
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Visualisierung der DNA-Reparaturproteine Interaktion durch Immunfluoreszenz
In diesem Artikel
Zusammenfassung
Nach DNA-Schäden aktivieren menschliche Zellen wichtige Reparaturwege, um die Integrität ihres Genoms wiederherzustellen. Hier beschreiben wir die Methode der indirekten Immunfluoreszenz als Mittel, um DNA-Reparaturproteine zu erkennen, ihre räumliche und zeitliche Rekrutierung zu analysieren und die Protein-Protein-Interaktion an den Stellen von DNA-Schäden zu begutachten.
Zusammenfassung
Säugetierzellen sind ständig Chemikalien, Strahlungen und natürlich vorkommenden metabolischen Nebenprodukten ausgesetzt, die bestimmte Arten von DNA-Beleidigungen erzeugen. Genotoxische Wirkstoffe können das DNA-Rückgrat beschädigen, es brechen oder die chemische Natur einzelner Basen verändern. Nach DNA-Beleidigung werden DNA-Schadensreaktionen (DDR) aktiviert und an der Reparatur beteiligte Proteine rekrutiert. Eine Vielzahl von Faktoren sind beteiligt, um die Art des Schadens zu erfassen und die entsprechende Reparaturreaktion zu aktivieren. Wenn DDR-Faktoren nicht richtig aktiviert und rekrutiert werden, kann dies zu genomischer Instabilität führen, die vielen menschlichen Pathologien einschließlich Krebs zugrunde liegt. Studien mit DDR-Proteinen können Einblicke in die genotoxische Arzneimittelreaktion und zelluläre Mechanismen der Arzneimittelresistenz liefern.
Es gibt zwei Hauptmethoden zur Visualisierung von Proteinen in vivo:direkte Beobachtung, indem das Protein von Interesse mit einem fluoreszierenden Protein getagt und ihm durch Live-Bildgebung oder indirekte Immunfluoreszenz auf festen Proben folgt. Während die Visualisierung fluoreszierender Proteine eine präzise Überwachung im Laufe der Zeit ermöglicht, kann die direkte Kennzeichnung in N- oder C-Terminus die Proteinlokalisierung oder -funktion beeinträchtigen. Die Beobachtung von Proteinen in ihrer unveränderten, endogenen Version wird bevorzugt. Wenn DNA-Reparaturproteine zur DNA-Beleidigung rekrutiert werden, erhöht sich ihre Konzentration lokal und sie bilden Gruppen oder "Foci", die durch indirekte Immunfluoreszenz mit spezifischen Antikörpern visualisiert werden können.
Obwohl der Nachweis von Proteinschwerpunkten keinen endgültigen Beweis für eine direkte Wechselwirkung liefert, weist die Kolokalisierung von Proteinen in Zellen darauf hin, dass sie sich an die Schadensstelle gruppieren und über die Abfolge der Ereignisse informieren können, die für eine komplexe Bildung erforderlich sind. Eine sorgfältige Analyse der räumlichen Überlappung von Brennpunkten in Zellen, die Wildtyp- oder mutierte Versionen eines Proteins exdrücken, kann wertvolle Hinweise auf funktionelle Domänen liefern, die für die DNA-Reparaturfunktion wichtig sind. Schließlich weist die Kolokalisierung von Proteinen auf mögliche direkte Wechselwirkungen hin, die durch Ko-Immunpräzipation in Zellen oder direkten Pulldown mit gereinigten Proteinen verifiziert werden können.
Einleitung
Menschliche Zellen sind ständig einer Vielzahl von DNA-schädigenden Wirkstoffen unterschiedlicher Herkunft ausgesetzt. Exogene Quellen bestehen hauptsächlich aus der Exposition gegenüber Strahlungen, Chemikalien (einschließlich Chemotherapeutika und einigen Antibiotika) und Viren, während die wichtigsten endogene Quellen Fehler bei der DNA-Replikation und oxidativem Stress umfassen. Die direkten Auswirkungen einer genotoxischen Exposition können je nach Stress und Expositionsdosis von einer modifizierten Basis bis hin zu einem potenziell tödlichen DNA-Doppelstrangbruch (DSB) reichen. Letztlich können nicht reparierte oder falsch reparierte DNA-Schäden zur Anhäufung von Mutationen, genomischen Umlagerungen, Genominstabilität und schließlich zu Karzinogenese1führen. Säugetierzellen haben komplexe Wege entwickelt, um bestimmte Arten von DNA-Schäden zu erkennen2,3 und reparieren sie rechtzeitig, synchronisiert mit dem Zellzyklusprogression.
Ionisierende Strahlung (IR) schädigt die DNA-Doppelhelix und erzeugt Doppelstrangbrüche (DSBs), eine der schädlichsten Formen von DNA-Schäden. Der MRN-Komplex (MRE11, RAD50, NBS1) fungiert als Sensor der DNA endet und aktiviert die Proteinkinase ataxia telangiectasia mutiert (ATM)4,5. Nach der ersten Aktivierung von ATM durch DNA-Ende löst ATM eine Kaskade von DDR-Ereignissen am Ort der Pause aus, die mit einem Schlüsselereignis die Phosphorylierung der Histonvariante H2AX6einlöst. Die H2AX-Phosphorylierung auf Rückstand S139 aktiviert sie in die Region H2AX und spannt Regionen bis zu Megabasen um die DNA-Läsion6,7,8,9. Dieses Ereignis erhöht die Zugänglichkeit der DNA, was zur Rekrutierung und Akkumulation anderer DNA-Reparaturproteine führt7. Da h2AX reichlich und spezifisch um d.B. heruminduziert ist, kann es mit spezifischen Antikörpern leicht visualisiert werden und wird häufig als Ersatzmarker für DSBs im DNA-Reparaturfeld verwendet. Sobald der Bruch signalisiert ist, aktivieren Zellen ihre DNA-Reparaturwege und verarbeiten die DNA-Schäden. Das Protein MDC1 (Mediator des DNA-Schadens-Checkpoint-Proteins101) bindet direkt mit AtM11 und auch mit NBS112,13. Es trägt dazu bei, die Konzentration des MRN-Komplexes am DSB zu erhöhen und eine positive ATM-Rückkopplungsschleife zu starten. H2AX wird nach der Reparatur des Bruchs schnell entfernt, so dass die DSB-Freigabe überwacht werden kann. Gefolgt von der Mikroskopie, bietet die Abnahme von H2AX im Laufe der Zeit eine indirekte Messung von Restbrüchen und DNA-Reparatureffizienz.
Eukaryotische Zellen können DSBs durch mehrere Wege reparieren, wobei die beiden wichtigsten nicht-homologe Endverbindung (NHEJ) und homologe Rekombination (HR)(Abbildung 1) sind. NHEJ liiert im Wesentlichen DNA-Doppelstrangenden ohne die Verwendung von erweiterter Homologie und arbeitet im gesamten Zellzyklus14,15. HR überwiegt in S- und G2-Phasen und wird ansonsten unterdrückt, da es ein Schwesterchromatid als homologe Schablone für die Reparatur14,16benötigt. Die Wahl des Weges zwischen NHEJ und HR hängt nicht nur von der physischen Nähe des Schwesterchromatids ab, sondern auch von der Ausdehnung der DNA-Endresektion17, die NHEJ hemmt.
Die homologieabhängige DSB-Reparatur initiiert durch nukleolytische Degradation des 5'-Strangs von den Bruchenden, um 3' einsträngige DNA-Schwänze (ssDNA) zu erzeugen, ein Prozess, der als 5'-3'-Resektion bezeichnet wird. Der MRN-Komplex initiiert die DNA-Endresektion und weitere Resektion wird in Kombination mit BLM/EXO1 (Bloom-Syndrom-Protein/Exonuklease 1) oder BLM/DNA2 (DNA-Replikation ATP-abhängige Helicase/Nuklease)18,19,20,21,22verarbeitet. Die DNA-Endresektion wird durch CtIP (CtBP-interagierendes Protein) durch seine direkte Interaktion mit dem MRN-Komplex23 und die Rekrutierung von BRCA1 (Brustkrebs Typ 1 Anfälligkeitsprotein)24,25verstärkt. Replikationsprotein A (RPA) bindet sich prompt an die ssDNA exponiert und wird dann durch das rekombinante Protein RAD51 zu einem Nukleoprotein-Filament verdrängt, das homologe Suche und Stranginvasion katalysiert26,27,28.
Die Einleitung der Resektion ist ein entscheidender Schritt für die Reparaturwegwahl. Sobald die Resektion eingeleitet wurde, werden die DNA-Enden zu schlechten Substraten für die Bindung durch Ku70/Ku80-Heterodimer (Komponente des NHEJ-Signalwegs) und Zellen werden HR17,29,30verschrieben. Der Ku70/Ku80 Heterodimer bindet an DSB-Enden und rekrutiert DNA-PKcs und p53 Binding Protein 1 (53BP1)29,30. 53BP1 wirkt als Hemmungshindernis in G1 und blockiert damit HR beigleichzeitigerFörderung von NHEJ 31,32, wird aber in der S-Phase in BRCA1-abhängiger Weise entfernt, so dass eine Resektion33,34möglich ist. Daher spielen 53BP1 und BRCA1 bei der DSB-Reparatur eine gegensätzliche Rolle, wobei 53BP1 ein NHEJ-Moderator ist, während BRCA1-Akte Pausen für die Reparatur durch HR ermöglichen.
Im Labor kann die DSB-Bildung durch ionisierende Strahlung (IR) induziert werden. Während dieses Beispiel eine hohe Dosis von 4 Gy verwendet, 1 Gy und 2 Gy auch eine signifikante Menge an DSBs erstellen, geeignet für die Analyse der Foci-Bildung durch reichliche Proteine. Es ist wichtig zu beachten, dass die Art und Dosis der verwendeten Strahlung zu verschiedenen Läsionen in der DNA und in der Zelle führen kann: Während IR DSBs induziert, kann es auch einzelne Strangbrüche oder Base-Modifikation verursachen (siehe35,36 für einen Verweis auf bestrahlung lineare Energieübertragung (LET) und Art der DNA-Schäden). Um die Kinetik der ionisierenden strahlungsinduzierten Brennweite (IRIF) Bildung und deren Clearance zu bestimmen, die auf die Reparatur des Schadens und die Umkehrung der aktivierten DDR8,9,37,38hinweist, kann die Brennpunktbildung zu verschiedenen Zeitpunkten nach ionisierender Strahlung überwacht werden. Timing der Aktivierung und Clearance aller wichtigen DNA-Schäden Proteine ist bekannt39, und viele werden als Ersatzmarker für Schlüsselereignisse untersucht. Beispielsweise wird pRPA, das eine hohe Affinität zu ssDNA besitzt, als Ersatz für die Bruchresektion verwendet, MRN-Proteine (MRE11, RAD50, NBS1) und Exonukleasen können auch verwendet werden, um die Resektionseffizienz zu bewerten. Während RAD51, BRCA1, BRCA2 (Brustkrebs Typ 2 Anfälligkeitsprotein) und PALB2 (Partner und Lokalisierer von BRCA2) überwacht werden können, um die HR-Effizienz zu bewerten, wird das Vorhandensein der Ku-Proteine oder 53BP1 als Marker von NHEJ(Abbildung 1)verwendet.
Während sich Proteine der DNA-Reparaturmaschinen gegenseitig bis zum Bruch rekrutieren und in Superkomplexen zusammensetzen, können DNA-Protein- und Protein-Protein-Wechselwirkungen abgeleitet werden, indem man ihrer individuellen Lokalisierung im Laufe der Zeit folgt und die Kolokalisierung von Proteinen analysiert, wie durch überlappende Signale in Zelle40,41,42visualisiert. In Zelllinien ermöglicht die Einführung von Punktmutationen oder das Löschen in bestimmten DNA-Reparaturgenen entweder durch Genombearbeitung oder durch Überexpression plasmidbasierter Mutanten die Untersuchung spezifischer Rückstände und ihre mögliche Rolle bei der Erkennung von DNA-Schäden (z. B. Kolokalisierung mit H2AX) oder komplexer Montage (Kolokalisierung mit einem anderen oder mehreren Proteinen) sowie deren Auswirkungen auf die DNA-Reparatur. Hier verwenden wir die indirekte Immunfluoreszenz als Mittel, um die Bildung und Auflösung von DSBs zu untersuchen, indem wir im Laufe der Zeit den H2AX-Brennpunkten folgen. Wir stellen auch ein Beispiel für die Foci-Bildung und Co-Lokalisierungsanalyse durch einen wichtigen Akteur in der DSB-Reparatur vor: p53 Binding Protein 1 (53BP1)32. Wie bereits erwähnt, 53BP1 gilt als zentral für DNA-Reparatur-Pfad Wahl. Nach 53BP1 Akkumulation und seine Co-Lokalisierung mit H2AX liefert wertvolle Informationen über Zellzyklusphase, DNA-Schadensakkumulation, und Weg verwendet, um DSBs zu reparieren. Der Zweck der indirekten Immunlokalisierung besteht darin, die Effizienz der REPARATUR von DNA-Schäden in Zelllinien nach IR wie in dieser Studie oder nach Exposition gegenüber verschiedenen Belastungen in der Zelle von der DNA-Vernetzung bis zur Blockierung der Replikationsgabel zu bewerten (eine Liste der DNA-schädlichen Mittel ist in Tabelle 1enthalten).

Abbildung 1: DNA Double Strand Breaks (DSB) Reparaturwege.
Die DSB-Reparatur umfasst zwei Hauptwege: Homologe Rekombination (HR, links) und Non-Homologous End-Joining (NHEJ, rechts). Nach der Pause werden Proteine aktiviert, um den Bruch zu markieren (H2AX), an der Endresektion (MRN) teilnehmen, die resezierte ssDNA (pRPA) beschichten, die Rekombination fördern (BRCA1, PALB2, BRCA2, RAD51) oder die Resektion begrenzen und NHEJ (53BP1) fördern. Andere Proteine nehmen an der Schadensreparatur teil, aber die aufgeführten Proteine werden routinemäßig mit indirekter Immunfluoreszenz gefolgt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
| DNA-schädigendes Mittel | Wirkmechanismus | Empfohlene Dosis |
| A-Strahlen/Röntgenstrahlen | Strahlung Bildung von doppelsträngigen Brüchen mit einigen unkontrollierten zellulären Effekten | 1-4 Gy |
| 36 Ar ionen | Strahlung Bildung von Doppelstrandedpausen | 270 keV/m |
| •Partikel | Strahlung Bildung von Doppelstrandedpausen | 116 keV/m |
| Bleomycin | Inhibitor der DNA-Synthese | 0,4-2 g/ml |
| Camptothecin | Inhibitor der Topoisomerase I | 10-200 nM |
| Cisplatin | Alkylierungsmittel (induzierenintrastrande Querverbindungen) | 0,25-2 m |
| Doxorubicin | Intercalating-Agent Inhibitor der Topoisomerase II | 10-200 nM |
| Etoposide | Inhibitor der Topoisomerase II | 10 m |
| Hydroxyurea | Inhibitor der DNA-Synthese (durch Ribonukleotid-Reduktase) | 10-200 m |
| Methylmethansulfonat | Alkylierungsmittel | 0,25-2 mM |
| Mitomycin C | Alkylierungsmittel | 0,25-2 m |
| Ultraviolettes (UV) Licht | Bildung von Thymidin-Dimeren (Erzeugung von Verzerrungen der DNA-Kette) | 50-100 mJ/cm2 |
Tabelle 1: Genotoxische Mittel. Beispiele für DNA-schädigende Mittel, deren Wirkmechanismus und der verursachte Schaden auf der Grundlage der vorgeschlagenen Arbeitskonzentration.
Protokoll
1. HeLa Zellkultur
- Rundglasabdeckungen mit 1 M HCl bei 50 °C für 16-18 Stunden vorbehandeln. Mit ddH2O abspülen und in 100% EtOH lagern.
- Zellkulturmedium vorbereiten: 10% (v/v) FBS zu DMEM medium hinzufügen.
- 4,0 x 104 Zellen/gut in steriler 12-Well-Platte mit einem 18 mm runden Glasdeckel bei 37 °C und 5%CO2 in einem befeuchteten Inkubator anbauen. Wachsen Sie Zellen bis zu 80% Koninfluenza (ca. 24 Stunden).
2. Zellbehandlung mit Strahlung (IR)
- Um die Bildung von Doppelstrangbrüchen zu induzieren, setzen Sie die Zellen einer 4 Gy-Bestrahlung aus (Kontrolle: Keine Bestrahlung, t=0). In der Gamma-Zelle -40 entspricht dies 4,54 min bei 0,815 Gy pro min.
- Inkubieren Sie Zellen bei 37 °C und 5%CO2 in einem befeuchteten Inkubator für die entsprechende Zeitdauer (hier gewählte Zeitpunkte t=1, 2, 4, 16 h).
3. Kernextraktion und Zellfixierung
- Lagerlösungen vorbereiten: 0,2 M PIPES (pH 6,8), 5 M NaCl, 2 M Saccharose, 1 M MgCl2, 0,1 M EGTA (pH 8,0).
- Nukleare Extraktionspuffer (NEB) vorbereiten: 10 mM PIPES (pH 6.8), 100 mM NaCl, 300 mM Saccharose, 3 mM MgCl2,1 mM EGTA (pH 8,0) und 0,5% (v/v) Triton X-100 in ddH2O. Mix vollständig auflösen.
- 4% (v/v) Paraformaldehyd (PFA) vorbereiten: 10 ml 16% PFA wässrige Lösung in 30 ml PBS auflösen. Mischen, bis vollständig aufgelöst.
- Zu einem geeigneten Zeitpunkt (t=0, 1, 2, 4, 16 h), Waschen Sie Zellen zweimal mit 1 ml PBS. Entfernen Sie PBS vollständig.
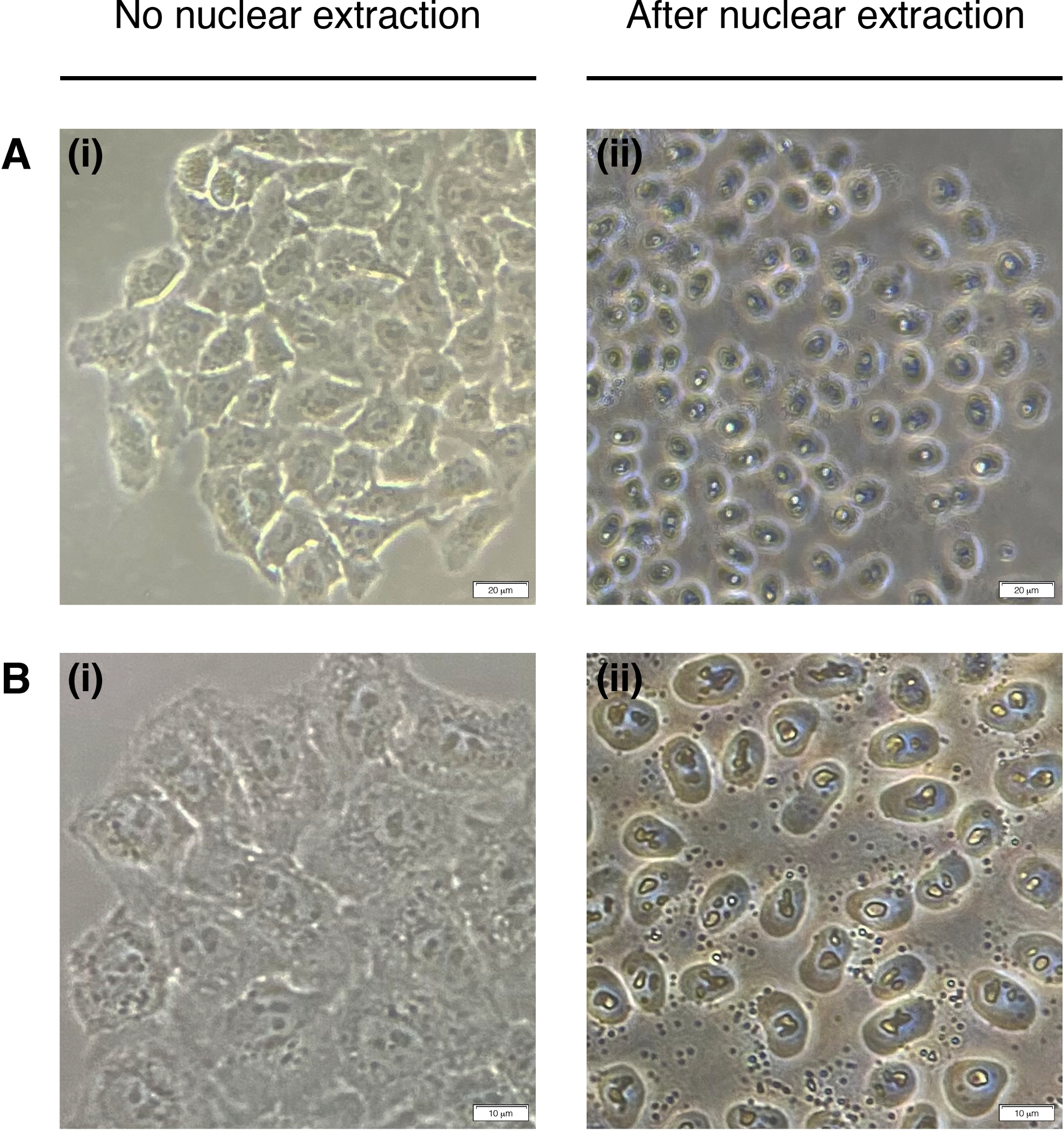
- Fügen Sie jedem Brunnen 200 L NEB für die Zellkernextraktion hinzu (Zytoplasma ist abgebaut, nur Kern restiert) (Abbildung 2). 2 Minuten bei Raumtemperatur inkubieren und vollständig entfernen.
HINWEIS: Nicht mehr als 2 Minuten.

Abbildung 2: Kerngewinnung.
Repräsentative Bilder von Zellen vor (links) und Post (rechts) kernadierungsischer Extraktion. Zytoplasma sollte verdaut werden, aber die Kernstruktur sollte nach der Extraktion intakt bleiben (rechts). (A) 20-fache Vergrößerung; Skala bar = 20 m und (B) 40x Vergrößerung; Skalenbalken = 10 m. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Waschen Sie Zellen mit 1 ml PBS. Entfernen Sie PBS vollständig. Fügen Sie PBS sorgfältig hinzu, Zellen sind bei diesem Schritt sehr zerbrechlich.
- Fügen Sie jedem Bohrkörper 200 l von 4% (v/v) PFA für die Zellfixierung hinzu. 10 Minuten bei 4 °C inkubieren. Entfernen Sie PFA vollständig.
- Fügen Sie 1 ml PBS hinzu.
HINWEIS: Zellen können in PBS bei 4 °C gelagert werden.
4. Immunfluoreszenzfärbung
- Vorbereiten der Blockierlösung: 5% BSA (w/v) in PBS auflösen und 0,3% (v/v) Triton X-100 hinzufügen. Mischen, bis vollständig aufgelöst.
- Verdünnungspuffer vorbereiten: 1% BSA (w/v) in PBS auflösen und 0,3% (v/v) Triton X-100 hinzufügen. Mischen, bis vollständig aufgelöst.
- Zum Blockieren fügen Sie jedem Bohrwert 200 L Blockierlösung hinzu. 2 Stunden bei Raumtemperatur oder 16-18 Stunden bei 4 °C inkubieren.
HINWEIS: Wenn Ziegenantikörper verwendet werden, fügen Sie 5% Ziegenserum zur Blockierlösung hinzu. - Primären Antikörper im Verdünnungspuffer (1:500; siehe Tabelle 2 für Antikörperliste) und Wirbel bis gut gemischt verdünnen.
HINWEIS: Wenn Ziegenantikörper verwendet wird, fügen Sie 1% Ziegenserum zum Verdünnungspuffer hinzu. - In einer Feuchtigkeits-/Inkubationsbox ein Stück Parafilm aufkleben. Fügen Sie 10 L primären Antikörper hinzu (in einem einzigen Tropfen). Richten Sie eine Kante des Deckels mit dem Tropfen aus und senken Sie langsam auf den Parafilm, damit sich die Flüssigkeit im ganzen ausbreitet (Blasen vermeiden, wenn möglich). 2 Stunden bei Raumtemperatur inkubieren.
- Waschen Sie Deckellipsen dreimal in PBS für 1 Minute.
- Sekundärantikörper im Verdünnungspuffer (Endkonzentration: 2 g/ml) und Wirbel bis gut vermischt verdünnen.
- Tragen Sie 10 L sekundären Antikörper n. A. auf, wie für die primären Antikörper beschrieben. 2 Stunden bei Raumtemperatur inkubieren.
HINWEIS: Schützen Sie sich vor Licht.
| Antikörper | Unternehmen | Verweis | Quelle |
| 53BP1 | Zellsignalisierung | 4937 | Kaninchen |
| Anti-Maus IgG H&L (Alexa Fluor 647) | Abcam | ab150103 | Esel |
| Anti-Phospho-Histon H2A. X (Ser139), Klon JBW301 | Millipore | 05-636 | Maus |
| Anti-Rabbit IgG H&L (Alexa Fluor 488) | Abcam | ab150081 | Ziege |
Tabelle 2: Verwendete Antikörper. Liste der in dieser Studie verwendeten Antikörper.
- Waschen Sie Deckellipsen dreimal in PBS für 1 Minute.
- Abdeckungen mit H2O 1 Minute waschen.
- Gegenfleck-DNA mit DAPI: 10 l 300 nM DAPI (wie für Antikörper beschrieben) auftragen, 30 Minuten bei Raumtemperatur inkubieren und dann mit einem Glyzerin-basierten Montagemedium auf Glasschlitten montieren. Alternativ können Sie einen Tropfen (10 l) kommerzieller Antifade-Montagemedien, die DAPI enthalten, auf ein Schiebeblatt geben und einen Deckelzettel auftragen. Drücken Sie vorsichtig den Deckelund entfernen Sie überschüssige Flüssigkeit um sie herum mit einem Papiertuch.
- Versiegeln Sie Deckellipsen mit transparentem Nagellack und lassen Sie sie für 20 Minuten trocknen.
- Dias bei 4 °C lagern.
5. Bildaufnahme
- Legen Sie einen Tropfen Tauchöl auf die 60x Objektivlinse. Verwenden Sie DAPI, um die Kerne durch das Augenstück zu lokalisieren.
- Für die XYZ-Bildaufnahme, öffnen Sie die Erfassungssoftware und wählen Sie Parameter aus: Scannertyp: Galvano; Scanner-Modus: Roundtrip; Bildgröße: 512 x 512; PMT-Modus: VBF; PMT-Durchschnitt: Rahmen (4-mal); PMT-Sequenzseimscan: Zeile.
- Wählen Sie den Farbstoff und die Detektoren:
Kanal (CH1), Farbstoff (DAPI), Detektor (SD1)
Kanal (CH2), Farbstoff (Alexa Fluor 488), Detektor (HSD3)
Kanal (CH3), Farbstoff (Alexa Fluor 647), Detektor (HSD4) - Wählen Sie ON in "Z".
- Passen Sie das Livebild an. Drücken Sie die Live-Taste im Live-Fenster.
- Passen Sie den Fokus an und stellen Sie die Laserintensität (%), Empfindlichkeit (HV), Verstärkung und Versatz auf dem Werkzeugfenster "PMT" ein.
- Passen Sie die Laserintensität (%): für Helligkeit und Bleichen an. Je höher die Laserintensität, desto stärker das Signal, aber die Probe wird photobleicht.
- Einstellen der Empfindlichkeit (HV): Geräuschpegel. Je höher der HV, desto stärker das Signal, aber das Bild wird laut, wenn zu hoch.
HINWEIS: Halten Sie die Spannung immer konstant. - Passen Sie den Offset: Hintergrundebene an.
- Wählen Sie Start/Ende (15 Slices) für Z-Stacks aus.
- Passen Sie den Fokus an und stellen Sie die Laserintensität (%), Empfindlichkeit (HV), Verstärkung und Versatz auf dem Werkzeugfenster "PMT" ein.
- Starten Sie die Erfassung.
- Wählen Sie den Ordner zum Speichern von Bildern aus. Drücken Sie die LSM-Starttaste, um mit dem Aufnehmen des Bildes zu beginnen. Drücken Sie die Taste Series Done, um die Bildaufnahme abzuschließen.
6. Datenanalyse
- Öffnen Sie zur Bildanalyse die Analysesoftware.
- Drücken Sie das Werkzeugfenster Batch, wählen Sie die zu analysierenden Bilder aus, und wählen Sie den Ausgabeordner aus.
- Drücken Sie das Werkzeugfenster Analyse, und wählen Sie Projektion aus (zeigt die maximale Intensitätsprojektion aus 15 Slices an).
- Wählen Sie unter Eingabe-/Ausgabeeinstellungden erstellten Stapel aus.
- Drücken Sie Prozess für die Verarbeitung von Bildern.
- Exportieren Sie Bilder als .tiff-Dateien.
- Für die Quantifizierung von Kernschwerpunkten, öffnen Sie CellProfiler.
- Öffnen Sie die Pipeline für die Quantifizierung von H2AX und 53BP1 (siehe Ergänzende Informationen).
- Zeichnen Sie Daten mithilfe einer Tabellensoftware.
- Öffnen Sie CellProfiler für die Analyse der Co-Lokalisierung.
- Öffnen Sie die Colokalisierungspipeline (siehe Ergänzende Informationen). Eine Tabellenkalkulationsdatei wird erstellt und am bevorzugten Speicherort gespeichert. Diagramme selbst werden jedoch nicht automatisch gespeichert. Es wird empfohlen, eine Momentaufnahme der Fenster zu machen, um die Ergebnisse aufzuzeichnen.
- Zeichnen Sie Daten mithilfe einer Tabellensoftware.
Ergebnisse
Am Tag 2 oder 24 h nach der Aussaat von Zellen auf Deckellips haben die Zellen eine Teilung durchlaufen und sind zu 80% konfluent. Spezifische Knockdowns oder Mutationen in DNA-Reparaturproteinen können die Verdoppelungszeit erhöhen oder Zellen für eine genotoxische Behandlung sensibilisieren, und die Saatdichte sowie die Behandlungsdosen sollten entsprechend angepasst werden. Um die besten Arbeitsbedingungen zu bestimmen, kann der Zeitpunkt der Reaktion auf DNA-Schäden durch Western-Blotting von H2AX im Laufe der Ze...
Diskussion
Die Analyse des Timings und der Effizienz der Reparatur von DNA-Schäden durch Mikroskopie hat sich als wesentlich erwiesen, um zu verstehen, wie die DNA-Reparaturmaschinerie funktioniert, in normalen Zellen und in menschlichen Pathologien wie Krebs.
Die Entwicklung spezifischer Antikörper, die den Nachweis aktivierter Proteine in ihrer phosphorylierten Version ermöglichen (z. B. H2AX, pRPA, pRAD50 und andere10,23,
Offenlegungen
Die Autoren haben nichts zu verraten.
Danksagungen
Diese Arbeit wurde durch ein Stipendium der San Antonio Area Foundation unterstützt. Das Mays Cancer Center wird vom NCI Cancer Center Support Core Grant P30 CA054174 unterstützt. Wir danken Stephen Holloway für seine Hilfe bei der Beschaffung der Reagenzien und dem Sung-Labor für die Bereitstellung von Platz- und Mikroskopiekapazitäten.
Materialien
| Name | Company | Catalog Number | Comments |
| 16% (v/v) paraformaldehyde (PFA) aqueous solution | Electron Microscopy Sciences | 15710 | Microscopy quality of the PFA ensures best images. If using "home-made PFA", filter prior to use. |
| Bovine serum albumin (BSA) | Sigma-Aldrich | A3059 | Heat-shock fraction is recommended, to avoid precipitation/background. |
| Coverglass #1, 18 mm round (coverslips) | Neuvitro | NC0308920 | Coverslips need to be cleaned and sterilized prior using, with HCl or ethanol. |
| DMEM, High Glucose [(+) 4.5 g/L D-Glucose, (+) L-Glutamine, (-) Sodium Pyruvate] | Gibco | 11965118 | Adjust the growing media to the needs of cell line used. |
| DPBS, no calcium, no magnesium | Gibco | 14190144 | PBS for tissue culture. |
| Ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) | Research Products International | E57060 | Nuclear extraction buffer. |
| Fetal Bovine Serum (FBS) | Life Technologies | 104370028 | The quality of FBS can be assessed by testing gH2AX foci formation. If traces of genotoxic endotoxin are present in the batch, gH2AX will be positive in the absence of stress. |
| Magnesium chloride (MgCl2) | Research Products International | M24000 | Nuclear extraction buffer. |
| Piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) | Research Products International | P40150 | Nuclear extraction buffer. |
| SlowFade Diamond Antifade Mountant with DAPI | Invitrogen | S36973 | 300 nM DAPI with VECTASHIELD Antifade Mounting Medium can be used instead. |
| Sodium chloride (NaCl) | Research Products International | S23020 | Nuclear extraction buffer. |
| Sucrose | Research Products International | S24060 | Nuclear extraction buffer. |
| Superfrost Plus Microscope Slides | Fisherbrand | 1255015 | Polysine Slides can be used instead. |
| TC-Treated Multiple Well Plates, size 12 wells | Costar | 3513 | Seeding on coverslips is done in 12-wells plate. |
| Triton X-100 | AmericanBio | AB02025 | Nuclear extraction buffer. |
| TrypLE Express Enzyme (1X), No Phenol Red | Gibco | 12604021 | Trypsin-EDTA can be used instead. |
| Trypsin-EDTA (0.5%), No Phenol Red | Gibco | 15400054 | TrypLE can be used instead. |
Referenzen
- Prakash, R., Zhang, Y., Feng, W., Jasin, M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harbor Perspectives in Biology. 7 (4), 016600 (2015).
- Jalan, M., Olsen, K. S., Powell, S. N. Emerging Roles of RAD52 in Genome Maintenance. Cancers (Basel). 11 (7), (2019).
- Oh, J., Symington, L. S. Role of the Mre11 Complex in Preserving Genome Integrity. Genes (Basel). 9 (12), (2018).
- Uziel, T., et al. Requirement of the MRN complex for ATM activation by DNA damage. The EMBO Journal. 22 (20), 5612-5621 (2003).
- Lee, J. H., Paull, T. T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 308 (5721), 551-554 (2005).
- Rogakou, E. P., Pilch, D. R., Orr, A. H., Ivanova, V. S., Bonner, W. M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. Journal of Biological Chemistry. 273, 5858-5868 (1998).
- Kinner, A., Wu, W., Staudt, C., Iliakis, G. γ-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Research. 36 (17), 5678-5694 (2008).
- Martin, O. A., Pilch, D. R., Redon, C., Bonner, W. M. Histone H2AX in DNA damage repair. Cancer Biology & Therapy. 2 (3), 233-235 (2003).
- Rogakou, E. P., Boon, C., Redon, C., Bonner, W. M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. Journal of Cell Biology. 146 (5), 905-916 (1999).
- Stucki, M., et al. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 123 (7), 1213-1226 (2005).
- Lou, Z., et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Molecular Cell. 21 (2), 187-200 (2006).
- Chapman, J. R., Jackson, S. P. Phospho-dependent interaction between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Reports. 9 (8), 795-801 (2008).
- Melander, F., et al. Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. Journal of Cell Biology. 181 (2), 213-226 (2008).
- Branzei, D., Foiani, M. Regulation of DNA repair throughout the cell cycle. Nature Review. Molecular Cell Biology. 9 (4), 297-308 (2008).
- Chiruvella, K. K., Liang, Z., Wilson, T. E. Repair of double-strand breaks by end joining. Cold Spring Harbor Perspectives in Biology. 5 (5), 012757 (2013).
- Mehta, A., Haber, J. E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harbor Perspectives in Biology. 6 (9), 016428 (2014).
- Symington, L. S., Gautier, J. Double-strand break end resection and repair pathway choice. Annual Review of Genetics. 45, 247-271 (2011).
- Huertas, P. DNA resection in eukaryotes: deciding how to fix the break. Nature Structural & Molecular Biology. 17 (1), 11-16 (2010).
- Nimonkar, A. V., et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes & Development. 25 (4), 350-362 (2011).
- Garcia, V., Phelps, S. E. L., Gray, S., Neale, M. J. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature. 479 (7372), 241-244 (2011).
- Sturzenegger, A., et al. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. Journal of Biological Chemistry. 289 (39), 27314-27326 (2014).
- Daley, J. M., Niu, H., Miller, A. S., Sung, P. Biochemical mechanism of DSB end resection and its regulation. DNA Repair. 32, 66-74 (2015).
- Sartori, A. A., et al. Human CtIP promotes DNA end resection. Nature. 450 (7169), 509-514 (2007).
- Chen, L., Nievera, C. J., Lee, A. Y. L., Wu, X. Cell cycle-dependent complex formation of BRCA1-CtIP-MRN is important for DNA double-strand break repair. Journal of Biological Chemistry. 283, 7713-7720 (2008).
- Yun, M. H., Hiom, K. CtIP-BRCA1 modulates the choice of DNA double-strand break repair pathway throughout the cell cycle. Nature. 459 (7245), 460-463 (2009).
- Sung, P., Klein, H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nature Review. Molecular Cell Biology. 7, 739-750 (2006).
- San Filippo, J., Sung, P., Klein, H. Mechanisms of eukaryotic homologous recombination. Annual Review of Biochemistry. 77, 229-257 (2008).
- Jasin, M., Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harbor Perspectives in Biology. 5 (11), 012740 (2013).
- Dynan, W. S., Yoo, S. Interaction of Ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Research. 26 (7), 1551-1559 (1998).
- Lieber, M. R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annual Review of Biochemistry. 79, 181-211 (2010).
- Cejka, P. DNA end resection: nucleases team up with the right partners to initiate homologous recombination. Journal of Biological Chemistry. 290 (38), 22931-22938 (2015).
- Mirman, Z., de Lange, T. 53BP1: a DSB escort. Genes & Development. 34, 7-23 (2020).
- Cao, L., et al. A selective requirement for 53BP1 in the biological response to genomic instability induces by BRCA1 deficiency. Molecular Cell. 35 (4), 534-541 (2009).
- Zimmermann, M., de Lange, T. 53BP1: Pro choice in DNA repair. Trends in Cell Biology. 24 (2), 108-117 (2014).
- Mavragani, I. V., Nikitaki, Z., Kalospyros, S. A., Georgakilas, A. G. Ionizing Radiation and Complex DNA Damage: From Prediction to Detection Challenges and Biological Significance. Cancers (Basel). 11 (11), (2019).
- Nikitaki, Z., et al. Measurement of complex DNA damage induction and repair in human cellular systems after exposure to ionizing radiations of varying linear energy transfer (LET). Free Radical Research. 50, 64-78 (2016).
- Redon, C., et al. Histone H2A variants H2AX and H2AZ. Current Opinion in Genetics & Development. 12 (2), 162-169 (2002).
- Fernandez-Capetillo, O., Lee, A., Nussenzweig, M., Nussenzweig, A. H2AX: the histone guardian of the genome. DNA Repair. 3 (8-9), 959-967 (2004).
- Paull, T. T., et al. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Current Biology. 10 (15), 886-895 (2000).
- Sy, S. M. H., Huen, M. S. Y., Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proceedings of the National Academy of Sciences. 106 (17), 7155-7160 (2009).
- Buisson, R., Masson, J. Y. PALB2 self-interaction controls homologous recombination. Nucleic Acids Research. 40 (20), 10312-10323 (2012).
- Belotserkovskaya, R., et al. PALB2 chromatin recruitment restores homologous recombination in BRCA1-deficient cells depleted of 53BP1. Nature Communications. 11 (1), 819 (2020).
- Betts, J. A., et al. Long noncoding RNAs CUPID1 and CUPID2 mediate breast cancer risk at 11q13 by modulating the response to DNA damage. American Journal of Human Genetics. 101 (2), 255-266 (2017).
- Dray, E., et al. Molecular basis for enhancement of the meiotic DMC1 recombinase by RAD51 associated protein 1 (RAD51AP1). Proceedings of the National Academy of Sciences. 108 (9), 3560-3565 (2011).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten