计和能量评分函数来评估这些突变对稳定性和结合亲和力的影响。然后,可以使用评估阶段得到的一组得分最高的配体序列来计算氨基酸偏好。这种策略有可能以有效的方式处理非常大量的配体序列。因此,与从湿实验室方法中通常可以处理的更有限数量的序列计算的序列相比,它可以提供更完整和一致的氨基酸偏好推断。
Rosetta 分子建模套件14 的 Pepspec 应用程序是一种工具,可将序列多样化作为其肽设计模式的关键步骤。此应用需要受体蛋白的结构或模型,其中结合肽的长度低至单个氨基酸残基,用作后续步骤的锚点。然后,结合肽的序列被延伸(如有必要)并多样化,以产生大量推定的肽配体。然后通过柔性骨架肽对接评估这些肽的结合亲和力,以选择具有最佳预测结合分数的肽。尽管此应用程序的主要输出是在设计阶段结束时选择的最佳候选肽,但在此阶段接受的更大的肽集也可用于计算目标结合位点的氨基酸偏好。氨基酸偏好计算为配体序列每个位置的每个氨基酸残基的频率,表示为位置权重矩阵 (PWM) 或更直观的序列标志。
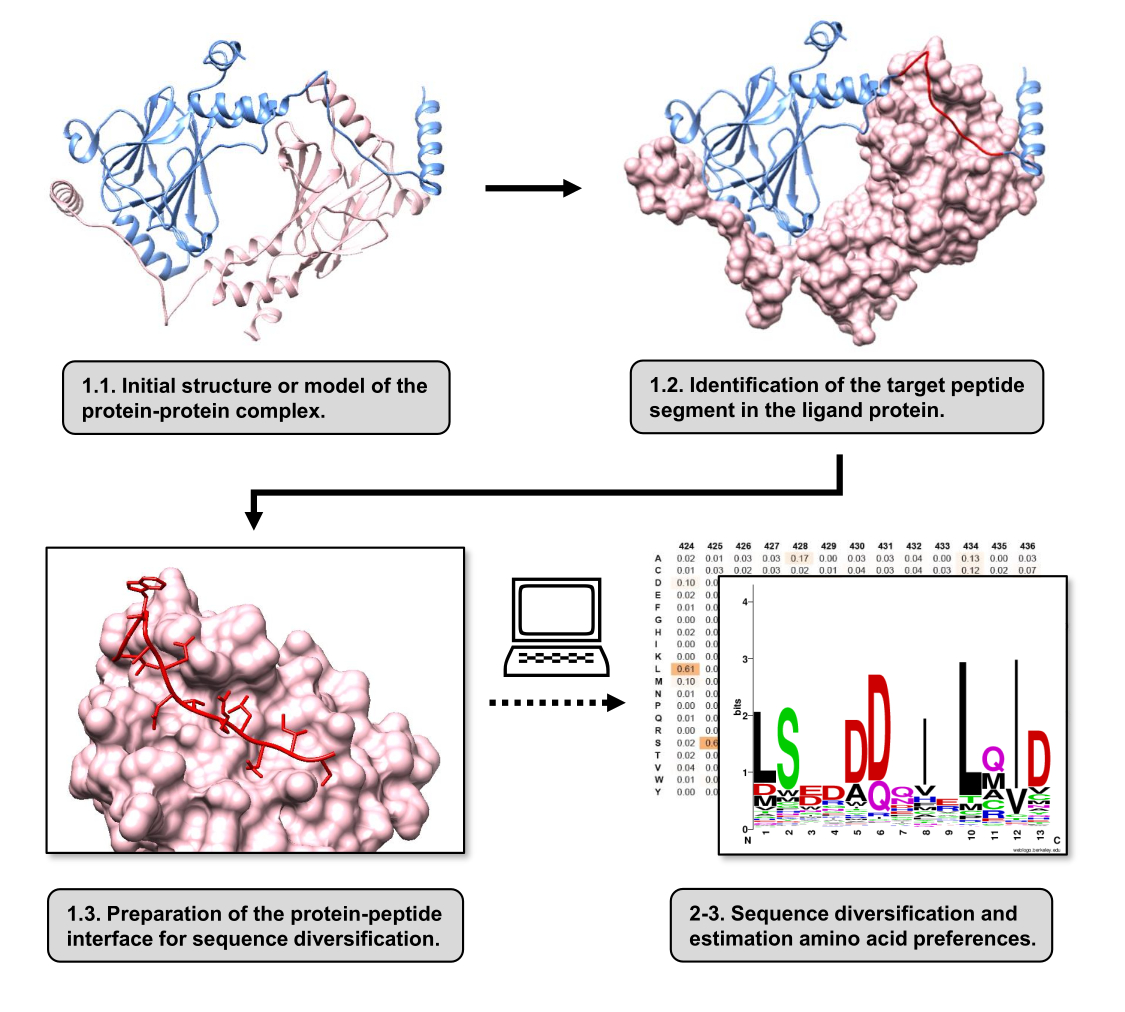
在本文中,我们描述了一种方案,用于估计参与 PPI 的受体蛋白结合表面的氨基酸偏好。该方案侧重于 PPI,其中已知蛋白质-配体的线性片段与受体蛋白结合,因此该场景可以建模为蛋白质-肽界面。在这种情况下,来自配体的保守基序通常与受体结合位点中的特定口袋相互作用,尽管 PPI 中涉及的整个配体片段可能包含不太保守的区域。 图 1 显示了总结该协议主要步骤的流程图。该方案从蛋白质-蛋白质复合物的 3D 结构开始,进一步将配体蛋白还原到潜在的最佳相互作用片段,保持受体蛋白完整。通过使用 BUDE Alanine Scan 服务器15 推断出相互作用最好的片段,该服务器进行计算丙氨酸扫描诱变以识别两种相互作用蛋白之间的热点残基。在这种方法中,来自配体的残基被丙氨酸单独取代,然后使用复合物自由能或稳定性 (ΔΔG) 的估计变化来推断相应残基与目标 PPI 的相关性。一旦推断出相互作用最好的片段,其与受体蛋白的复合物就被用作提交给 Pepspec 的碱基结构以进行序列多样化。

图 1:本研究中提出的协议的主要步骤概述。 编号与 protocol 部分中的步骤编号匹配。图是用文中描述的例子中使用的蛋白质-蛋白质复合物制作的。在该复合物中,被视为受体的蛋白质链显示为粉红色,而被视为配体的链显示为浅蓝色,其预测的最佳相互作用片段以红色突出显示。 请单击此处查看此图的较大版本。
{kind=link}
建议的方案的局限性之一是需要蛋白质-肽界面的解析结构。该方案可以选择从目标蛋白质 - 肽界面的模型开始,尽管此处未描述具体的建模步骤。此外,尽管该协议可以在运行任何操作系统的个人计算机上执行,但涉及 Rosetta 应用程序的步骤需要 Linux 环境。由于 Pepspec 通常执行大量迭代,因此强烈建议将计算机集群用于序列多样化步骤。
通过估计 IRF5(人干扰素调节因子 (IRF) 家族的成员)的出价表面的氨基酸偏好来说明建议的方案的应用。我们选择这种蛋白质作为示例,因为在其激活过程中,两个亚基结合形成一个二聚体,其结构非常明显16。在 IRF 二聚体中,结合可以建模为蛋白质-肽界面,其中一个亚基提供结合表面,另一个亚基通过包含称为 pLxIS 的短保守基序的区域相互作用17,18。此外,与 IRF 亚基的结合是多特异性的;因此,它们可以与其他称为共激活因子的细胞蛋白形成同源二聚体、异二聚体和复合物18。
研究方案
1. 蛋白质-肽界面的初始制备
- 下载蛋白质-蛋白质复合物的结构
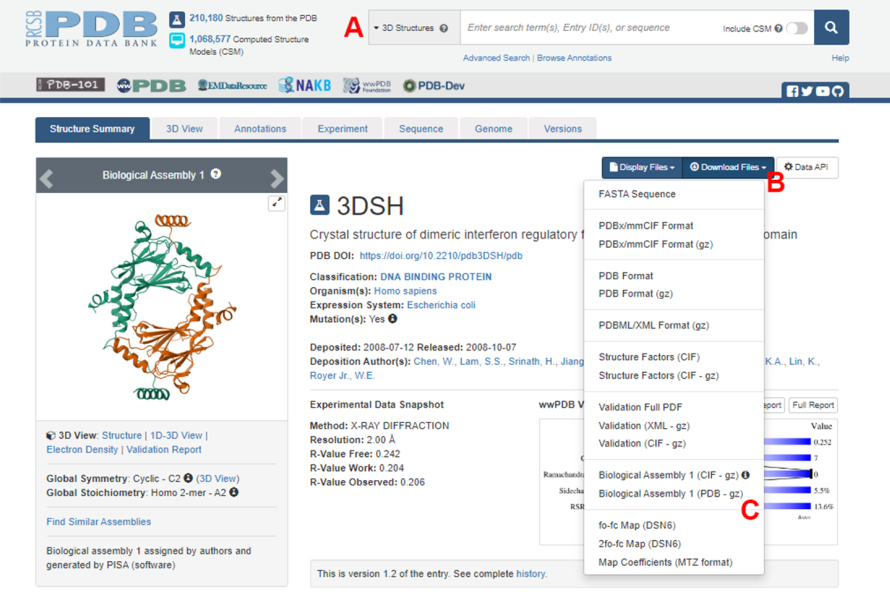
- 导航到蛋白质数据库 (PDB) 主页 (https://www.rcsb.org/),然后在主搜索框中键入蛋白质-蛋白质复合物结构的 PDB ID(图 2A)。在这项工作中用作示例的 IRF5 二聚体结构的 PDB ID 为 3DSH19。
- 在所需结构的主页中,单击"下载文件"(图 2B),然后单击"生物组装 1"(PDB - gz)(图 2C)。
注:在 PDB 数据库中,由相同单体形成的许多蛋白质复合物的结构表示为生物组装体,其中只有一个单体(不对称单元)的结构存储在 PDB 文件中。多聚体的结构,在本例中为 IRF5 二聚体,必须下载为包含两个不对称单元实例的生物组装体。为了方便该协议的下一步,首先将两个单体分开,并为它们分配不同的链 ID。 - 在 UCSF Chimera20 中打开下载的结构,然后单击 Tools > Structure Editing > Change Chain IDs。在此示例中,生物组件中的两条链都命名为 A。将第二条链(标记为 #0.2)重命名为 B,然后单击 OK。
- 单击 Model Panel > Favorites (收藏夹),然后选择包含两条链的模型。点击 Group/Ungroup 按钮将每条链分离到不同的模型中。然后,选择两个模型并单击 复制/合并 按钮。输入组合模型的新名称,选中 Close Source Models,然后单击 OK。
- 单击 Select > Chain(选择链 )并确认二聚体中的每个链现在都由不同的字母标识,即 A 和 B。
- 使用 File > Save PDB 将编辑后的结构保存到不同的 PDB 文件中,该文件将在协议的后续步骤中使用(此处使用名称 IRF5_dimer.pdb )。

图 2:本研究中用作代表性示例的结构的蛋白质数据库 (PDB) 页面。 (A) 介绍目标结构的 PDB 登录码的搜索框。(B) 用于下载多种格式结构的菜单。(C) 当结构保存为不对称单位时,下载生物组装体的选项(有关详细信息,请参阅步骤 1.1.2)。 请单击此处查看此图的较大版本。
{kind=link}
- 识别配体蛋白中的目标片段
- 导航到 BUDE Alanine Scan 服务器 (https://pragmaticproteindesign.bio.ed.ac.uk/balas/)。单击 Structure Upload 下的 Choose File 按钮,然后选择在步骤 1.1.6 中保存的 PDB 文件。
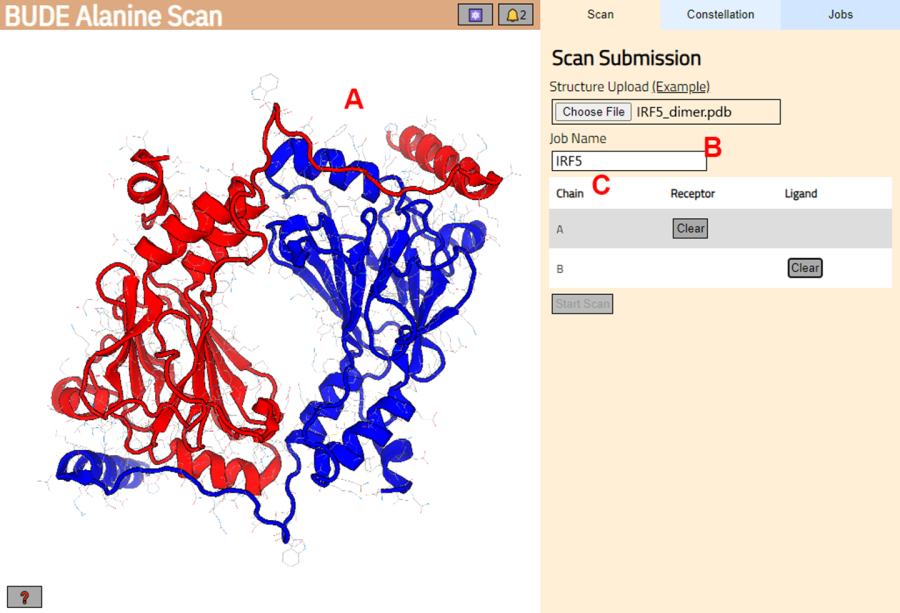
- 在下一页上,检查结构是否已正确加载(图 3A)并在服务器中输入作业的名称(图 3B)。
- 设置来自 PDB 的链,该链将被视为受体 (A) 和配体 (B)(图 3C)。然后,单击 Start Scan 按钮提交作业。
- 作业完成后,单击 Show Results 打开结果页面(图 4)。
注:在结果页面中,来自配体结构的残基根据其估计的自由能变化 (ΔΔG) 着色,而值较高的残基则为红色。 - 从残基列表中,选择预测能更好地与目标结合表面相互作用的残基延伸。确保这些残基聚集在自由能差 (ΔΔG) 的较高值上。在本例中,选择了残基 Leu424 和 Ser436 之间的片段(在 图 4 的右侧面板中用红色框突出显示)。
- 准备用于序列多样化的蛋白质-肽界面
- 在 Chimera 中打开步骤 1.1.6 中保存的 PDB 文件,并检查目标亚基的结构中没有缺失的原子或键。
- 删除所有与原始结构共结晶的小分子、离子和溶剂。为此,请单击 Select > Residues(选择残基 ),然后选择除标准氨基酸以外的所有分子。然后,单击 Actions > Atoms/Bonds 和 Delete。
- 将配体链裁剪为步骤 1.2.5 中选择的最佳相互作用片段。为此,请单击 Favorites 和 Sequence ,然后单击被视为配体 (B) 的链。在 Sequence (序列) 面板中,拖动鼠标以选择除位置 424 和 436 之间的残基之外的所有残基。要删除这些残基,请单击 Actions > Atoms/Bonds 和 Delete。
- 使用 File > Save PDB 将编辑后的结构保存到不同的 PDB 文件中,该文件将在协议的后续步骤中使用(此处使用名称 IRF5_interface.pdb )。

图 3:BUDE Alanine Scan 服务器中受体和配体的选择。 (A) 蛋白质-蛋白质复合物的图形表示。(B) 用于输入服务器中作业名称的文本框。(C) 面板以交互方式选择将被视为受体和配体的链(有关详细信息,请参阅步骤 1.2)。 请单击此处查看此图的较大版本。
{kind=link}

图 4:BUDE Alanine Scan 服务器的结果页面。 配体序列中潜在的相互作用最好的片段用红色框表示。在左侧面板中,预测能量贡献较高的残基 (Leu433) 以绿色突出显示。 请单击此处查看此图的较大版本。
{kind=link}
2. 序列多样化
注意:在以下步骤中, rosetta_main 是指主 Rosetta 安装目录,该目录通常位于 /opt/rosetta_src__bundle/main/,其中 表示已安装的 Rosetta 版本。此外,还假定 Rosetta 应用程序可以在系统范围内访问;如果不是这种情况,则必须提供可执行文件的完整路径。从源编译时,这些可执行文件位于 /rosetta_main/source/bin/ 目录中。
- 氨基酸侧链的初步优化
- 将步骤 1.3.4 中保存的编辑结构复制到 Rosetta 应用程序可访问的 Linux 位置。
- 在序列多样化之前,使用 Rosetta 的 FixBB 应用程序对碱基结构的所有氨基酸侧链进行重新打包。在此操作中,所有氨基酸侧链的方向都经过优化,以最大限度地减少能量并提高复合物的稳定性。为此,请运行以下命令:

注意:此命令将输出一个以原始结构命名的 PDB 文件,并带有一个额外的数字后缀(在本例中为 IRF5_interface_0001.pdb )。 - 为了便于协议的下一步,请使用以下命令重命名带有 _repack 后缀的重新打包的 PDB 文件:
mv IRF5_interface_0001.pdb IRF5_repack.pdb
- 序列多样化
- 在设计模式下运行 Pepspec 以使用以下命令执行实际的序列多样化步骤:

以下是常规选项:- -s 表示输入文件(在步骤 2.1.3 中生成的重新打包的 PDB 文件)。
- -o 表示用于命名输出文件的前缀。
- - database 指示 Rosetta 3 主数据库的路径。
- -ex1、-ex2 和 extrachi_cutoff 是 rotamer 库选项(有关详细信息,请参阅 Pepspec 文档)。
- -overwrite 指示应用程序覆盖先前迭代生成的可能预先存在的输出。
- -pepspec:pep_chain 表示被视为配体的 PDB 链(在本例中为 'b')。
- -pepspec:native_pep_anchor 表示用作锚点的氨基酸残基(在本例中,配体肽第 10 位的 Leu 残基)。
- -pepspec:n_peptides 表示要输出的肽结构的数量。
- -pepspec:no_prepack_prot 告诉应用程序跳过 input base 结构中的重新打包(因为之前在步骤 2.1 中执行过)。
注:主要的 Pepspec 输出是一个目录,其中包含设计阶段产生的肽的 PDB 文件,使用带有 .pdbs 后缀的输出前缀命名(示例中为 IRF5.pdbs )。此外,Pepspec 将作为序列多样化步骤的一部分测试的所有公认的肽序列及其相应的 Rosetta 能量分数输出到以输出前缀命名的制表符分隔的文本文件中,其中 .spec 后缀(示例中为 IRF5.spec )。由于这项工作中描述的协议旨在估计氨基酸偏好而不是实际的肽设计,因此下一步使用 IRF5.spec 而不是 .pdbs 目录中的 PDB 结构。
- 在设计模式下运行 Pepspec 以使用以下命令执行实际的序列多样化步骤:
3. 氨基酸偏好的估计
- 计算 PWM
- 要生成 PWM,请使用 Rosetta 套件中包含的 gen_pepspec_pwm.py 脚本。要运行此脚本,请使用以下命令:

哪里:- IRF5.spec 是在步骤 2.2 中生成的 Pepspec 输出文件。
- -1 表示序列中没有额外的 N 端残基,因此 PWM 中的位置从 1 开始。
- 0.2 指示脚本仅考虑 Pepspec 输出中得分最高的 20% 肽段(默认值为 0.1,对应于 10%)
- interface_score 指示脚本根据界面分数对肽段进行排名,界面分数是 Pepspec 输出文件中包含的各种 Rosetta 分数之一。
注意:此脚本生成两个输出文件,一个用于计算的 PWM(带有 .pwm 后缀),另一个用于用于计算 PWM 的肽子集的序列(带有 .seq 后缀)。这些文件的名称还包括用于排名的分数和肽的分数。在此示例中,这些文件分别命名为 IRF5_interface_score_0.2.pwm 和 IRF5_interface_score_0.2.seq。
- 要生成 PWM,请使用 Rosetta 套件中包含的 gen_pepspec_pwm.py 脚本。要运行此脚本,请使用以下命令:
- 生成序列徽标
- 导航到 WebLogo 服务器 (https://weblogo.berkeley.edu/logo.cgi)21,然后单击 Upload Sequence Data 旁边的 Choose File 按钮。上传包含步骤 3.1.1 中生成的肽序列的文件(本例中为 IRF5_interface_score_0.2.seq)。
- 根据输入长度选择所需的徽标格式和大小。该示例使用 PDF 格式,大小为 15 厘米 x 12 厘米。点击 Create Logo。
结果
在本文中,我们描述了一种预测 IRF5 结合表面氨基酸偏好的方案,IRF5 是称为人干扰素调节因子的转录因子家族的成员。这些蛋白质是先天性和适应性免疫反应的调节因子,参与多种免疫细胞的分化和激活。IRF 亚基具有高度可塑性和多特异性结合表面,能够与其他细胞蛋白形成同源二聚体、异源二聚体和复合物 17,18。二聚化被?...
讨论
本文描述了一种基于计算机序列多样化来估计潜在多特异性结合位点的氨基酸偏好的方案。很少有计算工具被开发出来来估计蛋白质-肽界面的氨基酸偏好 14,25,26。这些工具具有预测性质,但它们在用于执行预测的计算算法和为提高准确性而实施的校正方面有所不同。在这项工作中,我们使用了 Rosetta ?...
披露声明
作者没有什么可披露的。
致谢
感谢 Sistema Nacional de Investigación (SNI)(拨款号 SNI-043-2023 和 SNI-170-2021)、巴拿马国家科学秘书、技术和创新 (SENACYT) 和人类研究机构 (IFARHU) 的财政支持。作者要感谢 Miguel Rodríguez 博士仔细审阅手稿。
材料
| Name | Company | Catalog Number | Comments |
| BUDE Alanine Scan Server | University of Edinburgh | https://pragmaticproteindesign.bio.ed.ac.uk/balas/ | doi: 10.1021/acschembio.9b00560 |
| Rosetta Modeling Software | Rosetta Commons | https://www.rosettacommons.org/software | doi: 10.1002/prot.22851 |
| UCSF Chimera | University of California San Francisco | https://www.cgl.ucsf.edu/chimera/ | doi: 10.1002/jcc.20084 |
参考文献
- Kim, P. M., Lu, L. J., Xia, Y., Gerstein, M. B. Relating three-dimensional structures to protein networks provides evolutionary insights. Science. 314 (5807), 1938-1941 (2006).
- Schreiber, G., Keating, A. E. Protein binding specificity versus promiscuity. Current Opinion in Structural Biology. 21 (1), 50-61 (2011).
- Erijman, A., Aizner, Y., Shifman, J. M. Multispecific recognition: Mechanism, evolution, and design. Biochemistry. 50 (5), 602-611 (2011).
- Fromer, M., Shifman, J. M. Tradeoff between stability and multispecificity in the design of promiscuous proteins. PLoS Computational Biology. 5 (12), e1000627 (2009).
- Xie, T., Zmyslowski, A. M., Zhang, Y., Radhakrishnan, I. Structural basis for multispecificity of MRG domains. Structure. 23 (6), 1049-1057 (2015).
- Hendler, A., et al. Human SIRT1 multispecificity is modulated by active-site vicinity substitutions during natural evolution. Molecular Biology and Evolution. 38 (2), 545-556 (2021).
- Teilum, K., Olsen, J. G., Kragelund, B. B. On the specificity of protein-protein interactions in the context of disorder. The Biochemical Journal. 478 (11), 2035-2050 (2021).
- Pelay-Gimeno, M., Glas, A., Koch, O., Grossmann, T. N. Structure-based design of inhibitors of protein-protein interactions: Mimicking peptide binding epitopes. Angewandte Chemie (International ed. in English). 54 (31), 8896-8927 (2015).
- Wang, Y., Xue, P., Cao, M., Yu, T., Lane, S. T., Zhao, H. Directed evolution: Methodologies and applications. Chemical Reviews. 121 (20), 12384-12444 (2021).
- Liu, J., Cropp, T. A. Rational protein sequence diversification by multi-codon scanning mutagenesis. Methods in Molecular Biology. 978, 217-228 (2013).
- Wei, H., Li, X. Deep mutational scanning: A versatile tool in systematically mapping genotypes to phenotypes. Frontiers in Genetics. 14, 1087267 (2023).
- Bratulic, S., Badran, A. H. Modern methods for laboratory diversification of biomolecules. Current Opinion in Chemical Biology. 41, 50-60 (2017).
- Humphris, E. L., Kortemme, T. Prediction of protein-protein interface sequence diversity using flexible backbone computational protein design. Structure. 16 (12), 1777-1788 (2008).
- King, C. A., Bradley, P. Structure-based prediction of protein-peptide specificity in Rosetta. Proteins. 78 (16), 3437-3449 (2010).
- Ibarra, A. A., et al. Predicting and experimentally validating hot-spot residues at protein-protein interfaces. ACS Chemical Biology. 14 (10), 2252-2263 (2019).
- Chen, W., Srinath, H., Lam, S. S., Schiffer, C. A., Royer, W. E., Lin, K. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. Journal of Molecular Biology. 379 (2), 251-260 (2008).
- Mancino, A., Natoli, G. Specificity and function of IRF family transcription factors: Insights from genomics. Journal of Interferon & Cytokine Research. 36 (7), 462-469 (2016).
- Schwanke, H., Stempel, M., Brinkmann, M. M. Of keeping and tipping the balance: Host regulation and viral modulation of IRF3-dependent IFNB1 expression. Viruses. 12 (7), 33 (2020).
- Chen, W., et al. Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nature Structural & Molecular Biology. 15 (11), 1213-1220 (2008).
- Pettersen, E. F., et al. UCSF Chimera-A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25, 1605-1612 (2004).
- Crooks, G. E., Hon, G., Chandonia, J. -. M., Brenner, S. E. WebLogo: a sequence logo generator. Genome Research. 14 (6), 1188-1190 (2004).
- Panne, D., McWhirter, S. M., Maniatis, T., Harrison, S. C. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. The Journal of Biological Chemistry. 282 (31), 22816-22822 (2007).
- Weihrauch, D., et al. An IRF5 decoy peptide reduces myocardial inflammation and fibrosis and improves endothelial cell function in tight-skin mice. PloS One. 11 (4), e0151999 (2016).
- Mori, M., Yoneyama, M., Ito, T., Takahashi, K., Inagaki, F., Fujita, T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. The Journal of Biological Chemistry. 279 (11), 9698-9702 (2004).
- Smith, C. A., Kortemme, T. Predicting the tolerated sequences for proteins and protein interfaces using RosettaBackrub flexible backbone design. PloS One. 6 (7), e20451 (2011).
- Rubenstein, A. B., Pethe, M. A., Khare, S. D. MFPred: Rapid and accurate prediction of protein-peptide recognition multispecificity using self-consistent mean field theory. PLoS Computational Biology. 13 (6), e1005614 (2017).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。