
Method Article
Hybride De-Novo-Genomassemblierung zur Erzeugung vollständiger Genome von Harnbakterien unter Verwendung von Kurz- und Langzeitsequenzierungstechnologien
In diesem Artikel
Zusammenfassung
Dieses Protokoll beschreibt einen umfassenden Ansatz für die Kultivierung, Sequenzierung und De-novo-Hybridgenomassemblierung von Harnbakterien. Es bietet ein reproduzierbares Verfahren zur Erzeugung vollständiger, zirkulärer Genomsequenzen, die bei der Untersuchung sowohl chromosomaler als auch extrachromosomaler genetischer Elemente nützlich sind, die zur Kolonisierung im Urin, zur Pathogenese und zur Verbreitung antimikrobieller Resistenzen beitragen.
Zusammenfassung
Vollständige Genomsequenzen liefern wertvolle Daten für das Verständnis der genetischen Vielfalt und der einzigartigen Kolonisationsfaktoren von Urinmikroben. Diese Daten können mobile genetische Elemente wie Plasmide und extrachromosomale Phagen umfassen, die zur Verbreitung antimikrobieller Resistenzen beitragen und die Behandlung von Harnwegsinfektionen (UTI) weiter erschweren. Vollständige, geschlossene Genome bieten nicht nur eine feine Auflösung der Genomstruktur, sondern ermöglichen auch detaillierte vergleichende Genomik- und Evolutionsanalysen. Die Generierung vollständiger Genome de novo ist aufgrund der Einschränkungen der verfügbaren Sequenzierungstechnologie seit langem eine herausfordernde Aufgabe. Paired-End Next Generation Sequencing (NGS) erzeugt qualitativ hochwertige Kurzlesevorgänge, die oft zu genauen, aber fragmentierten Genomanordnungen führen. Im Gegenteil, die Nanoporensequenzierung liefert lange Lesevorgänge von geringerer Qualität, die normalerweise zu fehleranfälligen kompletten Baugruppen führen. Solche Fehler können genomweite Assoziationsstudien behindern oder irreführende Ergebnisse der Variantenanalyse liefern. Daher haben sich hybride Ansätze, die sowohl kurze als auch lange Lesevorgänge kombinieren, als zuverlässige Methoden herausgestellt, um hochgenaue geschlossene Bakteriengenome zu erhalten. Hierin wird eine umfassende Methode für die Kultur verschiedener Harnbakterien, die Speziesidentifizierung durch 16S rRNA-Gensequenzierung, die Extraktion genomischer DNA (gDNA) und die Generierung von kurzen und langen Lesevorgängen durch NGS- bzw. Nanopore-Plattformen beschrieben. Darüber hinaus beschreibt diese Methode eine bioinformatische Pipeline von Qualitätskontroll-, Assemblierungs- und Genvorhersagealgorithmen zur Erzeugung annotierter vollständiger Genomsequenzen. Die Kombination bioinformatischer Werkzeuge ermöglicht die Auswahl hochwertiger Lesedaten für die hybride Genomassemblierung und die nachgelagerte Analyse. Der in diesem Protokoll beschriebene gestraffte Ansatz für die hybride De-novo-Genomanordnung kann für die Verwendung in allen kultivierbaren Bakterien angepasst werden.
Einleitung
Das Urin-Mikrobiom ist ein aufstrebendes Forschungsgebiet, das ein jahrzehntelanges Missverständnis zerschlagen hat, dass die Harnwege bei gesunden Menschen steril sind. Mitglieder der Harnmikrobiota können dazu dienen, die Harnumgebung auszugleichen und Harnwegsinfektionen (UTI) zu verhindern1,2. Uropathogene Bakterien dringen in die Harnwege ein und nutzen verschiedene Virulenzmechanismen, um die ansässige Mikrobiota zu verdrängen, das Urothel zu besiedeln, Immunreaktionen zu entgehen und Umweltbelastungen entgegenzuwirken3,4. Urin ist ein relativ nährstoffarmes Medium, das durch hohe Osmolarität, begrenzte Stickstoff- und Kohlenhydratverfügbarkeit, geringe Sauerstoffversorgung und niedrigen pH-Wert5,6,7gekennzeichnet ist. Urin gilt auch als antimikrobiell, bestehend aus hohen Konzentrationen von inhibitorischem Harnstoff und antimikrobiellen Peptiden wie dem humanen Cathelicidin LL-378. Die Untersuchung von Mechanismen, die sowohl von ansässigen Bakterien als auch von Uropathogenen zur Besiedlung der Harnwege eingesetzt werden, ist entscheidend für das weitere Verständnis der Gesundheit der Harnwege und die Entwicklung neuer Strategien für die Behandlung mit Harnwegsinfektionen. Da das Versagen antimikrobieller Therapien an vorderster Front immer häufiger auftritt, wird es immer wichtiger, die Verbreitung mobiler genetischer Elemente zu überwachen, die antimikrobielle Resistenzdeterminanten in Populationen von Harnbakterien tragen9,10.
Um Genotypen und Phänotypen von Harnbakterien zu untersuchen, ist ihre erfolgreiche Kultur und anschließende Sequenzierung des gesamten Genoms (WGS) unerlässlich. Kulturabhängige Methoden sind notwendig, um lebensfähige Mikroben in Urinproben nachzuweisen und zu identifizieren11. Bei der klinischen Standard-Urinkultur werden Urin auf 5% Schafblutagar (BAP) und MacConkey-Agar plattiert und bei 35 °C für 24 h aerob inkubiert12. Mit einer Nachweisschwelle von ≥105 KBE/ml13werden jedoch viele Mitglieder der Urinmikrobiota durch diese Methode nicht berichtet. Verbesserte Kultivierungstechniken wie Enhanced Quantitative Urine Culture (EQUC)11 verwenden verschiedene Kombinationen unterschiedlicher Urinvolumina, Inkubationszeiten, Kulturmedien und atmosphärischer Bedingungen, um Mikroben zu identifizieren, die häufig von der Standardurinkultur übersehen werden. In diesem Protokoll wird eine modifizierte Version von EQUC beschrieben, hier als Modifiziertes Enhanced Urine Culture-Protokoll bezeichnet, das die Kultivierung verschiedener Harnbakterien und Uropathogene unter Verwendung selektiver Medien und optimaler atmosphärischer Bedingungen ermöglicht, aber nicht von Natur aus quantitativ ist. Die erfolgreiche Isolierung von Harnbakterien ermöglicht die Extraktion genomischer DNA (gDNA) für nachgeschaltete WGS und Genomassemblierung.
Genom-Assemblierungen, insbesondere komplette Assemblierungen, ermöglichen die Entdeckung genetischer Faktoren, die zur Kolonisierung, Nischenerhaltung und Virulenz sowohl bei ansässigen Mikrobiota als auch bei uropathogenen Bakterien beitragen können. Entwürfe von Genomassemblys enthalten eine Vielzahl von zusammenhängenden Sequenzen (Contigs), die Sequenzierungsfehler enthalten können und keine Orientierungsinformationen enthalten. In einer vollständigen Genomanordnung wurden sowohl die Orientierung als auch die Genauigkeit jedes Basenpaares verifiziert14. Darüber hinaus bietet die Gewinnung vollständiger Genomsequenzen Einblicke in die Genomstruktur, die genetische Vielfalt und die mobilen genetischen Elemente15. Kurze Lektüren allein können das Vorhandensein oder Fehlen wichtiger Gene identifizieren, aber möglicherweise nicht ihren genomischen Kontextbestimmen 16. Mit der Ermöglichung von Long-Read-Sequenzierungstechnologien wie Oxford Nanopore und PacBio erfordert die Erzeugung geschlossener De-novo-Assemblierungen bakterieller Genome keine anstrengenden Methoden wie das manuelle Schließen von De-novo-Assemblierungen durch Multiplex-PCR17,18. Die Kombination von Next Generation Short-Read-Sequencing und Nanopore Long-Read-Sequencing-Technologien ermöglicht die einfache Generierung genauer, vollständiger und geschlossener bakterieller Genom-Assemblierungen zu relativ geringen Kosten19. Die Short-Read-Sequenzierung erzeugt genaue, aber fragmentierte Genomanordnungen, die im Allgemeinen aus durchschnittlich 40-100 Contigs bestehen, während die Nanoporensequenzierung lange Lesevorgänge von etwa 5-100 kb Länge erzeugt, die weniger genau sind, aber als Gerüste dienen können, um Contigs zu verbinden und die genomische Syntenie aufzulösen. Hybride Ansätze, die sowohl Short-Read- als auch Long-Read-Technologien verwenden, können genaue und vollständige Bakteriengenome erzeugen19.
Hier wird ein umfassendes Protokoll für die Isolierung und Identifizierung von Bakterien aus menschlichem Urin, genomische DNA-Extraktion, Sequenzierung und vollständige Genomassemblierung unter Verwendung eines hybriden Assemblierungsansatzes beschrieben. Dieses Protokoll legt besonderen Wert auf die Schritte, die notwendig sind, um Lesevorgänge richtig zu modifizieren, die durch Short-Read- und Long-Read-Sequenzierung für die genaue Montage eines geschlossenen bakteriellen Chromosoms und extrachromosomaler Elemente wie Plasmide erzeugt werden.
Protokoll
Bakterien wurden aus Urin kultiviert, der von einwilligenden Frauen im Rahmen der vom institutionellen Überprüfungsausschuss genehmigten Studien 19MR0011 (UTD) und STU 032016-006 (UTSW) gesammelt wurde.
1. Modifizierte verstärkte Urinkultur
HINWEIS: Alle Kulturschritte müssen unter sterilen Bedingungen durchgeführt werden. Sterilisieren Sie alle Instrumente, Lösungen und Medien. Reinigen Sie den Arbeitsbereich mit 70% Ethanol, stellen Sie dann einen Bunsenbrenner auf und arbeiten Sie sorgfältig in der Nähe der Flamme, um die Wahrscheinlichkeit einer Kontamination zu verringern. Alternativ kann eine Biosicherheitswerkbank der Klasse II verwendet werden, um eine sterile Umgebung aufrechtzuerhalten. Tragen Sie eine geeignete persönliche Schutzausrüstung (PSA), um eine Exposition gegenüber potenziell pathogenen Mikroben zu vermeiden.
- Beschichtung von glycerinbestücktem Urin und Kolonieisolierung
- Auftauen von glycerinbestücktem Urin bei Raumtemperatur (RT). Nach dem Auftauen die Probe für 5 s schwenken, um sie zu mischen. In sterilen Mikrozentrifugenröhrchen 1:3 und 1:30 Verdünnungen des Urins in steriler 1x phosphatgepufferter Kochsalzlösung (PBS) auf ein Endvolumen von 100 μL vorbereiten.
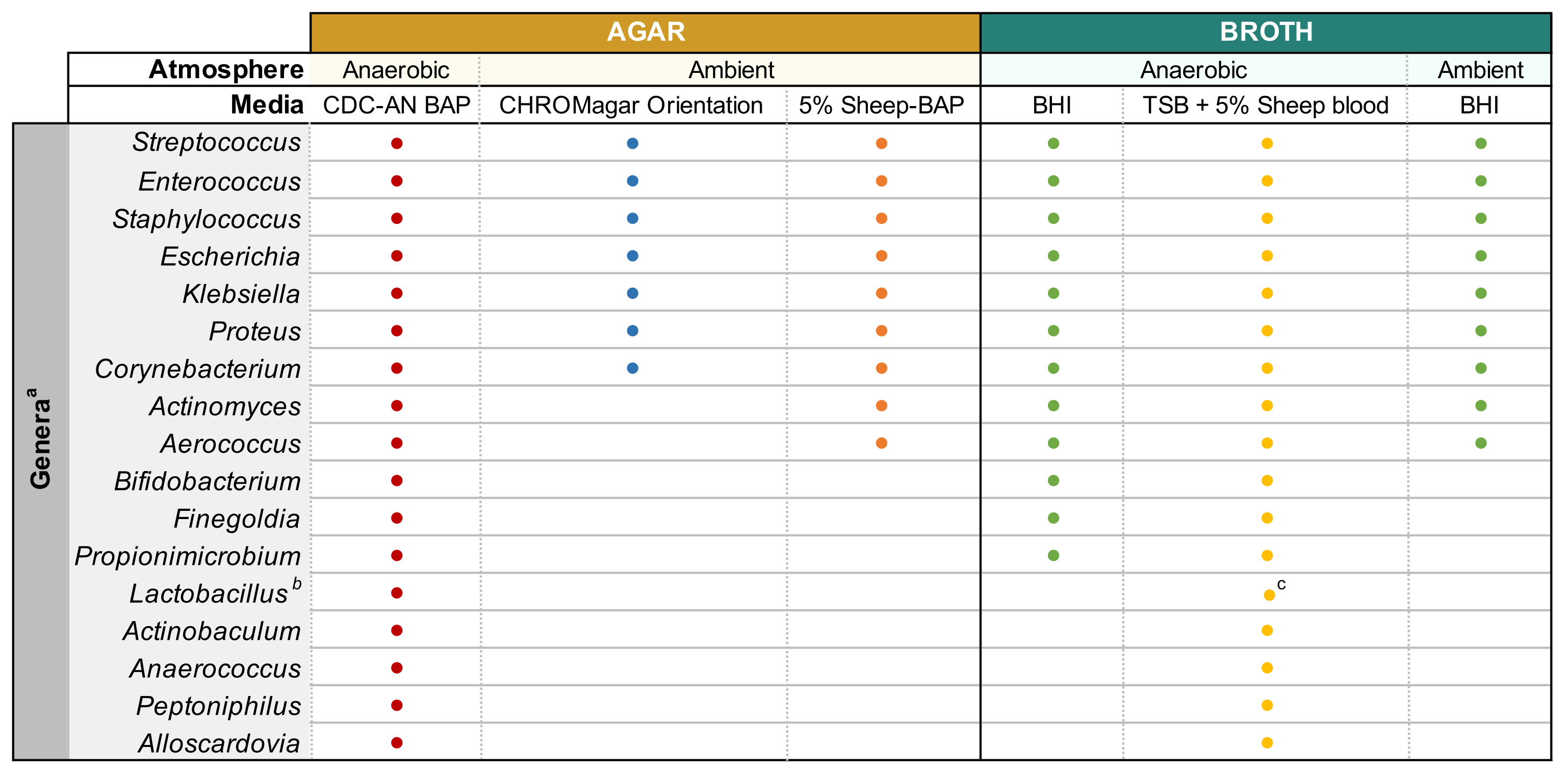
HINWEIS: Glycerin-gefüllter Urin wird durch Mischen von 500 μL unverdünntem Urin und 500 μL 50% sterilem Glycerin in Kryovialen und Lagerung bei -80 °C hergestellt. - Agarplatten bei 37 °C vor Gebrauch 15 min vorwärmen. Siehe Abbildung 1 für Medientypen und Kulturbedingungen, die für gewöhnliche Bakteriengattungen im Urin geeignet sind. Mischen Sie den verdünnten Urin gut durch Pipettieren vor dem Plattieren, platten Sie 100 μL des verdünnten Urins auf die gewünschte Agarplatte und verteilen Sie die Probe mit sterilen Glasperlen. Platte 100 μL des 1x PBS-Verdünnungsmittels auf einer separaten Platte als keine Wachstumskontrolle.
HINWEIS: Beim Versuch, häufige uropathogene Arten (z. B. Escherichia coli, Klebsiella spp., Enterococcus faecalis usw.) zu kultivieren, wird empfohlen, chromogenes Agar (Materialtabelle) zu verwenden, da es eine einfache Identifizierung uropathogener Bakterienarten ermöglicht (Abbildung 1). Colistin-Nalidixinsäure (CNA) oder MRS-Agar sind nützlich für die Isolierung anspruchsvoller Gram-positiver Spezies (z. B. Lactobacillus spp.)aus Urin, von dem bekannt ist, dass er gramnegative Uropathogene enthält, die die anspruchsvollen Spezies in nicht-selektiven Agaren übertreffen können. - Inkubieren Sie die Platte invertiert unter den gewünschten atmosphärischen Bedingungen bei 35 °C für einen Zeitraum von 24 h für Uropathogene und 3-5 Tage für anspruchsvolle Bakterien (Abbildung 1).
- Entfernen Sie nach der Inkubationszeit die Platten aus dem Inkubator. Wählen Sie von jeder Platte die Kolonien aus, die eine einzigartige Farbe, Morphologie oder hämolytische Muster aufweisen.
- Streifen Sie die Bakterienkolonie mit einer sterilen Schleife auf das entsprechende Agar und inkubieren Sie die Platte umgekehrt für 2-5 Tage in der gewünschten Atmosphäre, um gut isolierte Kolonien zu erhalten.
HINWEIS: Wenn BAP für die Primärkultur verwendet wird, kann das Patchen von Kolonien auf chromogenem Agar nützliche Informationen über die Heterogenität der Bakterienpopulation in der Probe liefern.
- Auftauen von glycerinbestücktem Urin bei Raumtemperatur (RT). Nach dem Auftauen die Probe für 5 s schwenken, um sie zu mischen. In sterilen Mikrozentrifugenröhrchen 1:3 und 1:30 Verdünnungen des Urins in steriler 1x phosphatgepufferter Kochsalzlösung (PBS) auf ein Endvolumen von 100 μL vorbereiten.
- Kultivierung in flüssiger Brühe und Glycerinstrumpf-Bakterienisolaten
- Sobald die isolierten Kolonien, die der Morphologie der Elternkolonie entsprechen, erhalten sind, wählen Sie eine einzelne Kolonie und impfen Sie sie mit einer sterilen Impfschleife in 3 ml flüssige Brühe. Siehe Abbildung 1 für Brühe, die das Wachstum gewöhnlicher Mikrobiota-Gattungen im Urin unterstützen kann. Verschließen Sie die Agarplatten mit Parafolie und lagern Sie sie bei 4 °C für 2-4 Tage. Inkubieren Sie flüssige Kulturen unter den gewünschten atmosphärischen Bedingungen für 1-5 Tage, bis die Kultur sichtbar trüb ist.
- Nachdem das Wachstum beobachtet wurde, wirbeln Sie die Kultur und fügen Sie dann 1 ml der Übernachtkultur zu 500 μL sterilem 50% Glycerin in einem 2 ml Kryovial hinzu; verschließen und durch Inversion schonend mischen. Bereiten Sie für jede Kolonie zwei Glycerinvorräte vor (einer dient als Backup) und lagern Sie sie bei -80 °C.
2. Identifizierung von Bakterienarten durch 16S rRNA Gen Sanger Sequenzierung
HINWEIS: Die mikrobielle Identität kann alternativ mit Matrix-Assisted Laser Desorption Ionization Time of Flight Mass Spectrometry (MALDI-TOF)20bestätigt werden.
- Kolonie-Polymerase-Kettenreaktion (PCR)
- Bereiten Sie eine 25 μL der PCR-Reaktion in PCR-Röhrchen vor, indem Sie 12,5 μL 2x Taq Polymerase Master Mix, 0,5 μL 10 μM 8F Primer, 0,5 μL 10 μM 1492R Primer (Materialtabelle) und 11,5 μL nukleasefreies Wasser21hinzufügen.
HINWEIS: Wenn Sie eine PCR für mehrere Proben durchführen, stellen Sie eine Reaktionsmastermischung aus Taq-Polymerase-Mischung, Primern und sterilem nukleasefreiem Wasser her. Dann aliquot 25 μL in jedes PCR-Röhrchen. - Um eine Kolonie-PCR durchzuführen, streichen Sie eine gut isolierte Kolonie mit einem sterilen Zahnstocher oder einer Pipettenspitze aus dem erneuten Streifen. Resuspendieren Sie die Kolonie in der in Schritt 2.1.1 hergestellten PCR-Reaktionsmischung. Vorsichtig mischen. Sammeln Sie die Flüssigkeit am Boden des Röhrchens durch eine schnelle Drehung bei 2000 x g.
HINWEIS: Stellen Sie sicher, dass die Probe frei von Luftblasen ist. Fügen Sie eine NTC-Probe (No-Template Control) hinzu, die nur die PCR-Reaktionsmischung enthält. - Legen Sie die Probenröhrchen in den Thermocycler und führen Sie das folgende Programm aus: 95 °C für 3 min; 40 Zyklen von: 95 °C für 30 s, 51 °C für 30 s und 72 °C für 1 min 30 s; 72 °C für 10 min; bei 10 °C halten.
- Bereiten Sie eine 25 μL der PCR-Reaktion in PCR-Röhrchen vor, indem Sie 12,5 μL 2x Taq Polymerase Master Mix, 0,5 μL 10 μM 8F Primer, 0,5 μL 10 μM 1492R Primer (Materialtabelle) und 11,5 μL nukleasefreies Wasser21hinzufügen.
- Gelextraktion und Artbestimmung
- Überprüfen Sie nach Abschluss des PCR-Laufs das PCR-Produkt auf einem 1% igen Agarosegel, das in einem 0,5-fachen Tris-Borat-EDTA (FSME)-Puffer hergestellt wird. Vor dem Gießen des Gels Ethidiumbromid (EtBr) hinzufügen. Dann gießen Sie das Gel mit Kämmen für Vertiefungen, die mindestens 20 μL Probenvolumen enthalten.
VORSICHT: EtBr ist ein Interkalationsmittel, das im Verdacht steht, krebserregend zu sein. Tragen Sie beim Umgang immer Handschuhe und PSA und entsorgen Sie EtBr-haltige Materialien gemäß den Richtlinien der Institution. - Wenn das Gel gesetzt ist, legen Sie das Gel in den mit 0,5x FSME-Puffer gefüllten Elektrophoresetank und entfernen Sie den Kamm. Laden Sie die 1 kb Leiter in die erste Vertiefung und 10-20 μL der PCR-Reaktion in nachfolgende Vertiefungen. Laufen Sie mit 100-140 V, bis es gelöst ist. Visualisieren Sie das Gel unter UV-Licht und bestätigen Sie das Vorhandensein einer klar definierten Bande bei ~ 1,5 kb, die in der NTC-Vertiefung fehlt.
ACHTUNG: UV-Strahlen sind schädlich für Haut und Augen, verwenden Sie bei der Visualisierung des Gels einen geeigneten Schutz und tragen Sie eine entsprechende PSA.
HINWEIS: Colony PCR kann für einige Bakterien erfolglos sein; Die Fortsetzung der PCR aus isolierter gDNA ist eine alternative Option22. - Schneiden Sie die ~ 1,5 kb-Bänder mit einem Rasierer aus und übertragen Sie die Gelstecklinge in saubere Mikrozentrifugenröhrchen. Fahren Sie mit dem Gelextraktionsprotokoll gemäß den Anweisungen des Herstellers fort (Tabelle der Materialien). Messen Sie die Konzentration der gereinigten DNA mit einem Mikrovolumenspektrophotometer.
HINWEIS: Eine Konzentration >10 ng/μL ist wünschenswert, und A260/280 zwischen 1,7-2,0 ist akzeptabel. - Bereiten Sie zwei Sanger-Sequenzierungsreaktionen für jede Probe vor, eine mit dem 8F und die andere mit dem 1492R-Primer in nukleasefreiem Wasser gemäß den Richtlinien eines beliebigen Sanger-Sequenzierungsdienstes.
- Sobald die Sequenzierungsdaten eingegangen sind, laden Sie die DNA-Sequenzen auf die NCBI Basic Local Alignment Search Tool (BLAST) Website (blast.ncbi.nlm.nih.gov/Blast.cgi) hoch, wählen Sie Nucleotide BLAST (Blastn), wählen Sie die rRNA/ITS-Datenbank 16S ribosomale RNA-Sequenzen (Bakterien und Archaeen) und führen Sie das Megablast-Programm aus. Das Isolat kann durch den Treffer von höchster Qualität auf eine Referenz aus der Datenbank identifiziert werden.
HINWEIS: Einige Bakterienarten weisen eine hohe Identität in ihren 16S-rRNA-Sequenzen auf und sind mit dieser Methode allein möglicherweise nicht zu unterscheiden. Die Artbildung erfordert DNA-Homologie und biochemische Analysen, um Mitglieder derselben Gattung sicher zu unterscheiden23.
- Überprüfen Sie nach Abschluss des PCR-Laufs das PCR-Produkt auf einem 1% igen Agarosegel, das in einem 0,5-fachen Tris-Borat-EDTA (FSME)-Puffer hergestellt wird. Vor dem Gießen des Gels Ethidiumbromid (EtBr) hinzufügen. Dann gießen Sie das Gel mit Kämmen für Vertiefungen, die mindestens 20 μL Probenvolumen enthalten.
3. Extraktion genomischer DNA (gDNA)
HINWEIS: Dieser Abschnitt verwendet Reagenzien und Spinsäulen, die im gDNA-Extraktionskit enthalten sind, auf das in der Materialtabelle verwiesen wird, für die ertragreiche Extraktion hochwertiger genomischer DNA aus verschiedenen Bakterienarten. Im Folgenden finden Sie empfohlene Änderungen und Anweisungen.
- Bereiten Sie Kit-Reagenzien gemäß den Anweisungen des Herstellers vor.
- Bereiten Sie 3-10 ml Kulturen in geeigneter steriler Brühe vor (Abbildung 1), indem Bakterien aus gut isolierten Kolonien in die Medien geimpft und bei der in Abbildung 1 angegebenen Temperatur und dem atmosphärischen Druck inkubiert werden, bis ein ausreichendes Wachstum beobachtet wird.
- Nach der Inkubation wird die optische Dichte bei 600 nm (OD600)der Kultur mit einem Spektralphotometer24gemessen.
- Bereiten Sie die Probe für die Quantifizierung vor, indem Sie die Übernachtkulturen im Verhältnis 1:10 verdünnen. Fügen Sie auch einen Rohling des sterilen Kulturmediums zur Messung bei. Berechnen Sie die optische Dichte, indem Sie den Blindwert vom Probenwert subtrahieren und mit dem Verdünnungsfaktor von zehn multiplizieren.
- Berechnen Sie anhand der OD600-Messung und eines für die Spezies vorab festgelegten OD600 zu KBE / ml-Verhältnisses, wie viele Milliliter Kultur erforderlich sind, um 2 x 109 Zellen zu erhalten.
- Zentrifugieren Sie das benötigte Kulturvolumen für 5 min bei 5000 x g auf Pellet. Den Überstand absaugen und das Pellet in 200 μL kaltem TE-Puffer resuspenieren (zu Beginn des Verfahrens auf Eis vorkühlen).
- Zentrifugieren Sie die Probe für 2 min bei 5000 x g. Entfernen Sie den Überstand und resuspenieren Sie dann das Pellet in 180 μL Enzymatic Lysis Buffer (ELB) und fügen Sie 20 μL vorgekochte RNase A (10 mg / ml) hinzu. Für eine effiziente Lyse von Gram-positiven Bakterien werden 18 μL Mutanolysin (25 kU/ml) zugegeben. Wirbeln Sie gut und inkubieren Sie dann die Proben bei 37 ° C auf dem Rotator für 2 h.
HINWEIS: Es wird empfohlen, die im Protokoll des Herstellers beschriebene ELB sowohl für Gram-positive als auch für Gram-negative Bakterien zu verwenden. - Gehen Sie gemäß den Anweisungen des Herstellers vor.
HINWEIS: Wiederholen Sie die Elutionsschritte ein- oder zweimal, um bei Bedarf eine zusätzliche gDNA-Ausbeute zu erhalten. - Beurteilen Sie die Qualität der extrahierten gDNA gemäß Abschnitt 4 und lagern Sie gDNA bei 4 °C, wenn es innerhalb von 1 Woche verwendet wird. Alternativ können Sie gDNA zur Langzeitlagerung bei -20 °C halten.
4. Bewertung der Qualität der extrahierten gDNA
- Um die Qualität durch Gelelektrophorese zu beurteilen, wird 1% Agarosegel wie in Unterabschnitt 2.2 beschrieben hergestellt. Bereiten Sie die Probe in einem sauberen Röhrchen vor: Mischen Sie 1-2 μL extrahierte gDNA und 3 μL 2x Ladefarbstoff auf Parafilm. Führen Sie das Gel nach dem Laden aus und visualisieren Sie es dann unter UV-Licht.
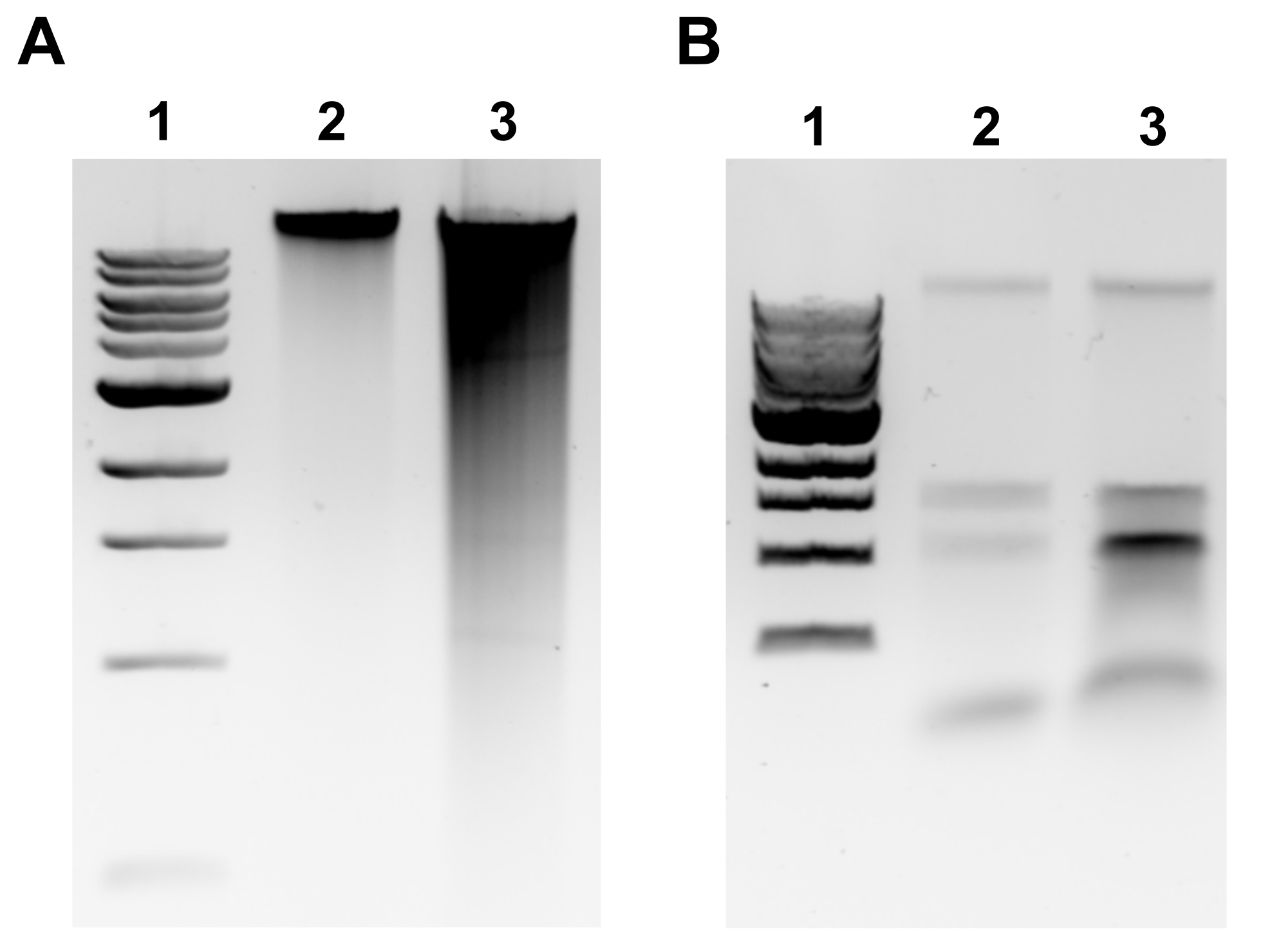
HINWEIS: Eine erfolgreiche gDNA-Extraktion zeigt sich in einem diskreten Band an der Oberseite des Gels und einer minimalen Verschmierung (Abbildung 2A). Verschmieren ist ein Hinweis auf eine Scherung. Wenn kein gDNA-Band erkennbar ist und/oder die Verschmierung erheblich ist, wiederholen Sie die gDNA-Extraktion. Erwägen Sie, die Inkubationszeiten in RNase A und Proteinase K zu verkürzen. Wenn zwei Banden um 1,5-3 kb beobachtet werden, deutet dies auf eine RNA-Kontamination hin (Abbildung 2B). Bereiten Sie frische RNase A zu und wiederholen Sie die Extraktion. - Um die Qualität mittels Mikrovolumenspektrophotometer zu beurteilen, messen Sie die gDNA-Konzentration und das Absorptionsverhältnis A260/280 mit einem Mikrovolumenspektrophotometer. Konzentrationen >50 ng/μL und A260/280 zwischen 1,7-2,0 sind akzeptabel.
HINWEIS: Eine niedrige gDNA-Ausbeute kann auf geringen Input, hohen Input, Kontamination von Nukleasen und unzureichende Lyse zurückzuführen sein. Absorptionsverhältnisse oberhalb des Bereichs weisen auf eine RNA-Kontamination hin. Wiederholen Sie die Extraktion, wenn die gDNA-Qualität schlecht ist. - Um die Qualität durch Fluorometer zu beurteilen, befolgen Sie die Anweisungen des Herstellers zur Quantifizierung der gDNA-Konzentration mit hochempfindlichem Assay-Kit und Fluorometer-Instrument (Materialtabelle). Konzentration >50 ng/μL ist wünschenswert.
5. Paired-End-Sequenzierung der nächsten Generation und Bibliotheksvorbereitung
HINWEIS: Short-Read-Sequenzierung kann auf verschiedenen Instrumenten mit unterschiedlichen Leselängen und Orientierungen durchgeführt werden. 150 bp (300 Zyklus) Paired-End-Sequenzierung wird für bakterielle WGS empfohlen. Sowohl die Vorbereitung als auch die Sequenzierung der Bibliothek können an Kerneinrichtungen oder kommerzielle Labors ausgelagert werden.
- Erstellen Sie die Sequenzierungsbibliothek gemäß den Anweisungen des Herstellers (Materialverzeichnis). Befolgen Sie die vom Hersteller empfohlene endgültige Belastungsbibliothekskonzentration. Eine empfohlene Änderung besteht jedoch darin, die gepoolte Bibliothek mit 1,8 pM zu laden, um eine optimale Lesegenerierung auf NextSeq-Geräten zu erzielen.
- Obwohl optional, verwenden Sie einen Bioanalyzer (Table of Materials), um die Verteilung der gepoolten Bibliotheksfragmente zu bewerten und sicherzustellen, dass die Fragmentgröße im Durchschnitt 600 bp beträgt.
6. Vorbereitung der Nanoporen-MinION-Sequenzierungsbibliothek
- Bereiten Sie die Sequenzierungsbibliothek gemäß dem Protokoll des Herstellers vor (Materialtabelle). Die Verwendung von zwei Barcode-Erweiterungskits ermöglicht das Multiplexen von bis zu 24 Proben auf einer einzigen Durchflusszelle. Es wird empfohlen, die Bibliotheksvorbereitung in zwei Teilen durchzuführen, 12 Proben gleichzeitig, wenn 24 Proben gemultiplext werden. Alle 24 Proben können wie unten beschrieben gepoolt werden.
HINWEIS: Proben können nach Abschluss der Native Barcode Ligation über Nacht bei 4 °C gelagert werden - dies bietet bei Bedarf einen Haltepunkt im Protokoll. Am Ende des Abschnitts Native Barcode Ligation des Bibliotheksvorbereitungsprotokolls wird empfohlen, äquimolare Mengen jeder Probe bis zur maximal möglichen DNA-Masse (ng) zu bündeln.- Um dies zu tun, quantifizieren Sie alle Proben nach Barcode-Ligation mit einem Fluorometer (Table of Materials) gemäß den Anweisungen des Herstellers. Schätzen Sie das Volumen der Probe mit der niedrigsten dsDNA-Konzentration und berechnen Sie dann die gesamte dsDNA, die in dieser Probe gefunden wurde. Verwenden Sie diese Zahl, um die Äquimolarmengen aller anderen Proben zu bestimmen, die zusammengelegt werden.
HINWEIS: Da die äquimolare Berechnung die Menge der gepoolten dsDNA maximiert und somit einen Pool mit hohem Volumen (>65 μL) ergibt, ist eine Bereinigung erforderlich, um den Pool zu konzentrieren.
- Um dies zu tun, quantifizieren Sie alle Proben nach Barcode-Ligation mit einem Fluorometer (Table of Materials) gemäß den Anweisungen des Herstellers. Schätzen Sie das Volumen der Probe mit der niedrigsten dsDNA-Konzentration und berechnen Sie dann die gesamte dsDNA, die in dieser Probe gefunden wurde. Verwenden Sie diese Zahl, um die Äquimolarmengen aller anderen Proben zu bestimmen, die zusammengelegt werden.
- dsDNA-Poolbereinigung und -konzentration
- Fügen Sie dem DNA-Pool 2,5-faches Volumen paramagnetischer Perlen(Table of Materials)hinzu und streichen Sie dann vorsichtig mit dem Röhrchen, um den Inhalt zu mischen. Legen Sie das Röhrchen für 5 min bei RT in den Rotator. Drehen Sie die Probe bei 2000 x g nach unten und pelletieren Sie es auf einem Magneten.
- Fügen Sie 250 μL frisch zubereitetes 70% Ethanol (in nukleasefreiem Wasser) hinzu und achten Sie darauf, das Pellet nicht zu stören. Aspirieren Sie das Ethanol und wiederholen Sie die Ethanolwäsche einmal.
- Nach der zweiten Aspiration drehen Sie die Probe mit 2000 x g nach unten und legen Sie sie wieder auf den Magneten. Alle Reste von Ethanol pipettieren und die Probe etwa 30 s trocknen lassen.
- Entfernen Sie das Röhrchen vom Magneten und resuspenieren Sie das Pellet in 60-70 μL nukleasefreiem Wasser. Inkubieren Sie bei RT für 2 Min. Pelletieren Sie die Probe auf dem Magneten, bis die Elute klar ist, und entfernen Sie dann die Elute und übertragen Sie sie in ein sauberes 1,5 ml Mikrozentrifugenröhrchen.
- Quantifizieren Sie den konzentrierten Pool mit einem Fluorometer und bereiten Sie dann ein Aliquot vor, um mit dem Adapterligationsschritt fortzufahren: Bereiten Sie 700 ng der Probe in 65 μL Endvolumen vor. Halten Sie den Rest des Pools bei 4 ° C für einen zweiten Lauf, der nach Abschluss des ersten Laufs abgeschlossen ist.
- Fahren Sie mit der Adapterligatur gemäß den Anweisungen des Herstellers fort und laden Sie die Probe auf die Durchflusszelle. Starten Sie den Sequenzierungslauf.
HINWEIS: Saugluft und ~200 μL Speicherpuffer aus dem Ansauganschluss der Durchflusszelle vor der Probenbeladung. Dies ist entscheidend für die erfolgreiche Durchflusszellenprimierung und Probenbeladung. Verwenden Sie eine p1000-Pipette und Spitzen, wenn Sie Lösungen durch den Grundierungsanschluss der Durchflusszelle ziehen und ablegen.
- Sequenzieren Sie die Bibliothek gemäß den Anweisungen des Herstellers.
- Öffnen Sie die Betriebssoftware für die Sequenzierung und klicken Sie auf Start. Geben Sie einen Namen für das Experiment ein, eine empfohlene Nomenklatur enthält das Ausführungsdatum und den Namen des Benutzers. Klicken Sie auf Weiter zur Kit-Auswahl,wählen Sie das entsprechende Library Prep Kit und Barcode Expansion Pack(s) aus und klicken Sie dann auf Weiter zu Ausführungsoptionen.
- Passen Sie die Lauflänge auf 48 h an, wenn Sie planen, genügend Bibliothek für einen zweiten Lauf vorzubereiten (andernfalls belassen Sie standardmäßig 72 h). Klicken Sie auf Weiter zu Basecalling.
- Aktivieren Sie die Basecalling-Option Config: Fast Basecalling und stellen Sie sicher, dass Barcoding auf Enabled gesetzt ist, damit die ausgegebenen FASTQ-Dateien von den Barcode-Sequenzen abgeschnitten und basierend auf Barcode in separate Verzeichnisse demultiplex werden. Klicken Sie auf Weiter zur Ausgabe.
- Wählen Sie aus, wo Ausgabesequenzierungsdaten gespeichert werden sollen. Erwarten Sie ungefähr 30-50 GB Daten, wenn Sie nur fastQ-Ausgabe speichern, und >500 GB Daten, wenn Sie auch fast5-Ausgabe speichern. Deaktivieren Sie die Filteroption Qscore: 7 | Readlength: Ungefiltert, wenn Sie mit der in Abschnitt 7.2 beschriebenen Filterung fortfahren möchten, andernfalls lassen Sie sie aktiviert und passen Sie readlength auf 200 an.
- Klicken Sie auf Weiter, um Setup auszuführen, und überprüfen Sie alle Einstellungen. Wenn die Einstellungen korrekt sind, klicken Sie auf Start,andernfalls klicken Sie auf Zurück und nehmen Sie die erforderlichen Anpassungen vor.
- Auf Wunsch kann die Durchflusszelle gemäß den Anweisungen des Herstellers gewaschen und mit dem verbleibenden Pool nachgeladen werden. Wiederholen Sie die Schritte in 6.2 für den verbleibenden Pool, sobald der erste Durchlauf abgeschlossen ist und die Durchflusszelle gewaschen wurde.
HINWEIS: Stellen Sie beim Einrichten des zweiten Laufs die Bias-Spannung auf -250 mV gemäß den Empfehlungen des Herstellers für Durchflusszellen ein, die zuvor in Läufen über 48 h verwendet wurden.
7. Bewertung und Vorbereitung von Lesevorgängen
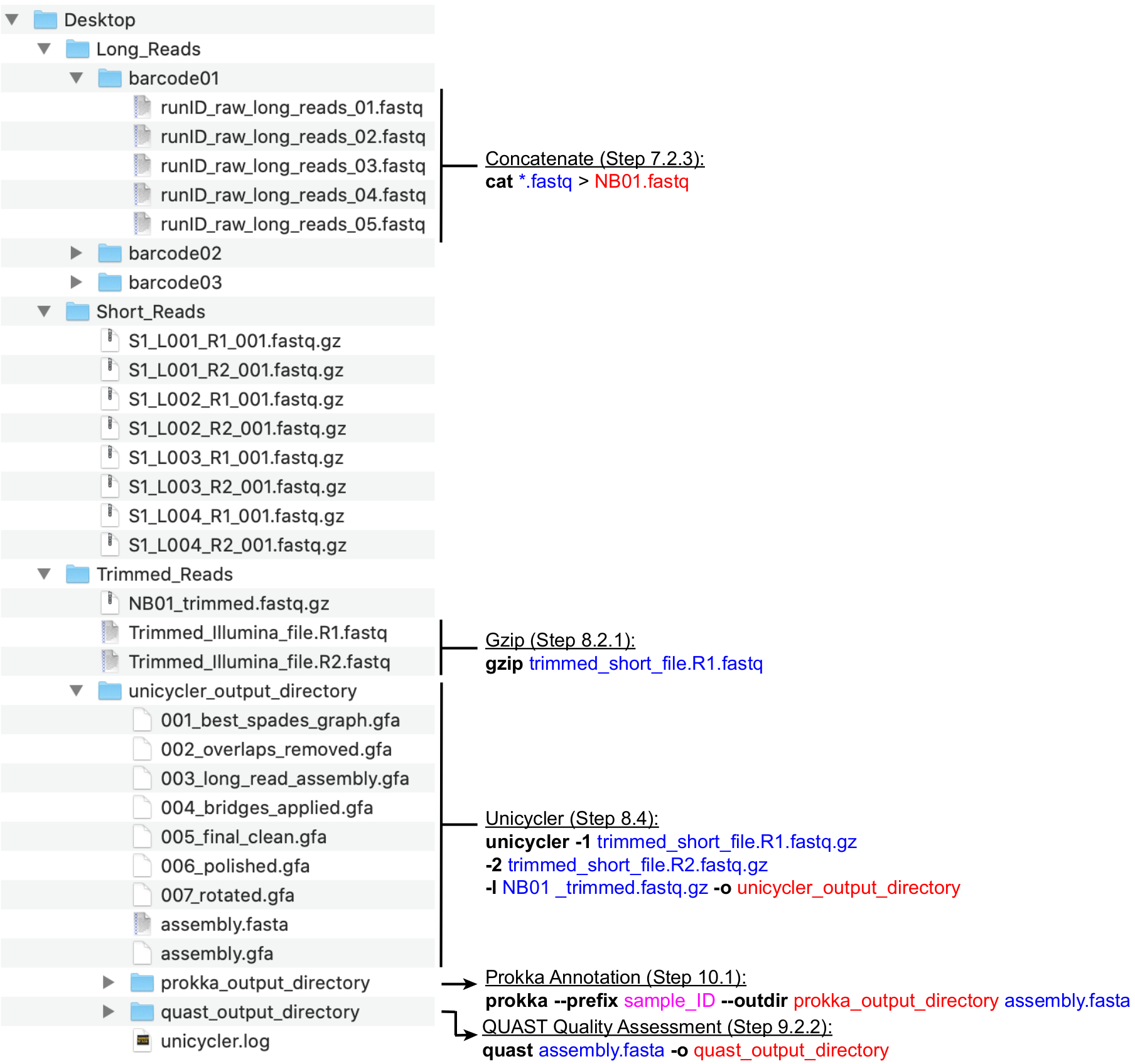
HINWEIS: Eine empfohlene Verzeichnisstruktur ist in Abbildung 4dargestellt. Erstellen Sie die auf dem Desktopgefundenen Verzeichnisse, d. h. Long_Reads, Short_Reads und Trimmed_Reads, bevor Sie mit den folgenden Berechnungsschritten fortfahren.
- Kurze Lektüren (Abbildung 3)
HINWEIS: Kurze Lesevorgänge werden im FASTQ-Format generiert. Die Dateien enthalten maximal 4000 Lesevorgänge pro FASTQ. Diese sind oft gezippt (.gz Archiv) und in mehreren Dateien organisiert. Je nach Plattform werden Barcodes typischerweise beschnitten. Einige Programme akzeptieren Dateien im gezippten Format, andere erfordern möglicherweise deren Extraktion vor dem Import. Die Lesevorgänge müssen die Qualitätskontrollschritte (QC) bestehen, um die Datengenauigkeit während der Genomassemblierung sicherzustellen. Wenn CLC Genomics Workbench nicht verfügbar ist, können alternative Programme zum Trimmen und QC-Kurzlesevorgänge wie Trimmomatic verwendet werden.25 oder Trim Galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) zum Trimmen und FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) zur Bewertung der Lesequalität. Es wird empfohlen, die durchschnittliche short read coverage, geschätzt durch Multiplikation der Anzahl der Lesevorgänge mit der durchschnittlichen Lesedauer und Dividierung durch die Genomgröße, >100x.- Öffnen Sie die Genomics Workbench-Software (Table of Materials) und importieren Sie alle gepaarten FASTQ-Dateien mit kurzer Lektüre. Gepaarte Dateien werden automatisch generiert.
- Erstellen Sie einen neuen Ordner unter CLC_Data, indem Sie in der oberen Symbolleiste auf Neu klicken und Ordner... , um die Dateien zu speichern. Benennen Sie den Ordner wie gewünscht, eine empfohlene Konvention ist die Verwendung der Beispiel-ID. Speichern Sie die gesamte Ausgabe aus den folgenden Schritten in diesem Ordner.
- Klicken Sie in der oberen Symbolleiste auf die Schaltfläche Importieren und wählen Sie Illumina... Navigieren Sie zu allen Short-Read-Dateien, die dem Beispiel entsprechen, und wählen Sie sie aus. Stellen Sie sicher, dass die Option gepaarte Lesevorgänge ausgewählt ist, und deaktivieren Sie die Option Fehlgeschlagene Lesevorgänge entfernen. Klicken Sie auf Weiter, wählen Sie Speichernund klicken Sie erneut auf Weiter. Speichern Sie die importierten Dateien in dem neuen Ordner, der im vorherigen Schritt erstellt wurde, und klicken Sie auf Fertig stellen.
- Erstellen Sie eine Sequenzliste aller gepaarten Dateien für das Isolat. Dadurch werden gelesene Daten in einer einzigen Datei verkettet, um die Analyse zu vereinfachen.
- Klicken Sie in der oberen Symbolleiste auf die Schaltfläche Neu und wählen Sie Sequenzliste... Wählen Sie in der Verzeichnisliste auf der linken Seite die zu verkettenden Dateien aus, und verschieben Sie diese mithilfe der Pfeile in die Liste der ausgewählten Dateien auf der rechten Seite. Klicken Sie auf Weiter, wählen Sie Speichernund klicken Sie erneut auf Weiter. Speichern Sie die Sequenzliste und klicken Sie auf Fertig stellen.
- Sobald die Sequenzliste generiert wurde, benennen Sie sie sofort mit der Beispiel-ID um.
- Führen Sie das QC for Sequencing Reads-Tool in der Sequenzliste aus: Mit diesem Verfahren werden die allgemeinen Qualitätsparameter der Lesevorgänge bewertet, die durch Short-Read-NGS generiert werden.
- Suchen Sie im Toolbox-Menü (unteres linkes Fenster) nach dem QC for Sequencing Reads-Tool. Doppelklicken Sie auf das Werkzeug, wählen Sie dann die zu analysierende Sequenzliste aus und klicken Sie auf Weiter.
- Stellen Sie sicher, dass alle Ausgabeoptionen aktiviert sind, und wählen Sie unter Ergebnisbehandlung die Option Speichern aus. Klicken Sie auf Weiter und geben Sie an, die Ausgabedateien zu speichern, und klicken Sie dann auf Fertig stellen.
- Führen Sie das Werkzeug "Lesevorgänge zuschneiden" in der Sequenzliste aus: Das Zuschneiden erfolgt basierend auf Qualität, Länge und Mehrdeutigkeit. Bei diesem Prozess wird davon ausgegangen, dass die bei der Sequenzierung verwendeten Barcodes vor diesem Schritt beschnitten wurden.
- Suchen Sie in der Toolbox (unteres linkes Fenster) nach dem Werkzeug Lesevorgänge zuschneiden. Doppelklicken Sie auf Reads trimmen, wählen Sie dann die zu analysierende Sequenzliste aus und klicken Sie auf Weiter.
- Qualitätskürzung: Legen Sie die Qualitätsfaktorgrenze auf 0,01 fest und lassen Sie mehrdeutige Nukleotide bei 2. Klicken Sie auf Weiter.
HINWEIS: Die Parameter können nach Ermessen des Benutzers angepasst werden. Dies sind die empfohlenen Einstellungen. - Deaktivieren Sie Automatisches Durchsuchen des Adapters (nur tun Sie dies, wenn adapter vor dem Import in CLC aus den Lesevorgängen beschnitten wurden). Klicken Sie auf Weiter und aktivieren Sie Discard Reads Below Length, verwenden Sie default 15.
- Klicken Sie auf Weiter, aktivieren Sie Bericht erstellen, und wählen Sie dann Speichern aus. Klicken Sie auf Weiter und geben Sie an, wo die Ausgabedateien gespeichert werden sollen. Klicken Sie auf Fertig stellen.
- Exportieren Sie die Liste der getrimmten Sequenzen: Die nachfolgende Hybridmontage und -analyse wird außerhalb von CLC abgeschlossen und erfordert den Export von getrimmten Kurzlesedateien.
- Wählen Sie in der Verzeichnisnavigation oben links die in Schritt 7.1.4 generierte zugeschnittene Datei aus und klicken Sie dann in der oberen Symbolleiste auf Exportieren. Wählen Sie Fastq als Exportdateityp und klicken Sie auf Weiter. Aktivieren Sie Das Kontrollkästchen Liste der gepaarten Sequenzen in zwei Dateien exportieren. Klicken Sie dann auf Weiter und wählen Sie das Verzeichnis Trimmed_Reads, in das die Dateien exportiert werden sollen. Klicken Sie auf Fertig stellen. Stellen Sie sicher, dass die gekürzten Short-Read-Dateien erfolgreich als zwei Dateien (R1 und R2) mit der Erweiterung .fastqexportiert wurden.
HINWEIS: Die Liste der zugeschnittenen Sequenzen muss in zwei Dateien exportiert werden, die von CLC in der Regel als R1 und R2 bezeichnet werden. Dies ist von entscheidender Bedeutung, da die nachgelagerte Hybridbaugruppe eine kurz lesbare Dateneingabe erfordert, um als solche eingerichtet zu werden. - Benennen Sie die exportierten Dateien um, bitte verzichten Sie auf die Verwendung von Leerzeichen und Sonderzeichen in Dateinamen. Der Einfachheit halber wird ein empfohlenes Format trimmed_short_file. R1.fastq.
- Wählen Sie in der Verzeichnisnavigation oben links die in Schritt 7.1.4 generierte zugeschnittene Datei aus und klicken Sie dann in der oberen Symbolleiste auf Exportieren. Wählen Sie Fastq als Exportdateityp und klicken Sie auf Weiter. Aktivieren Sie Das Kontrollkästchen Liste der gepaarten Sequenzen in zwei Dateien exportieren. Klicken Sie dann auf Weiter und wählen Sie das Verzeichnis Trimmed_Reads, in das die Dateien exportiert werden sollen. Klicken Sie auf Fertig stellen. Stellen Sie sicher, dass die gekürzten Short-Read-Dateien erfolgreich als zwei Dateien (R1 und R2) mit der Erweiterung .fastqexportiert wurden.
- Öffnen Sie die Genomics Workbench-Software (Table of Materials) und importieren Sie alle gepaarten FASTQ-Dateien mit kurzer Lektüre. Gepaarte Dateien werden automatisch generiert.
- Lange (MinION) Lesevorgänge (Abbildung 3)
HINWEIS: Die folgende Pipeline für die Vorbereitung von Long (MinION) Sequenzierungslesevorgängen für die Hybridmontage verwendet die NanoFilt- und Nanostat-Programme26, die über die Befehlszeile ausgeführt werden. Installieren Sie die Tools, bevor Sie fortfahren, und machen Sie sich mit den Grundlagen von UNIX vertraut, um diese Befehle auszuführen. Standardterminals und Bash Shell werden empfohlen. Eine Lektionsanleitung für gängige Terminalbefehle und -verwendung finden Sie unter Software Carpentry27. Die folgenden Anweisungen gehen davon aus, dass die generierten Dateien mit der Barcode-Nomenklatur (NB01, NB02 usw.) benannt und im Verzeichnis Long_Reads gespeichert werden. Alternativ kann die Lesefilterung mit MinKNOW beim Einrichten des Sequenzierungslaufs durchgeführt werden. Es wird empfohlen, die durchschnittliche lange Leseabdeckung >100x zu betragen. Die empfohlene durchschnittliche Lesedauer beträgt >2000 bp; Daher ist die Anzahl der erforderlichen langen Lesevorgänge niedriger als die Anzahl der kurzen Lesevorgänge.- Erstellen Sie für jeden im Lauf verwendeten Barcode (barcode01, barcode02 usw.) neue Verzeichnisse innerhalb des Long_Reads-Verzeichnisses (Abbildung 4). Kopieren Sie alle FASTQ-Dateien, die jedem Barcode entsprechen, in den entsprechenden Ordner. Kombinieren Sie alle .fastq-Dateien für jeden Barcode aus jedem Lauf.
- Öffnen Sie Terminal und navigieren Sie mit dem Befehl cd zu den Barcode-Verzeichnissen im Verzeichnis Long_Reads: cd Desktop/Long_Reads/barcode01
- Verketten Sie alle .fastq-Dateien pro Barcode zu einer einzigen .fastq-Datei, indem Sie den folgenden Befehl ausführen: cat *.fastq > NB01.fastq
HINWEIS: Dieser Befehl kombiniert alle Lesevorgänge aus jeder der FASTQ-Dateien in einem großen, einzigen FASTQ mit dem Namen NB01.fastq. - Verwenden Sie NanoStat, um die Lesequalität der Probe zu bewerten, indem Sie den folgenden Befehl ausführen: NanoStat --fastq NB01.fastq
- Zeichnen Sie die Ergebnisse auf, indem Sie die Ausgabe zur späteren Bezugnahme in eine Text- oder Word-Datei kopieren.
- Verwenden Sie NanoFilt, um MinION-Lesevorgänge zu filtern und Lesevorgänge mit Q < 7 und einer Länge < 200 zu verwerfen, indem Sie den Folgenden ausführen: NanoFilt -q 7 -l 200 bp NB01.fastq | gzip > NB01 _trimmed.fastq.gz
- Führen Sie NanoStat für die in Schritt 7.2.6 generierte getrimmte Datei aus, indem Sie den folgenden Befehl ausführen: NanoStat --fastq NB01 _trimmed.fastq.gz
- Zeichnen Sie die Ergebnisse auf, indem Sie die Ausgabe in eine Text- oder Word-Datei kopieren und mit den Ergebnissen aus Schritt 7.2.4 vergleichen, um sicherzustellen, dass die Filterung erfolgreich war (Tabelle 1).
- Wiederholen Sie die Schritte 7.2.2 bis 7.2.8 für jeden Barcode, der im Sequenzierungslauf verwendet wird.
HINWEIS: Die in Schritt 7.2.6 generierte Datei NB01_trimmed.fastq.gz wird für die Hybridassembly verwendet.
8. Hybridgenom-Assemblierung generieren
HINWEIS: Die folgende Montagepipeline verwendet Unicycler19,28,29,30, um kurze und lange Lesevorgänge zu kombinieren, die in den Abschnitten 7.1 und 7.2 vorbereitet wurden (Abbildung 3). Installieren Sie Unicycler und seine Abhängigkeiten und führen Sie die folgenden Befehle aus. Es wird davon ausgegangen, dass Dateien mit kurzer Lesezeit, die in Schritt 7.1.5 exportiert wurden, trimmed_short_file benannt sind. R1.fastq und trimmed_short_file. R2.fastq für Einfachheit.
- Organisieren Sie die Dateien mit kurzer Lesegeschwindigkeit und die Dateien mit langer Lesedauer in einem einzigen Verzeichnis mit dem Namen Trimmed_Reads. Das Verzeichnis muss Folgendes enthalten:
- Eine .fastq.gz Datei für abgeschnittene lange Lesevorgänge (generiert in Schritt 7.2.6).
- Zwei .fastq-Dateien (R1 und R2) für abgeschnittene kurze Lesevorgänge (generiert in Schritt 7.1.5).
- Navigieren Sie mit dem Befehl cd im Terminal zu dem Verzeichnis Trimmed_Reads, in dem die gelesenen Dateien gespeichert sind: cd Desktop/Trimmed_Reads
- Sobald Sie sich im richtigen Verzeichnis befinden, komprimieren Sie die beiden kurz gelesenen Dateien so, dass sie auch im .fastq.gz-Format sind, indem Sie den folgenden Befehl ausführen: gzip trimmed_short_file. R1.fastq
- Wiederholen Sie Schritt 8.2 für R1 und R2. Überprüfen Sie, ob alle gelesenen Dateien jetzt im Fastq.gz Format vorliegen, und stellen Sie sicher, dass alle Dateien mit demselben Isolat übereinstimmen.
- Starten Sie die Hybridassembly mit Unicycler, indem Sie den folgenden Befehl ausführen:
Einradfahrer -1 trimmed_short_file. R1.fastq.gz -2 trimmed_short_file. R2.fastq.gz -l NB01 _trimmed.fastq.gz -o unicycler_output_directory
HINWEIS: -o gibt das Verzeichnis an, in dem die Unicycler-Ausgabe gespeichert wird, Unicycler erstellt dieses Verzeichnis, sobald der Befehl ausgeführt wird; Generieren Sie das Verzeichnis nicht vorher. Die Laufzeit variiert je nach Rechenleistung des verwendeten Computers sowie der Genomgröße und der Anzahl der Lesevorgänge. Dies kann zwischen 4 Stunden und 1 oder 2 Tagen dauern. Dieses Protokoll wurde auf einer CentOS Linux 7 Maschine mit 250 Gb RAM, Intel Xeon (R) CPU mit 2,5 GHz 12 praktischen Kernen und 48 virtuellen Kernen durchgeführt. Alternativ können PCs mit 16 Gb RAM und 2,6 GHz 6-Core-Prozessoren diese Assemblys mit einer längeren Verarbeitungszeit berechnen. - Wenn der Lauf abgeschlossen ist, überprüfen Sie die Einrad.log Datei, um sicherzustellen, dass keine Fehler auftreten - notieren Sie die Anzahl, Größe und den Status (vollständig, unvollständig) der generierten Contigs.
- Wenn unvollständige Contigs identifiziert werden (im Unicycler-Protokoll als unvollständig gekennzeichnet), führen Sie Unicycler im Fettdruckmodus erneut aus, indem Sie dem Befehl in Schritt 8.4 das folgende Flag hinzufügen: --mode bold.
HINWEIS: Der Fettdruckmodus senkt den Qualitätsschwellenwert, der für lange Lesebrücken während der Montage akzeptiert wird. dies kann zu einer vollständigen Baugruppe führen, aber die Montagequalität kann verringert werden. Es wird empfohlen, den Fettmodus nur bei Bedarf und als vorläufigen Nachweis für den Contig-Beitritt zu verwenden, der später durch die PCR bestätigt wird.
- Wenn unvollständige Contigs identifiziert werden (im Unicycler-Protokoll als unvollständig gekennzeichnet), führen Sie Unicycler im Fettdruckmodus erneut aus, indem Sie dem Befehl in Schritt 8.4 das folgende Flag hinzufügen: --mode bold.
9. Beurteilung der Montagequalität
HINWEIS: Das folgende Protokoll verwendet Bandage31 und QUAST32, zwei Programme, die vor der Verwendung eingerichtet werden müssen (Abbildung 2 und Abbildung 4). Bandage erfordert keine Installation nach dem Herunterladen und QUAST erfordert Vertrautheit mit der grundlegenden Befehlszeilenverwendung. Es wird auch empfohlen, die Vollständigkeit des Genoms mit Benchmarking Universal Single-Copy Orthologs (BUSCO)33zu bewerten.
- Verband: Klicken Sie auf Datei. Wählen Sie dann Diagramm laden und wählen Sie die Datei assembly.gfa aus, die in unicycler_output_directory gespeichert wurde, die von Unicycler in Schritt 8.4 generiert wurde. Klicken Sie nach dem Laden auf der linken Symbolleiste auf die Schaltfläche Diagramm zeichnen, und sehen Sie sich an, wie die Contigs (Knoten genannt) verbunden und organisiert sind, um zu bewerten, ob die Baugruppe vollständig ist (Abbildung 5).
HINWEIS: Vollständige Baugruppen werden durch einzelne Kreiskonturen dargestellt, die an beiden Enden miteinander verbunden sind (Abbildung 5A, B). Unvollständige Baugruppen haben mehrere Contigs miteinander verbunden oder sind linear (Abbildung 5C). Kleine lineare Contigs sind möglicherweise nicht unvollständig, da sie auf lineare extrachromosomale Elemente hinweisen können. Die Abdeckung, auch Tiefe genannt, wird in Bandage notiert und stellt die relative Häufigkeit der Contigs zum Chromosom dar, normalisiert in Unicycler auf 1x. - QUAST
- Navigieren Sie im Terminal zu dem Ordner, in dem die Unicycler-Ausgabe gespeichert ist, indem Sie den Befehl cd verwenden: cd Desktop/Trimmed_Reads/unicycler_output_directory
HINWEIS: Leerzeichen sind im Pfad zu dem Ort, an dem sich die Baugruppe befindet, nicht zulässig, d. h. keine Verzeichnisse, die zur Unicycler-Ausgabe führen, dürfen Leerzeichen in ihrem Namen haben. Alternativ können Sie die Datei assembly.fasta zum einfachen Zugriff auf den Desktop kopieren. - Führen Sie QUAST aus, indem Sie den folgenden Befehl ausführen: quast assembly.fasta -o quast_output_directory
- Überprüfen Sie die von QUAST generierten Berichte im Ausgabeverzeichnis quast_output_directory.
- Navigieren Sie im Terminal zu dem Ordner, in dem die Unicycler-Ausgabe gespeichert ist, indem Sie den Befehl cd verwenden: cd Desktop/Trimmed_Reads/unicycler_output_directory
10. Genom-Annotation
HINWEIS: Die folgende Anmerkungspipeline verwendet Prokka34, ein Befehlszeilentool, das vor der Verwendung installiert werden muss. Alternativ können Sie Prokka über die automatisierte GUI K-Base (Table of Materials) verwenden oder Genome über den Webserver RAST35 kommentieren. Wenn Genome in NCBI abgelegt werden, werden sie automatisch mit der Prokaryotic Genome Annotation Pipeline (PGAP)36annotiert.
- Navigieren Sie innerhalb des Terminals mit dem Befehl cd zu dem Ordner, in dem die Unicycler-Ausgabe gespeichert ist (siehe Schritt 9.2.1). Führen Sie dann Prokka aus, indem Sie den folgenden Befehl ausführen: prokka --Präfix sample_ID --outdir prokka_output_directory assembly.fasta
HINWEIS: --prefix benennt alle Ausgabedateien basierend auf dem angegebenen sample_ID. --outdir erstellt ein Ausgabeverzeichnis mit dem angegebenen Namen, in dem alle Prokka-Ausgabedateien gespeichert werden; Erstellen Sie vorher kein Ausgabeverzeichnis für Prokka. - Überprüfen Sie die Anmerkungen, indem Sie die .tsv-Tabelle öffnen und/oder die generierte .gff-Datei in eine Sequenzanalysesoftware hochladen, um die Anmerkungen zu visualisieren und zu analysieren (Abbildung 6).
- Spezifische Arten von Anmerkungen können in Abhängigkeit von genetischen Faktoren von Interesse generiert werden. Es wird empfohlen, mit den benutzerfreundlichen Tools auf dem Webserver des Zentrums für Genomische Epidemiologie (www.genomicepidemiology.org/) für die vorläufige Analyse37,38,39,40,41zubeginnen. Zusätzliche Werkzeuge zum Nachweis von CRISPR-cas-Systemen und Prophagen sind verfügbar (Abbildung 3)42,43.
11. Empfohlene Vorgehensweisen für die Datendemokratisierung
- Wenn möglich, hinterlegen Sie alle gelesenen Rohdaten sowie die zusammengestellten Genome in einem öffentlichen Repository wie ncbi Sequence Read Archive (SRA) und Genbank. Genome werden während des NCBI-Ablagerungsprozesses automatisch über die PGAP-Pipeline annotiert.
Ergebnisse
Dieses Protokoll wurde für die Kultivierung und Sequenzierung von Harnbakterien optimiert, die zu den in Abbildung 1aufgeführten Gattungen gehören. Nicht alle Harnbakterien sind mit dieser Methode kultivierbar. Kulturmedien und -bedingungen sind durch die Gattung in Abbildung 1spezifiziert. Exemplarische Gelelektrophorese-Bewertungen der gDNA-Integrität sind in Abbildung 2dargestellt. Ein Überblick über die Bioinformatik-Pipeline zur Sequenzierung von Leseverarbeitung, Genomassemblierung und Annotation ist in Abbildung 3 beschrieben. In Abbildung 4 wird ein Leitfaden für die rechnerische Verzeichnisstruktur bereitgestellt, um sowohl das Protokollverständnis zu vereinfachen als auch einen Rahmen für eine erfolgreiche Organisation bereitzustellen. Weiterhin sind repräsentative vollständige Genome von zwei Klebsiella spp., K. pneumoniae und K. oxytoca,enthalten, die durch dieses Protokoll erzeugt wurden. Eine Darstellung dieser Anordnungen ist in Abbildung 5 dargestellt und enthält auch ein zusätzliches unvollständiges Beispiel für das Genom von K. pneumoniae. Eine detaillierte Übersicht über jedes vollständig annotierte vollständige Genom ist in Abbildung 6 dargestellt. Schließlich wird in Tabelle 1 eine Zusammenfassung der Sequenzierungsstatistik bereitgestellt, um ein breites Verständnis der rohen und beschnittenen Daten zu bieten, die für die Erzeugung hochwertiger geschlossener Genomassemblys ausreichen. Zusätzlich vervollständigen Schlüsselparameter der beiden repräsentativen Klebsiella spp. Genome sind aufgelistet. Genome und Rohdaten wurden im Rahmen des BioProjekts PRJNA683049 in Genbank hinterlegt.

Abbildung 1: Modifizierte verstärkte Urinkultur verschiedener Harngattungen. Diagramm für die Agar- und Flüssigbrühe, die zur Kultivierung verschiedener Harngattungen verwendet werden kann. Es wird empfohlen, alle Kultivierungen bei 35 °C gemäß Unterabschnitt 1.1 durchzuführen. Kreise repräsentieren Medien, die für die Kultivierung einer bestimmten Gattung geeignet sind, Farben wurden willkürlich ausgewählt, um einen Medientyp von einem anderen zu unterscheiden. CDC-AN BAP (rot), CDC Anaerobe Schafblut-Agar; 5% Schaf-BAP (orange), Schafblut-Agar; BHI (grün), Gehirnherzinfusion; TSB (gelb), Tryptische Sojabrühe; CHROMagar Orientierung (blau). aGardnerella vaginalis sollte auf HBT Bilayer G. vaginalis Selektives Agar in mikroaerophiler Atmosphäre und unter besonderen Anforderungen an die Brühekultur kultiviert werden44. bLactobacillus iners sollten auf 5% Kaninchen-BAP-Platten und NYCIII-Brühe in mikroaerophiler Atmosphäre kultiviert werden. cLactobacillus spp. kann unter mikroaerophilen Bedingungen auf MRS kultiviert werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Genomische DNA-Extraktion Agarose Gel Bilder. Repräsentative Gelbilder, die die Ergebnisse der gDNA-Extraktion darstellen. (A) Bahn 1: 1 kb Leiter, Spur 2: intakte gDNA für erfolgreiche Extraktion, Spur 3: Verschmieren, die auf fragmentierte gDNA hinweist. (B) Bahn 1: 1 kb Leiter, Bahnen 2 & 3: rRNA-Kontamination gekennzeichnet durch zwei Bänder zwischen 1,5 kb und 3 kb. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

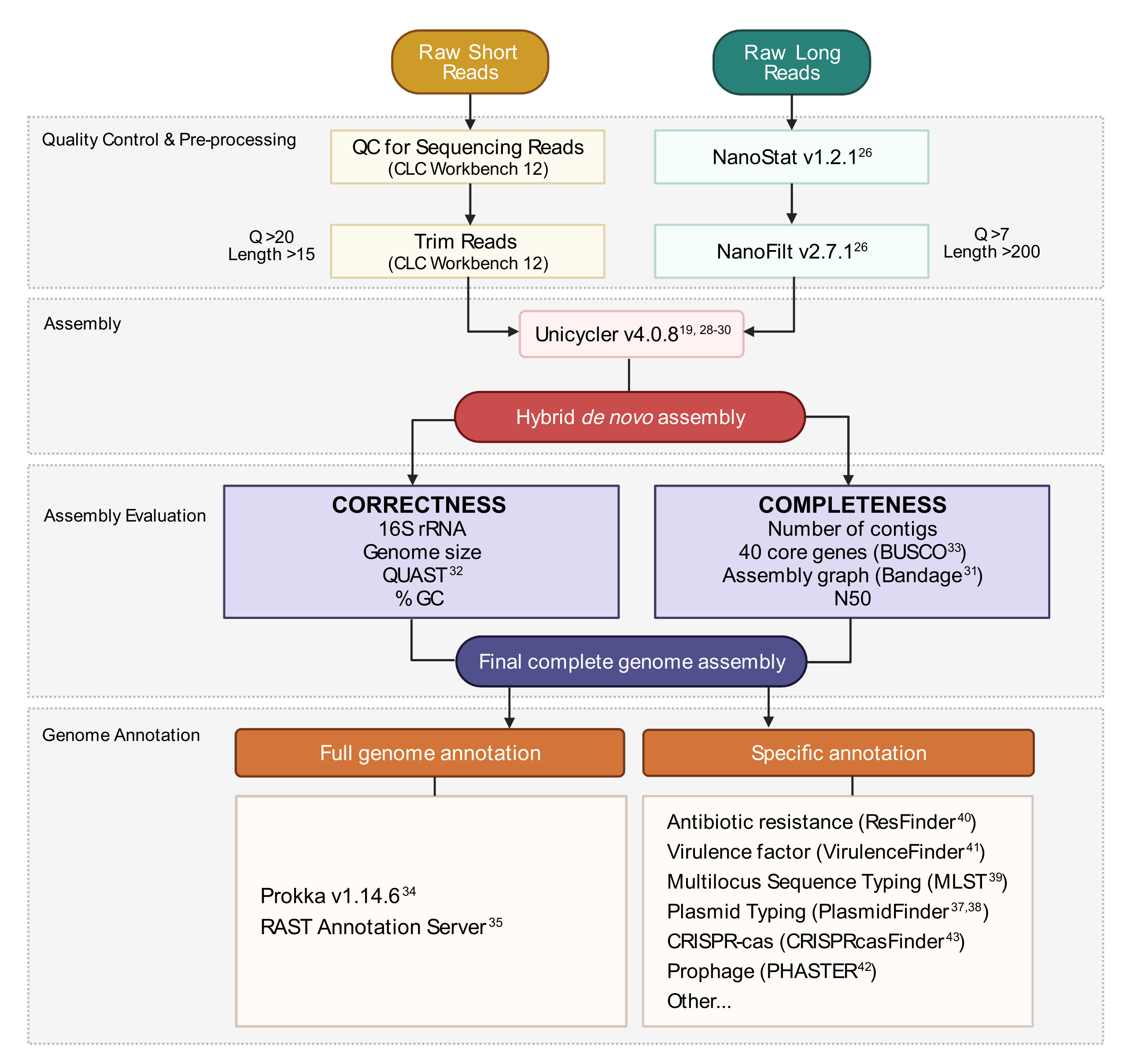
Abbildung 3: Workflow für die Hybridgenomassemblierung. Schematische Darstellung der Schritte von der Lesequalitätskontrolle über die Vorverarbeitung bis hin zur Montageanmerkung. Durch das Zuschneiden von Lesevorgängen werden mehrdeutige Lesevorgänge mit geringer Qualität entfernt. Q-Score- und Längenparameter werden angezeigt und stellen die Lesevorgänge dar, die beibehalten werden. Die Assemblierung verwendet sowohl kurze als auch lange Lesevorgänge, um eine hybride De-novo-Genomanordnung zu erzeugen. Die Montagequalität wird anhand der Vollständigkeit und Korrektheit mit vorgegebenen Werkzeugen und Parametern bewertet. Die endgültige Genomanordnung wird für alle Gene und spezifischen Orte von Interesse annotiert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4:Strukturhandbuch für Bioinformatik-Verzeichnisse. Ein Schema der empfohlenen Verzeichnis- und Dateiorganisation für die Verarbeitung von kurzen und langen Lesevorgängen, hybrider Assemblierung und Genomannotation und QC. Wichtige Befehlszeilen-Datenverarbeitungsschritte werden neben den entsprechenden Dateien und Verzeichnissen hervorgehoben. Auslösen von Befehlen und Flags (fett), Eingabedateien (blau), Ausgabedateien oder -verzeichnissen (rot), Benutzereingaben wie Dateibenennungskonvention (Magenta). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

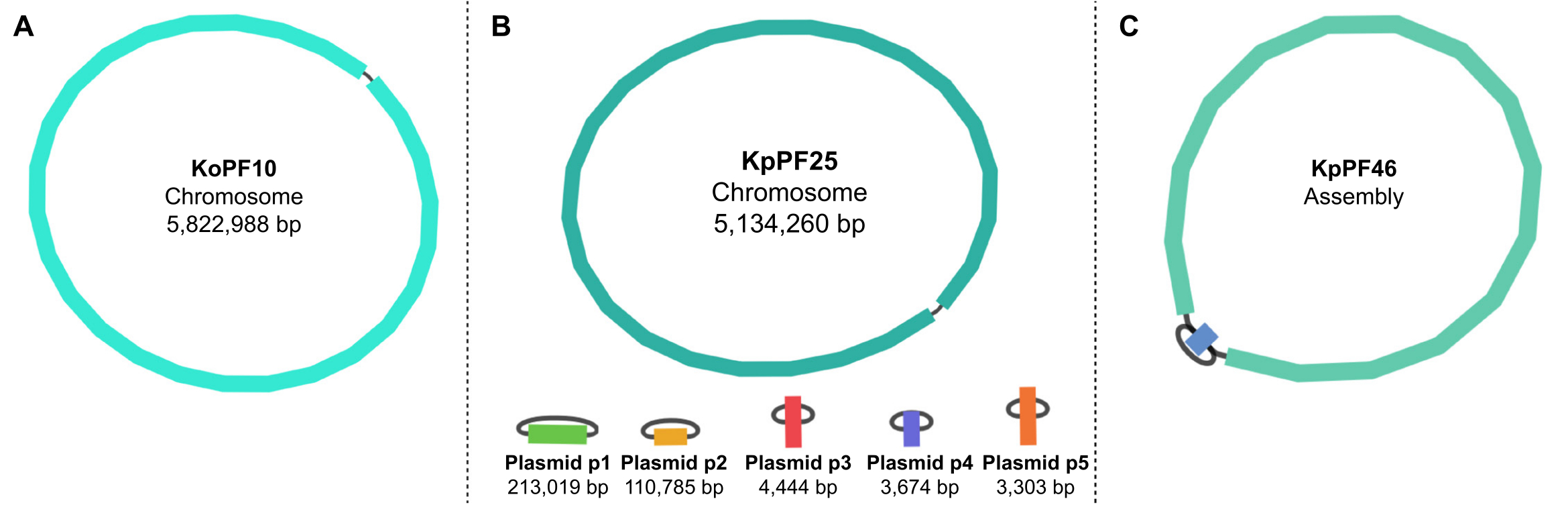
Abbildung 5: Genom-Assemblierungsgraphen nach Bandage. Repräsentative vollständige Genom-Assemblierungsgraphen von (A) Klebsiella oxytoca KoPF10 und (B) Klebsiella pneumoniae KpPF25 und unvollständige Genomassemblierung von (C) Klebsiella pneumoniae KpPF46. Das vollständige Genom von KoPF10 zeigt ein einziges geschlossenes Chromosom und das vollständige Genom von KpPF25 besteht aus einem geschlossenen Chromosom und fünf geschlossenen Plasmiden. Das unvollständige Chromosom von KpPF46 besteht aus zwei miteinander verbundenen Contigs. Unicycler hybrid de novo assembly erzeugt einen Montagegraphen, der durch Bandage visualisiert wird. Der Montagegraph liefert ein vereinfachtes Schema des Genoms, das geschlossene Chromosomen oder Plasmide durch einen Linker anzeigt, der zwei Enden eines einzelnen Kontinents verbindet. Das Vorhandensein von mehr als einem miteinander verbundenen Contig weist auf eine unvollständige Montage hin. Contig Größe und Tiefe können auch in Bandage vermerkt werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

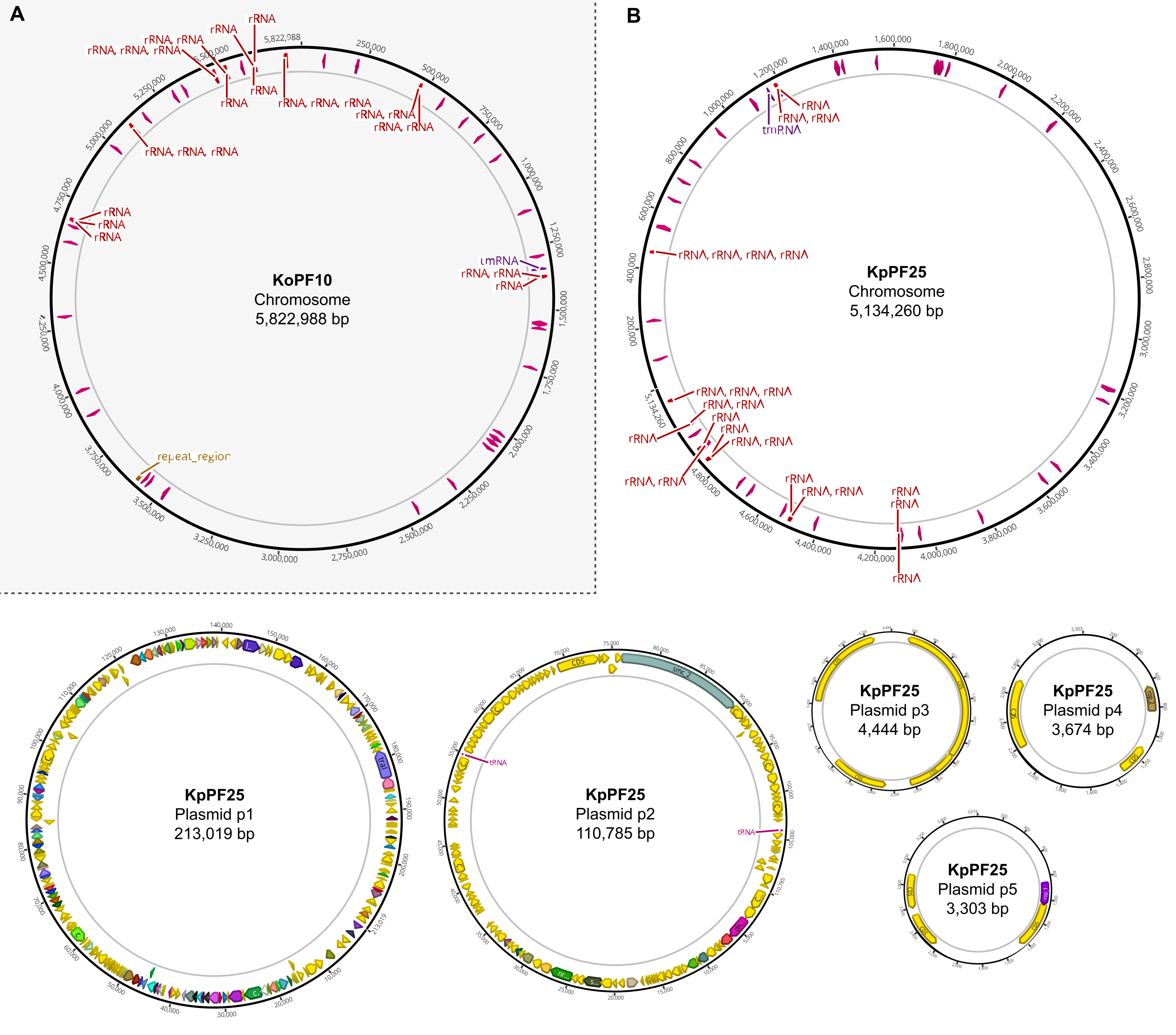
Abbildung 6: Vollständige Genomkarten von annotierten Hybridbaugruppen. Von Geneious Prime erstellte Montagekarten für das vollständige Genom von (A) K. oxytoca KoPF10 und (B) K. pneumoniae KpPF25, die annotierte Gene zeigen, die durch farbige Pfeile entlang der Plasmidrückgrats gekennzeichnet sind. Chromosomen zeigen der Einfachheit halber nur rRNA- und tRNA-Gene. Genomannotationen wurden mit Prokka durchgeführt, wie in Abschnitt 10 dieses Protokolls angegeben. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Tabelle 1: Repräsentativer Klebsiella spp.. vollständige Montageeigenschaften. Montageparameter des K. oxytoca-Stammes KoPF10 und des K. pneumoniae-Stammes KpPF25. Zugangsnummern für die hinterlegten Daten zu NCBI werden angegeben. Für beide Sequenzierungstechnologien wird die Anzahl der Lesevorgänge vor und nach dem Trimmen angegeben. N50 wird nur für lange Lesevorgänge bereitgestellt, da kurze Lesevorgänge eine kontrollierte Länge haben. Plasmid Replicon mit PlasmidFinder v2.1 Enteroebacteriaceae Datenbank mit Parametern auf 80% Identität und 60% Länge vorhergesagt. ein MLST, Multilocus Sequence Type. b CDS, Codierung von Sequenzen. c Plasmid-Replicon wurde mit der PlasmidFinder v2.1 Enterobacteriaceae-Datenbank mit Parametern von 80% Identität und 60% Länge vorhergesagt. d Oxford Nanopore Technologies (ONT) hinterlegte Lesedaten. e Illumina hinterlegte gelesene Daten. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Diskussion
Das hier beschriebene umfassende Hybridgenom-Assemblierungsprotokoll bietet einen optimierten Ansatz für die erfolgreiche Kultivierung verschiedener Harnmikrobiota und Uropathogene und die vollständige Assemblierung ihrer Genome. Erfolgreiche WGS von Bakteriengenomen beginnt mit der Isolierung verschiedener und manchmal anspruchsvoller Mikroben, um ihre genomische DNA zu extrahieren. Bis heute fehlt den bestehenden Urinkulturprotokollen entweder die notwendige Empfindlichkeit, um viele Harnspezies zu erkennen, oder sie beinhalten langwierige und umfangreiche Ansätze, die längere Zeit und Ressourcen erfordern11. Der beschriebene Modified Enhanced Urine Culture-Ansatz bietet ein vereinfachtes und dennoch umfassendes Protokoll für die erfolgreiche Isolierung von Bakterien, die zu 17 häufigen Harngattungen gehören, einschließlich potenziell pathogener oder nützlicher kommensaler Spezies und sowohl fakultativer als auch obligater aerober oder anaerober Bakterien. Dies wiederum liefert das notwendige Ausgangsmaterial für eine genaue Sequenzierung und Assemblierung bakterieller Genome und für kritische phänotypische Experimente, die zum Verständnis von Harngesundheit und -krankheit beitragen. Darüber hinaus bietet dieser modifizierte Kulturansatz eine genauere klinische Diagnose von lebensfähigen Mikroorganismen, die in Urinproben vorkommen, und ermöglicht deren Biobanking für zukünftige genomische Studien. Dieses Protokoll ist jedoch nicht ohne Einschränkungen. Es kann je nach Organismus lange Inkubationszeiten sowie die Verwendung von Ressourcen wie einer Hypoxiekammer oder kontrollierten Inkubatoren erfordern, die möglicherweise nicht ohne weiteres verfügbar sind. Die Verwendung von anaeroben GasPaks bietet eine alternative Lösung, aber diese sind kostspielig und erzeugen nicht immer eine nachhaltige und kontrollierte Umgebung. Schließlich können Kulturverzerrungen sowie die Probenvielfalt es bestimmten Organismen und Uropathogenen ermöglichen, anspruchsvolle Bakterien zu übertreffen. Trotz dieser Einschränkungen wird durch diesen Ansatz eine Kultur verschiedener Harnbakterien ermöglicht.
Die genomische Sequenzierung hat mit der Weiterentwicklung der Next Generation Sequencing-Technologien an Popularität gewonnen, die sowohl die Ausbeute als auch die Genauigkeit der Sequenzierungsdaten enorm erhöht haben14,15. In Verbindung mit der Entwicklung von Algorithmen zur Datenverarbeitung und De-novo-Assemblierung stehen Anfängern und Fachwissenschaftlern komplette Genomsequenzen gleichermaßen zur Verfügung15,45. Das Wissen über die gesamte Genomorganisation, das durch vollständige Genome bereitgestellt wird, bietet wichtige evolutionäre und biologische Erkenntnisse, einschließlich Genduplikation, Genverlust und horizontaler Gentransfer14. Darüber hinaus sind Gene, die für antimikrobielle Resistenz und Virulenz wichtig sind, oft auf mobilen Elementen lokalisiert, die typischerweise nicht in Entwurfsgenomassemgruppen aufgelöst werden15,16.
Das hierin enthaltene Protokoll folgt einem hybriden Ansatz für die Kombination von Sequenzierungsdaten von Short-Read- und Long-Read-Plattformen, um vollständige Genom-Assemblierungen zu erzeugen. Während es sich auf bakterielle Genome im Urin konzentriert, kann dieses Verfahren an verschiedene Bakterien aus verschiedenen Isolationsquellen angepasst werden. Kritische Schritte in diesem Ansatz umfassen die Anwendung einer adäquaten sterilen Technik und die Verwendung geeigneter Medien- und Kulturbedingungen für die Isolierung reiner Harnbakterien. Darüber hinaus ist die Extraktion intakter, ertragreicher gDNA unerlässlich, um Sequenzierungsdaten frei von kontaminierenden Lesevorgängen zu generieren, die den Montageerfolg beeinträchtigen können. Nachfolgende Bibliotheksvorbereitungsprotokolle sind entscheidend für die Generierung von Qualitätslesevorgängen von ausreichender Länge und Tiefe. Daher ist es von äußerster Wichtigkeit, gDNA bei der Bibliotheksvorbereitung insbesondere für die Long-Read-Sequenzierung sorgfältig zu behandeln, da der größte Vorteil dieser Technologie die Generierung von langen Lesevorgängen ohne theoretische obere Längenbegrenzung ist. Ebenfalls beschrieben sind Abschnitte für die geeignete Qualitätskontrolle (QC) der Sequenzierung von Lesevorgängen, die verrauschte Daten eliminiert und das Montageergebnis verbessert.
Trotz erfolgreicher DNA-Isolierung, Bibliotheksvorbereitung und Sequenzierung kann die Art der genomischen Architektur einiger Arten immer noch ein Hindernis für die Erzeugung einer geschlossenen Genomanordnung darstellen45,46. Sich wiederholende Sequenzen erschweren oft die Montageberechnung, und trotz langer gelesener Daten können diese Bereiche mit geringer Zuverlässigkeit oder gar nicht aufgelöst werden. Lange Lesevorgänge müssen also im Durchschnitt länger sein als die größte Wiederholungsregion im Genom oder die Abdeckung muss hoch sein (>100x)19. Einige Genome können unvollständig bleiben und manuelle Ansätze zur Vervollständigung erfordern. Dennoch bestehen hybrid zusammengesetzte unvollständige Genome typischerweise aus weniger Contigs als kurz gelesene Entwurfsgenomen. Das Anpassen der Standardparameter des Baugruppenalgorithmus oder das Befolgen strengerer Cutoffs für die Lese-QC kann hilfreich sein. Alternativ besteht ein vorgeschlagener Ansatz darin, lange Lesevorgänge auf die unvollständigen Regionen auf der Suche nach Beweisen für den wahrscheinlichsten Montagepfad abzubilden und dann den Pfad unter Verwendung von PCR und Sanger-Sequenzierung der amplifizierten Region zu bestätigen. Mapping-Lesevorgänge mit Minimap2 wird vorgeschlagen und Bandage bietet ein nützliches Werkzeug für die Visualisierung von zugeordneten Lesevorgängen entlang zusammengesetzter Contigs, die Beweise für die Contig-Verknüpfungliefern 47.
Eine zusätzliche Herausforderung bei der Generierung vollständiger Genome liegt in der Vertrautheit und dem Komfort mit Befehlszeilenwerkzeugen. Viele bioinformatische Werkzeuge werden entwickelt, um jedem Benutzer Rechenmöglichkeiten zu bieten. Ihre Verwendung hängt jedoch von einem Verständnis der Grundlagen von UNIX und der Programmierung ab. Dieses Protokoll zielt darauf ab, ausreichend detaillierte Anweisungen bereitzustellen, damit Personen ohne vorherige Befehlszeilenerfahrung geschlossene Genomassemblys generieren und kommentieren können.
Offenlegungen
Die Autoren haben nichts preiszugeben.
Danksagungen
Wir danken Dr. Moutusee Jubaida Islam und Dr. Luke Joyce für ihre Beiträge zu diesem Protokoll. Wir möchten auch die University of Texas am Dallas Genome Center für ihr Feedback und ihre Unterstützung danken. Diese Arbeit wurde von der Welch Foundation, Vergabenummer AT-2030-20200401 an N.J.D., von den National Institutes of Health, Preisnummer R01AI116610 an K.P., und vom Felecia and John Cain Chair in Women's Health, gehalten von P.E.Z.
Materialien
| Name | Company | Catalog Number | Comments |
| Equipment: | |||
| Bioanalyzer 2100 | Agilent | G29398A | Optional but recommended |
| Centrifuge | Eppendorf | -- | Any centrifuge for spinning conicals and microcentrifuge tubes (e.g. Models 5810R/5424R) |
| Electrophoresis | BioRad Laboratories | 1645070 | |
| Gel Imaging System | BioRad Laboratories | ChemiDoc models | |
| Incubator | ThermoFisher Scientific | -- | Any CO2 Incubator (e.g. Thermo Forma model 3110) |
| Magnetic Rack | New England BioLabs | S15095 | 12-tube rack |
| MinION | Oxford Nanopore Technologies | -- | |
| Nanodrop | ThermoFisher Scientific | ND-ONE-W | |
| NextSeq 500 | Illumina | SY-415-1002 | Other Illumina models are acceptable |
| Plate Reader | BioTek | -- | Synergy H1 |
| Qubit fluorometer | ThermoFisher Scientific | Q33238 | |
| Rotator | Benchmark Scientific | H2024 | |
| Thermocycler | ThermoFisher Scientific | -- | Any thermocycler for PCR reactions (e.g. ProFlex PCR system) |
| Materials: | |||
| 10X Phosphate Buffered Saline (PBS) | Fisher Scientific | BP3991 | |
| 10X TBE buffer | -- | -- | 1M Tris,1M Boric Acid,0.2M EDTA (pH 8.0) |
| 1429R primer | Sigma Aldrich (Custom oligos) | -- | GGTTACCTTGTTACGACTT |
| 1kb Ladder | VWR | 101228-494 | |
| 1M Tris-Cl (pH 7.5) | ThermoFisher Scientific | 15567027 | |
| 6x Loading dye | Fisher Scientific | NC0783588 | |
| 8F primer | Sigma Aldrich (Custom oligos) | -- | AGAGTTTGATCCTGGCTCAG |
| Agar | Fisher Scientific | BP1423-2 | |
| Agarose | BioRad Laboratories | 63001 | |
| AMPure XP Beads | Beckman Coulter | A63880 | |
| Anaerobe Pouch System - GasPak EZ | BD Diagnostic Systems | B260683 | |
| Boric Acid | Fisher Scientific | A73-500 | |
| Brain Heart Infusion Broth | BD Diagnostic Systems | 212304 | |
| CDC Anaerobe 5% Sheep Blood Agar | BD Diagnostic Systems | L007357 | |
| CHROMagar Orientation | BD Diagnostic Systems | PA-257481.04 | |
| DNeasy Blood & Tissue | QIAGEN | 69504 | |
| DreamTaq Master Mix | ThermoFisher Scientific | K1081 | |
| Dry Anaerobic Indicator Strips | BD Diagnostic Systems | 271051 | |
| EDTA | Fisher Scientific | S311-500 | |
| Ethanol 200 Proof | Sigma Aldrich | E7023 | For molecular biology |
| Ethidium Bromide | ThermoFisher Scientific | BP130210 | |
| Flow cell priming kit | Oxford Nanopore Technologies | EXP-FLP002 | |
| Flow cell wash kit | Oxford Nanopore Technologies | EXP-WSH003 | |
| Gel Extraction Miniprep Kit | BioBasic | BS654 | |
| Ligation sequencing kit | Oxford Nanopore Technologies | SQK-LSK109 | |
| Lysozyme | Research Products International Corp | L381005.05 | |
| Mutanolysin | Sigma Aldrich | M9901-5KU | |
| Native barcoding expansion 1-12 | Oxford Nanopore Technologies | EXP-NBD104 | |
| NEB Blunt/TA Ligase Master Mix | New England BioLabs | M0367L | |
| NEBNext FFPE DNA Repair Mix | New England BioLabs | M6630L | |
| NEBNext quick ligation buffer | New England BioLabs | B6058S | |
| NEBNext Ultra II End repair / dA-tailing module | New England BioLabs | E7546L | |
| Nextera DNA CD Indexes | Illumina | 20018708 | |
| Nextera DNA Flex Library Prep - (M) Tagmentation | Illumina | 20018705 | |
| Nuclease-free water | Sigma Aldrich | W4502 | |
| Qubit 1X dsDNA HS Assay Kit | ThermoFisher Scientific | Q33230 | |
| Qubit Assay Tubes | ThermoFisher Scientific | Q32856 | |
| Quick T4 DNA Ligase | New England BioLabs | E6056L | |
| R9 Flow cell | Oxford Nanopore Technologies | FLO-MIN106D | |
| RNase A | ThermoFisher Scientific | EN0531 | |
| Sheep Blood | Hemostat Laboratories | DS13250 | |
| TE buffer | -- | -- | 10mM Tris, 1mM EDTA (pH 8.0) |
| Triton X-100 | Sigma Aldrich | T8787 | |
| Tryptic Soy Broth | BD Diagnostic Systems | 211825 | |
| Software & Bioinformatic Tools: | |||
| Bandage | -- | -- | https://rrwick.github.io/Bandage/ |
| Center for Genomic Epidemiology | -- | -- | http://www.genomicepidemiology.org/ |
| CLC Genomics Workbench 12 | QIAGEN | -- | |
| CRISPRcasFinder | -- | -- | https://crisprcas.i2bc.paris-saclay.fr/ |
| FastQC | -- | -- | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Geneious Prime | Geneious | -- | |
| gVolante (BUSCO) | -- | -- | https://gvolante.riken.jp/ |
| Kbase Prokka Wrapper | -- | -- | https://kbase.us/applist/apps/ProkkaAnnotation/annotate_contigs/release |
| Minimap2 | -- | -- | https://github.com/lh3/minimap2 |
| MinKNOW | Oxford Nanopore Technologies | -- | |
| NanoFilt | -- | -- | https://github.com/wdecoster/nanofilt |
| NanoStat | -- | -- | https://github.com/wdecoster/nanostat |
| PHASTER | -- | -- | https://phaster.ca/ |
| Prokka | -- | -- | https://github.com/tseemann/prokka |
| QUAST | -- | -- | http://quast.sourceforge.net/quast |
| Trim Galore | -- | -- | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Trimmomatic | -- | -- | http://www.usadellab.org/cms/?page=trimmomatic |
| Unicycler | -- | -- | https://github.com/rrwick/Unicycler#necessary-read-length |
Referenzen
- Brubaker, L., Wolfe, A. The urinary microbiota: a paradigm shift for bladder disorders. Current Opinion in Obstetrics & Gynecology. 28 (5), 407-412 (2016).
- Neugent, M. L., Hulyalkar, N. V., Nguyen, V. H., Zimmern, P. E., De Nisco, N. J. Advances in understanding the human urinary microbiome and its potential role in urinary tract infection. mBio. 11 (2), (2020).
- Klein, R. D., Hultgren, S. J. Urinary tract infections: microbial pathogenesis, host-pathogen interactions and new treatment strategies. Nature Reviews. Microbiology. 18 (4), 211-226 (2020).
- Horsley, H., et al. Enterococcus faecalis subverts and invades the host urothelium in patients with chronic urinary tract infection. PLoS One. 8 (12), 83637 (2013).
- Reitzer, L., Zimmern, P. Rapid growth and metabolism of uropathogenic Escherichia coli in relation to urine composition. Clinical Microbiology Reviews. 33 (1), 00101-00119 (2019).
- Snyder, J. A., et al. Transcriptome of uropathogenic Escherichia coli during urinary tract infection. Infection and Immunity. 72 (11), 6373-6381 (2004).
- Ipe, D. S., Horton, E., Ulett, G. C. The basics of bacteriuria: Strategies of microbes for persistence in urine. Frontiers in Cellular and Infection Microbiology. 6, 14 (2016).
- Babikir, I. H., et al. The impact of cathelicidin, the human antimicrobial peptide LL-37 in urinary tract infections. BMC Infectious Diseases. 18 (1), 17 (2018).
- Jancel, T., Dudas, V. Management of uncomplicated urinary tract infections. The Western Journal of Medicine. 176 (1), 51-55 (2002).
- Ventola, C. L. The antibiotic resistance crisis: part 1: causes and threats. P & T. 40 (4), 277-283 (2015).
- Price, T. K., et al. The clinical urine culture: Enhanced techniques improve detection of clinically relevant microorganisms. Journal of Clinical Microbiology. 54 (5), 1216-1222 (2016).
- Kass, E. H. Asymptomatic infections of the urinary tract. Transactions of the Association of American Physicians. 69, 56-64 (1956).
- Garcia, L. S. . Clinical microbiology procedures handbook. 3rd edn. , (2010).
- Fraser, C. M., Eisen, J. A., Nelson, K. E., Paulsen, I. T., Salzberg, S. L. The value of complete microbial genome sequencing (you get what you pay for). Journal of Bacteriology. 184 (23), 6403-6405 (2002).
- Chen, Z., Erickson, D. L., Meng, J. Benchmarking hybrid assembly approaches for genomic analyses of bacterial pathogens using Illumina and Oxford Nanopore sequencing. BMC Genomics. 21 (1), 631 (2020).
- Greig, D. R., Dallman, T. J., Hopkins, K. L., Jenkins, C. MinION nanopore sequencing identifies the position and structure of bacterial antibiotic resistance determinants in a multidrug-resistant strain of enteroaggregative Escherichia coli. Microbial Genomics. 4 (10), 000213 (2018).
- Carraro, D. M., et al. PCR-assisted contig extension: stepwise strategy for bacterial genome closure. Biotechniques. 34 (3), 626-628 (2003).
- Tettelin, H., Radune, D., Kasif, S., Khouri, H., Salzberg, S. L. Optimized multiplex PCR: efficiently closing a whole-genome shotgun sequencing project. Genomics. 62 (3), 500-507 (1999).
- Wick, R. R., Judd, L. M., Gorrie, C. L., Holt, K. E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Computational Biology. 13 (6), 1005595 (2017).
- Singhal, N., Kumar, M., Kanaujia, P. K., Virdi, J. S. MALDI-TOF mass spectrometry: an emerging technology for microbial identification and diagnosis. Frontiers in Microbiology. 6, 791 (2015).
- Turner, S., Pryer, K. M., Miao, V. P., Palmer, J. D. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. The Journal of Eukaryotic Microbiology. 46 (4), 327-338 (1999).
- Weisburg, W. G., Barns, S. M., Pelletier, D. A., Lane, D. J. 16S ribosomal DNA amplification for phylogenetic study. Journal of Bacteriology. 173 (2), 697-703 (1991).
- Janda, J. M., Abbott, S. L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. Journal of Clinical Microbiology. 45 (9), 2761-2764 (2007).
- Stevenson, K., McVey, A. F., Clark, I. B. N., Swain, P. S., Pilizota, T. General calibration of microbial growth in microplate readers. Science Reports. 6, 38828 (2016).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- De Coster, W., D'Hert, S., Schultz, D. T., Cruts, M., Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 34 (15), 2666-2669 (2018).
- Wilson, G., et al. The UNIX Shell. Zenodo. , (2019).
- Bankevich, A., et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology. 19 (5), 455-477 (2012).
- Vaser, R., Sovic, I., Nagarajan, N., Sikic, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Research. 27 (5), 737-746 (2017).
- Walker, B. J., et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 9 (11), 112963 (2014).
- Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 31 (20), 3350-3352 (2015).
- Gurevich, A., Saveliev, V., Vyahhi, N., Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 29 (8), 1072-1075 (2013).
- Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31 (19), 3210-3212 (2015).
- Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 30 (14), 2068-2069 (2014).
- Aziz, R. K., et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 9, 75 (2008).
- Tatusova, T., et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Research. 44 (14), 6614-6624 (2016).
- Carattoli, A., Hasman, H. PlasmidFinder and In Silico pMLST: Identification and Typing of Plasmid Replicons in Whole-Genome Sequencing (WGS). Methods in Molecular Biology. 2075, 285-294 (2020).
- Carattoli, A., et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrobial Agents and Chemotherapy. 58 (7), 3895-3903 (2014).
- Larsen, M. V., et al. Multilocus sequence typing of total-genome-sequenced bacteria. Journal of Clinical Microbiology. 50 (4), 1355-1361 (2012).
- Bortolaia, V., et al. ResFinder 4.0 for predictions of phenotypes from genotypes. The Journal of Antimicrobial Chemotherapy. 75 (12), 3491-3500 (2020).
- Joensen, K. G., et al. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. Journal of Clinical Microbiology. 52 (5), 1501-1510 (2014).
- Arndt, D., et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Research. 44 (1), 16-21 (2016).
- Couvin, D., et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Research. 46 (1), 246-251 (2018).
- Totten, P. A., Amsel, R., Hale, J., Piot, P., Holmes, K. K. Selective differential human blood bilayer media for isolation of Gardnerella (Haemophilus) vaginalis. Journal of Clinical Microbiology. 15 (1), 141-147 (1982).
- Nagarajan, N., Pop, M. Sequence assembly demystified. Nat Reviews. Genetics. 14 (3), 157-167 (2013).
- Phillippy, A. M., Schatz, M. C., Pop, M. Genome assembly forensics: finding the elusive mis-assembly. Genome Biology. 9 (3), 55 (2008).
- . Unicycler Wiki Available from: https://github.com/rrwick/Unicycler/wiki (2017)
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten