Multiplex-Immunfluoreszenz kombiniert mit räumlicher Bildanalyse zur klinischen und biologischen Beurteilung der Tumormikroumgebung
In diesem Artikel
Zusammenfassung
In diesem Artikel wird ein Protokoll zur manuellen Tyramid-Signalverstärkung (TSA) Multiplex-Immunfluoreszenz (mIF) in Kombination mit Bildanalyse und räumlicher Analyse beschrieben. Dieses Protokoll kann mit formalinfixierten Paraffin-eingebetteten (FFPE) Schnitten für die Färbung von zwei bis sechs Antigenen pro Objektträger verwendet werden, je nachdem, welcher Objektträgerscanner im Labor verfügbar ist.
Zusammenfassung
Die Tumormikroumgebung (TME) setzt sich aus einer Vielzahl verschiedener Zelltypen zusammen, wie z.B. zytotoxischen Immunzellen und immunmodulatorischen Zellen. Abhängig von seiner Zusammensetzung und den Wechselwirkungen zwischen Krebszellen und peritumoralen Zellen kann der TME das Fortschreiten der Krebserkrankung beeinflussen. Die Charakterisierung von Tumoren und ihrer komplexen Mikroumgebung könnte das Verständnis von Krebserkrankungen verbessern und Wissenschaftlern und Klinikern helfen, neue Biomarker zu entdecken.
Wir haben kürzlich mehrere Multiplex-Immunfluoreszenz-Panels (mIF) auf Basis der Tyramid-Signalverstärkung (TSA) zur Charakterisierung des TME bei Darmkrebs, Kopf-Hals-Plattenepithelkarzinom, Melanom und Lungenkrebs entwickelt. Sobald die Färbung und das Scannen der entsprechenden Platten abgeschlossen sind, werden die Proben mit einer Bildanalysesoftware analysiert. Die räumliche Position und die Färbung jeder Zelle werden dann aus dieser Quantifizierungssoftware nach R exportiert. Wir haben R-Skripte entwickelt, die es uns ermöglichen, nicht nur die Dichte jedes Zelltyps in mehreren Tumorkompartimenten (z.B. dem Zentrum des Tumors, dem Rand des Tumors und dem Stroma) zu analysieren, sondern auch abstandsbasierte Analysen zwischen verschiedenen Zelltypen durchzuführen.
Dieser spezielle Workflow fügt der klassischen Dichteanalyse, die bereits routinemäßig für mehrere Marker durchgeführt wird, eine räumliche Dimension hinzu. Die mIF-Analyse könnte es Wissenschaftlern ermöglichen, die komplexe Interaktion zwischen Krebszellen und TME besser zu verstehen und neue prädiktive Biomarker für das Ansprechen auf Behandlungen, wie z. B. Immun-Checkpoint-Inhibitoren und zielgerichtete Therapien, zu entdecken.
Einleitung
Mit der Entwicklung zielgerichteter Therapien und Immun-Checkpoint-Inhibitoren ist es von größter Bedeutung geworden, die Wechselwirkungen zwischen Krebszellen und ihrer Tumormikroumgebung besser zu charakterisieren, und dies ist derzeit ein wichtiges Feld der translationalen Forschung. Das TME besteht aus einer Vielzahl verschiedener Zelltypen, mit einem Gleichgewicht aus zytotoxischen Immunzellen, die auf die Krebszellen abzielen, und immunmodulatorischen Zellen, die das Tumorwachstum und die Invasivität begünstigen könnten 1,2,3,4. Die Charakterisierung dieser komplexen Umgebung könnte das Verständnis von Krebserkrankungen verbessern und Wissenschaftlern und Klinikern helfen, neue prädiktive und prognostische Biomarker zu entdecken, um Patienten für eine zukünftige Behandlung besser auswählen zu können 5,6. Galon und sein Team haben zum Beispiel den Immunoscore entwickelt, eine reproduzierbare Scoring-Methode, die als prädiktiver Biomarker verwendet werden kann. Der Immunoscore wird anhand der Dichte der CD3+ und CD8+ T-Zellen am invasiven Rand und im Zentrum des Tumorsberechnet 7,8.
In den letzten Jahrzehnten wurden kommerzielle Lösungen für mIF entwickelt, die jedoch oft teuer sind und für bestimmte Antigenpanels ausgelegt sind. Um den Bedarf an spezifischen Antigen-Panels in der akademischen und translationalen Forschung zu überwinden, haben wir eine kostengünstige Methode zur Durchführung von mIF an FFPE-Tumorschnitten entwickelt, die die Färbung von zwei bis sechs Antigenen ermöglicht, die den Zellkernen zur Gegenfärbung von menschlichen und Mausproben hinzugefügt werden.
Sobald die gesamten Gewebeschnitte gefärbt und mit einem Fluoreszenz-Objektträgerscanner gescannt wurden, können die Proben mit mehreren Bildanalyseprogrammen analysiert werden, die große pyramidale Datensätze unterstützen. Schließlich können die Rohdaten in einer Umgebung für statistische Berechnungen und Grafiken wie der R-Software (v.4.0.2) verwendet werden, um dichte- und räumliche Analysen durchzuführen.
In diesem Manuskript wird ein für die Fünf-Marker-Färbung optimiertes Protokoll sowie Tricks und Tipps zur Optimierung neuer Tafeln vorgestellt. Darüber hinaus werden detaillierte Schritte der Bildanalyse und die für die statistische und räumliche Analyse verwendeten R-Funktionen erläutert.
Protokoll
Alle im vorliegenden Protokoll verwendeten Proben stammen aus einer Studie, die von den lokalen Ethikkommissionen genehmigt und von der zuständigen Behörde genehmigt wurde. Alle Studienteilnehmer gaben eine schriftliche Einverständniserklärung ab. Die Studie ist bei ClinicalTrials.gov registriert (NCT03608046).
1. Multiplex-Immunfluoreszenz
- FFPE-Schneiden
- Fixieren Sie das Gewebe in 4%igem Paraformaldehyd und betten Sie das fixierte Gewebe in Paraffin ein.
- Schneiden Sie 5 μm große Schnitte aus und legen Sie sie auf selbstklebende Objektträger.
- Trocknen Sie die Objektträger über Nacht bei Raumtemperatur (RT).
- Deparaffinierung und Hemmung endogener Peroxidasen
- Entwachsen Sie das Gewebe, indem Sie die Objektträger in Toluol (3x für je 5 min) und Methanol (3x für je 5 min) unter einen Abzug tauchen.
- Hemmung der endogenen Peroxidasen, indem die Objektträger 20 min lang unter Abzug in 3%iges Wasserstoffperoxid getaucht werden, das in Methanol verdünnt ist.
- Spülen Sie die Objektträger in destilliertem (d)H2O(1x für 3 min).
- Multiplex-Immunfluoreszenzfärbung
- Tauchen Sie die Objektträger in ein 300-ml-Färbegefäß mit 10 mM Citrat- (pH 6) oder EDTA-Puffer (pH 9), ergänzt mit 0,1 % TritonX-100.
HINWEIS: Der verwendete Puffer (pH 6 oder pH 9) hängt vom gefärbten Antigen ab (siehe Tabelle 1). - Stellen Sie das Färbeglas bei geschlossenem Deckel für 3-5 Minuten bei maximaler Leistung (z. B. 900 W) in die Mikrowelle, bis der Puffer zu kochen beginnt.
Anmerkungen: Die optimale Zeit zum Kochen hängt von der Mikrowelle und dem Volumen des Puffers ab. Möglicherweise sind Anpassungen erforderlich, um das perfekte Timing zu finden. Für einige zerbrechliche Antigene oder zerbrechliche und weniger haftende Proben (z. B. Organoide und Sphäroide) kann das Mikrowellensieden zu hart sein. In diesem Fall kann stattdessen ein Schnellkochtopf verwendet werden. - Halten Sie den Puffer auf einer Temperatur nahe dem Siedeplatz, indem Sie das geschlossene Färbegefäß 15 Minuten lang bei niedriger Leistung (z. B. 90 W) in die Mikrowelle stellen.
- Führen Sie den letzten Schritt des Erhitzens durch, indem Sie die Mikrowelle 90 s lang auf maximale Leistung stellen.
- Nehmen Sie das Glas aus der Mikrowelle und lassen Sie den Puffer 15 Minuten bei RT abkühlen.
- Spülen Sie die Objektträger 3x für je 5 min in dH2O und 1x für 5 min in trisgepufferter Kochsalzlösung mit 0,1 % Tween 20 (TBS-T).
- Entfernen Sie das TBS-T, indem Sie die Objektträger auf einem Papiertuch abtupfen
- Legen Sie die Objektträger (flach) auf eine Färbekammer oder einen Objektträgerkasten (siehe Materialtabelle).
- Umkreisen Sie das Gewebe mit einem hydrophoben Stift.
- Blockieren Sie die unspezifischen Bindungsstellen, indem Sie das Gewebe 30 Minuten lang mit 5 % Rinderserumalbumin (BSA) bedecken, das in TBS-T gelöst ist.
- Entfernen Sie den blockierenden Puffer, indem Sie die Objektträger auf ein Papiertuch tupfen.
Anmerkungen: Spülen Sie die Objektträger nach dem Blockierungsschritt nicht aus. - Inkubieren Sie das Gewebe 60 Minuten lang mit dem primären Antikörper (siehe Tabelle 1), der in 1 % BSA TBS-T verdünnt ist, indem Sie das Gewebe mit etwa 300 μl der Lösung bedecken.
- Spülen Sie die Objektträger 3x für jeweils 3 min mit TBS-T aus.
- Inkubieren Sie das Gewebe 40 Minuten lang mit Poly-HRP-Sekundärantikörpern (siehe Tabelle 1), indem Sie das Gewebe mit etwa 300 μl der Lösung bedecken.
- Spülen Sie die Objektträger 3x für 3 min mit TBS-T aus.
- Inkubieren Sie das Gewebe 10 min lang mit einem Fluorochrom-Tyramid-Reagenz (siehe Tabelle 1), das 200-fach in Boratpuffer (0,1 M Borat, pH 7,8, 3 M NaCl) verdünnt und mit 0,003 %H2O2ergänzt wird, indem das Gewebe mit etwa 300 μl der Lösung bedeckt wird.
- Spülen Sie die Objektträger 3x für 3 min mit TBS-T aus.
- Wiederholen Sie die Schritte 1.3.1-1.3.16, bis die gesamte TSA-Färbung durchgeführt wurde.
- Das Gewebe wird über Nacht bei 4 °C inkubiert, wobei der letzte Primärantikörper (siehe Tabelle 1) in 1 % BSA TBS-T verdünnt wird.
Anmerkungen: Da die Inkubation über Nacht erfolgt, ist es wichtig, die Färbekammerschale oder die Objektträgerbox abzudecken und dH2O auf ein Papiertuch am Boden der Box (unter den Objektträgern) zu geben, um sicherzustellen, dass das Gewebe während der Inkubation nicht austrocknet. - Spülen Sie das Gewebe 3x für je 5 min mit TBS-T aus.
- Inkubieren Sie das Gewebe für 120 min mit dem Sekundärantikörper (direkt gekoppelt mit Fluorochrom), der 200-fach in 1% BSA TBS-T verdünnt ist.
- Spülen Sie das Gewebe 3x für je 5 min mit TBS-T aus.
- Färben Sie die Zellkerne, indem Sie das Gewebe 5 Minuten lang in Bisbenzimid (20 mM) inkubieren, das 1.000-fach in 10% BSA TBS-T verdünnt ist.
HINWEIS: Bisbenzimid kann durch DAPI ersetzt werden, letzteres ist jedoch giftiger und muss unter einem Abzug vorsichtig gehandhabt werden. - Spülen Sie das Tuch 3x für je 3 min in dH2O aus.
- Montieren Sie die Objektträger mit einem Fluoreszenz-Eindeckmedium und Borosilikat-Deckgläsern.
- Tauchen Sie die Objektträger in ein 300-ml-Färbegefäß mit 10 mM Citrat- (pH 6) oder EDTA-Puffer (pH 9), ergänzt mit 0,1 % TritonX-100.
2. Scannen von Objektträgern
- Digitalisieren Sie die Objektträger, indem Sie sie auf einem Fluoreszenz-Objektträgerscanner mit 20-facher Vergrößerung scannen (Details zum Objektträgerscanner finden Sie in der Materialtabelle).
HINWEIS: Ein repräsentativer Scan eines optimalen Multiplex ist in Abbildung 1 dargestellt.
3. Bildanalyse
- Importieren Sie die Scans in eine Bildanalysesoftware (Datei > Bild öffnen).
- Gehen Sie zur Registerkarte Klassifikatoren und wählen Sie das DenseNet AI V2-Plugin aus.
- Trainieren Sie das DenseNet AI V2-Plugin zur Erkennung von Kernen, indem Sie etwa 500 Kerne in einem Bild umgeben.
- Trainieren Sie die KI auf mehreren anderen Objektträgern aus derselben Charge und verschiedenen Chargen der mIF-Färbung, indem Sie mehrere Zellkerne (50) auf mehreren Objektträgern (ca. 10) umgeben.
HINWEIS: Eine detaillierte Anleitung zur Verwendung des AI-Plugins finden Sie im Softwarehandbuch. Die Verwendung von KI für die Erkennung von Kernen ist optional. Andere Methoden zur Detektion von Kernen stehen je nach verwendeter Bildanalysesoftware zur Verfügung. - Speichern Sie die trainierte KI (Klassifikatoraktionen > Speichern).
- Wechseln Sie zur Registerkarte Anmerkungen , und erstellen Sie mit dem Stiftanmerkungswerkzeug eine Anmerkung für jede Region of Interest (ROI), z. B. die Mitte des Tumors und den Rand des Tumors.
- Entfernen Sie bei Bedarf die Bereiche mit Falten und die Bereiche, die verschwommen erscheinen, mit dem Ausschlussanmerkungswerkzeug.
HINWEIS: Die Hämatoxylin-Eosin-Färbung eines Abschnitts, der an den für die mIF verwendeten Abschnitt angrenzt, kann vor der mIF-Färbung durchgeführt werden, um sicherzustellen, dass Tumorzellen in der Probe vorhanden sind, und um Anatomopathologen, die ROIs zu bestimmen. - Gehen Sie zur Registerkarte Analyse und wählen Sie den HighPlex FL-Algorithmus aus (Einstellungen, Aktionen > > HighPlex FL laden).
- Wählen Sie die Registerkarte Farbauswahl aus, und wählen Sie den gewünschten Farbstoff aus.
- Wechseln Sie auf der Registerkarte " Nukleare Erkennung " zu " Nuklearer Segmentierungstyp" und wählen Sie "KI benutzerdefiniert" aus.
- Wählen Sie unter Nuclear Segmentation Classifier die KI aus, die in Schritt 3.5 gespeichert wurde.
- Wählen Sie auf der Registerkarte Membran- und Zytoplasmatnachweis den maximalen Zytoplasmaradius (in dieser Studie wurde 1,5 verwendet) und die Anzahl der Membranfarbstoffe aus.
- Wählen Sie für jeden Farbstoff den positiven Schwellenwert für den Kern, den positiven Schwellenwert für das Zytoplasma und den Schwellenwert für den positiven Membranspiegel aus.
HINWEIS: Der Schwellenwert ist für jede Färbung unterschiedlich und sollte für jede Charge von Objektträgern und jedes gefärbte Antigen angepasst werden. Mit dem Werkzeug "Ansichtseinstellungen" (Ansicht > Ansichtseinstellungen) können Sie einen geeigneten Schwellenwert auswählen, indem Sie den Intensitätswert am Ende der Intensitätsspitze (rechts) verwenden. - Wählen Sie für jeden Farbstoff die Werte für den Zellkern, die Membran und das Zytoplasma aus.
HINWEIS: Dieser Parameter ist wichtig, um eine Fehlalarmerkennung zu vermeiden, wenn zwei Zellen mit unterschiedlicher Färbung nahe beieinander liegen (Abbildung 2). - Speichern Sie den Algorithmus (Einstellungen, Aktionen > Speichern).
- Analysieren Sie die ROIs (Analyze > Annotation Layer).
- Wechseln Sie zur Registerkarte Ergebnisse und wählen Sie alle Daten in Objektdaten aus (Strg + A).
- Exportieren Sie die Daten in .csv Format (klicken Sie mit der rechten Maustaste > > Objektdaten exportieren . Csv).
HINWEIS: Diese Tabelle enthält die Position (Xmin, Xmax; Ymin, Ymax) und die Positivität jedes Markers jeder analysierten Zelle.
4. Bioinformatik mit R
HINWEIS: Ein R-Skript mit weiteren Details zu den folgenden Schritten ist auf GitHub verfügbar (benidovskaya/Ring: Pipeline for the analysis of multiplex immunofluorescence stainings. [github.com])
- Definieren Sie zunächst anhand der exportierten Tabelle die verschiedenen Zelltypen basierend auf den Kolokalisationsfärbungen. Definieren Sie z. B. zytotoxische T-Zellen durch doppelt positive CD3+/CD8+-Zellen.
- Rekonstruieren Sie dann ein vereinfachtes Bild der Folie in einem Punktdiagramm mit den Koordinaten, die aus der Bildanalysesoftware exportiert wurden, und ggplot2 (Abbildung 3). Anhand dieser Daten können verschiedene Arten von Analysen durchgeführt werden:
- Dichteanalyse
HINWEIS: Die einfachste Analyse ist eine Dichteanalyse.- Führen Sie eine Dichteanalyse für alle Zelltypen durch, indem Sie den gesamten Objektträger für Biopsien oder einen bestimmten Bereich des Gewebes verwenden. Berechnen Sie beispielsweise die Dichte der CD3+- und CD8+-T-Zellen im Zentrum des Tumors und am Rand des Tumors (Abbildung 4A-C).
- Um diese Dichten zu berechnen, verwenden Sie eine Bildanalysesoftware, um einen bestimmten Datenrahmen pro Probe mit dem Phänotyp und den Koordinaten jeder Zelle zu erstellen. Erstellen Sie durch eine Clustering-Funktion (k-nächster Nachbar) auf R ein Polygonobjekt unter Verwendung der Ränder der untersuchten Biopsie und berechnen Sie die Dichte der interessierenden Zelltypen darin.
HINWEIS: Dies ermöglicht den Vergleich der Dichten verschiedener Zelltypen zwischen verschiedenen Zuständen (z. B. unterschiedliche Zeitpunkte, Behandlungstypen, Gewebetypen und Ansprechen auf die Behandlung) und Lokalisationen (Zentrum des Tumors, Rand des Tumors, Stromafibrose und Nekrosebereich) je nach biologischer Hypothese. Aufgrund der hohen Nähe zwischen Krebszellen, peritumoralen Zellen und tumorinfiltrierenden Zellen kann die Bildanalysesoftware doppelt positive Zellen gleichzeitig als Immun- und Krebszellen erkennen. In diesem Fall muss man dieses Problem bioinformatisch korrigieren, indem man erwähnt, was diese doppelt positiven Zellen sind. In diesem Fall wurden CD3+CD8+Cytokeratin+-Zellen als zytotoxische Zellen markiert, da die Zytokeratin-Positivität auf die Tumorzellen zurückzuführen war, die die infiltrierenden Lymphozyten umgeben.
- Heatmaps
- Zeichnen Sie unter Verwendung der Dichte jedes Zelltyps aus verschiedenen Panels und durch Anwenden einer Normalisierung (z. B. Skalierung, Zentrierung) Heatmaps (Abbildung 5), die die Zellhäufigkeit in der Grundgesamtheit der Stichproben darstellen.
- Unter Verwendung von hierarchischem, unüberwachtem Clustering auf der Grundlage der Zelldichte werden Patienten mit ähnlichen TME-Zusammensetzungen geclustert und diese Cluster mit klinischen Parametern wie Ansprechen auf die Behandlung und Überleben korreliert.
HINWEIS: Heatmaps und Clusterisierung können problemlos mit dem R ComplexHeatmap-Paket9 durchgeführt werden.
- Räumliche Zellverteilung
- Berechnen Sie bioinformatisch die Abstände zwischen den Zellen (z. B. Immun- und Tumorzellen; Abbildung 6A, B) basierend auf den Zellkoordinaten, die durch die Bildanalyse bereitgestellt wurden. Verwenden Sie den Median und die mittleren Abstände zwischen den interessierenden Zelltypen, um die Zellnähe über alle Stichproben einer Kohorte hinweg zu vergleichen.
- Räumlich beschreibende Funktionen
- Verwenden Sie die Kreuztyp-G-Cross-Funktion des nächsten Nachbarn, die über das R spatstat-Paket10 verfügbar ist, um die Wahrscheinlichkeit zu bestimmen, dass eine Zelle von Interesse, X (z. B. eine Tumorzelle), die nächstgelegene Zelle, Y (z. B. eine T-Zelle), innerhalb eines bestimmten Radius um Zelle X trifft.
- Berechnen Sie die Fläche unter der empirischen Kurve, um einen Zahlenwert zu erhalten, der die Tumorinfiltration von CD3+ T-Zellen um die Tumorzellendarstellt 11 (Abbildung 6C). Verwenden Sie andere räumliche Beschreibungsfunktionen wie die F-Funktion oder die J-Funktion12.
- Immunoscore-Analyse
- Berechnen Sie den Immunoscore (I), der vom Team von Galon7,8 entwickelt wurde, indem Sie die Dichte der CD3+ und CD8+ T-Zellen im Zentrum des Tumors und den invasiven Rand des Tumors verwenden.
HINWEIS: Die Punktzahl reicht von I0 bis I4. Eine niedrige Dichte von CD3+ und CD8+ T-Zellen in der Mitte und am Rand des Tumors ist mit einem Score von I0 assoziiert, während eine hohe Dichte von CD3+ und CD8+ T-Zellen in beiden Regionen mit einem Score von I4 assoziiert ist. Kürzlich wurde die prognostische Wirkung des Immunocore in einer Studie mit Proben von 2.681 Darmkrebspatienten im Stadium I-III aus 14 Zentren in 13 Ländern validiert7. Für die Berechnung des Immunoscore ist jedoch eine operativ resezierte Probe erforderlich, die sowohl das Zentrum als auch den Rand des Tumors enthält. Für Biopsien, die in der Regel keinen Rand haben, wurde kürzlich ein biopsieangepasster Immunscore entwickelt13. - Um den an die Biopsie angepassten Immunscore zu berechnen, konvertieren Sie den Wert der CD3+- und CD8+-T-Zelldichte in ein Perzentil und verwenden Sie dann das mittlere Perzentil der CD3+- und CD8+-T-Zellen für die Einstufung in eine von drei Kategorien (d. h. niedrig, mittel und hoch)13.
- Berechnen Sie den Immunoscore (I), der vom Team von Galon7,8 entwickelt wurde, indem Sie die Dichte der CD3+ und CD8+ T-Zellen im Zentrum des Tumors und den invasiven Rand des Tumors verwenden.
- Hotspot-Analyse
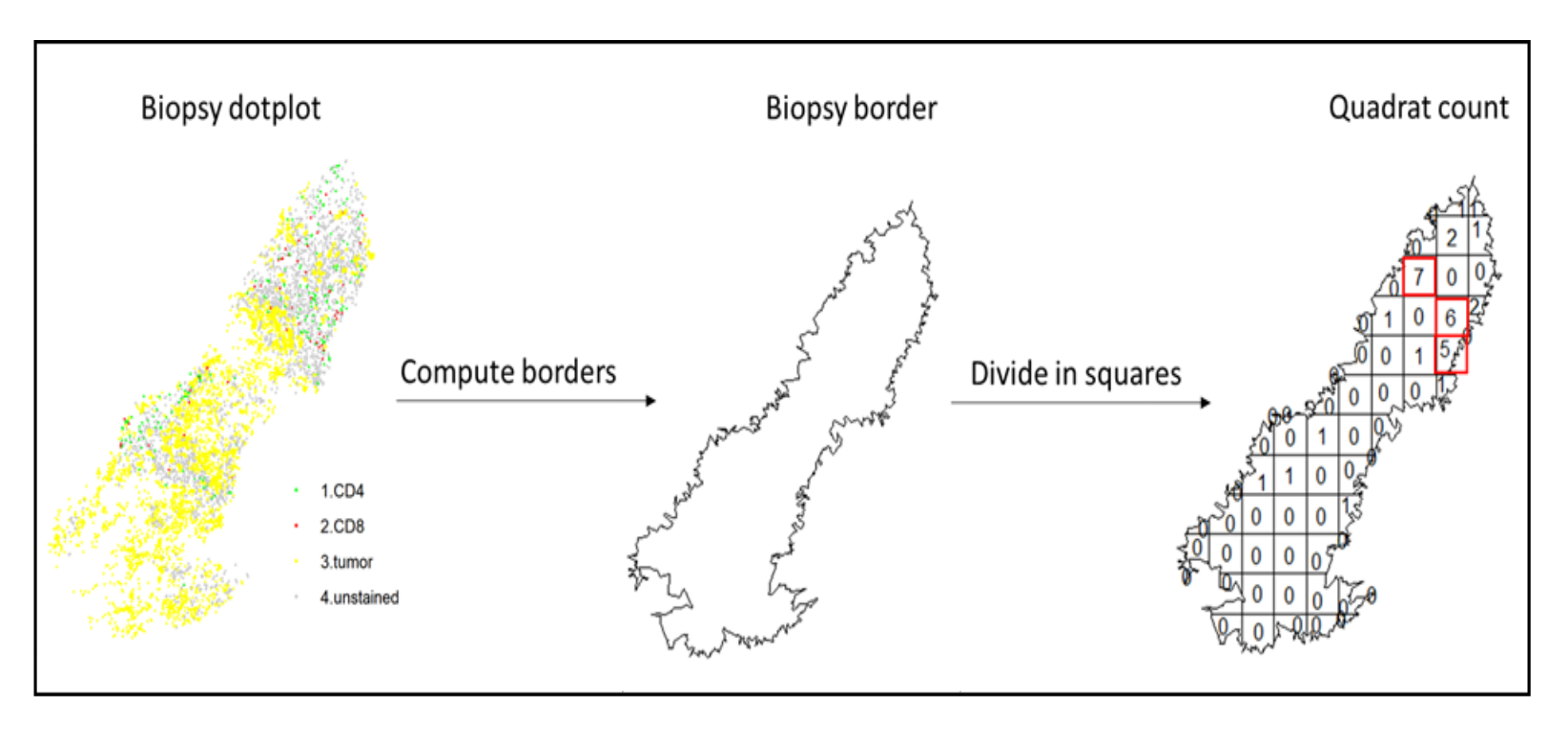
- Verwenden Sie die Hotspot-Analyse, indem Sie die Quadratzählfunktion (spatstat)10 verwenden, um die Dichten verschiedener Zelltypen im am stärksten infiltrierten Bereich des Gewebes zu vergleichen. Zum Beispiel ist es möglich, einen "Immunoscore-ähnlichen" Score zu berechnen, indem man den Wert der CD3- und CD8-T-Zelldichte der am stärksten infiltrierten Quadrate des Gewebes verwendet (Abbildung 7). Wenden Sie diese Methode für die Analyse aller Zelltypen mit einer inhomogenen Verteilung über das Gewebe an.
- Dichteanalyse
Repräsentative Ergebnisse
Nach diesem Protokoll sollten mehrere Parameter untersucht werden, um sicherzustellen, dass das Gewebe korrekt gefärbt ist. Erstens sollte die TSA-Färbung einen guten Dynamikumfang aufweisen, wenn während des Scanvorgangs niedrige Belichtungszeiten (typischerweise 2-100 ms) verwendet werden. Eine kurze Belichtungszeit impliziert, dass die Amplifikation während der Reaktion mit HRP korrekt durchgeführt wurde. Bei Antigene, die mit dem Sekundärantikörper direkt gekoppelt mit dem Fluorochrom gefärbt wurden, könnte die Expositionszeit viel länger sein, was zu Photobleaching (einer Abnahme der Signalintensität aufgrund einer langen Expositionszeit) führen könnte. Zweitens ist es wichtig zu überprüfen, ob jede Färbung ein hohes SNR aufweist. Ein hohes Hintergrundsignal mit einem niedrigen Antigensignal kann ein Hinweis darauf sein, dass der primäre Antikörper nicht spezifisch genug ist, dass die endogenen Peroxidasen nicht korrekt inaktiviert wurden oder dass ein Schritt des Protokolls nicht adäquat durchgeführt wurde. Drittens ist es je nach Diascanner und den für den Scan verwendeten Filtersätzen möglich, Überlappungen zwischen zwei Farben (z. B. AF555, AF594 und AF647) zu erkennen. Die Wahl der richtigen Filtersätze auf dem Scanner und die richtige Verdünnung der primären Antikörper sind entscheidend, um mögliche Kreuzdetektionen zu vermeiden. Die Qualitätskontrolle besteht aus der Erkennung einzelner gefärbter Zellen für jeden Marker auf der gescannten Datei. Schließlich ist es auch wichtig, für jede Färbecharge eine Positiv- und eine Negativkontrolle hinzuzufügen. Für Immunzellen ist die Mandel eine gute Positivkontrolle. Ein repräsentatives Ergebnis der optimalen Färbung ist in Abbildung 1 dargestellt.

Abbildung 1: Lokal fortgeschrittenes Rektumkarzinom, gefärbt durch Multiplex-Immunfluoreszenz. Abkürzungen: PD-1 = programmiertes Zelltodprotein 1; PD-L1 = Programmierter Todesligand 1; ROR-γ = RAR-verwandter Orphan-Rezeptor-Gamma; CD3 = Cluster der Differenzierung 3; hPanCK = humanes Pan-Zytokeratin. Jede Antigenfärbung wird in Graustufen gescannt, und die in der Abbildung dargestellten Farben sind Pseudofarben. Maßstabsleiste geringe Vergrößerung: 200 μm; Maßstabsleiste hohe Vergrößerung: 100 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 2: Zellkern- und Färbedetektion eines lokal fortgeschrittenen Rektumkarzinoms mit Hilfe einer Bildanalysesoftware. Wenn der Parameter für den Prozentsatz der Vollständigkeit nicht korrekt eingestellt ist, erkennt die Software zwei CD8+-Zellen (grüner Kreis), da sie nahe beieinander liegen, aber nur eine Zelle gefärbt ist. Die Verwendung einer Vollständigkeit von 70 % hilft, diese falsch positive Erkennung zu vermeiden. Grün = hPanCK; Gelb = CD3; Orange = CD8. Maßstabsleiste: 100 μm Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

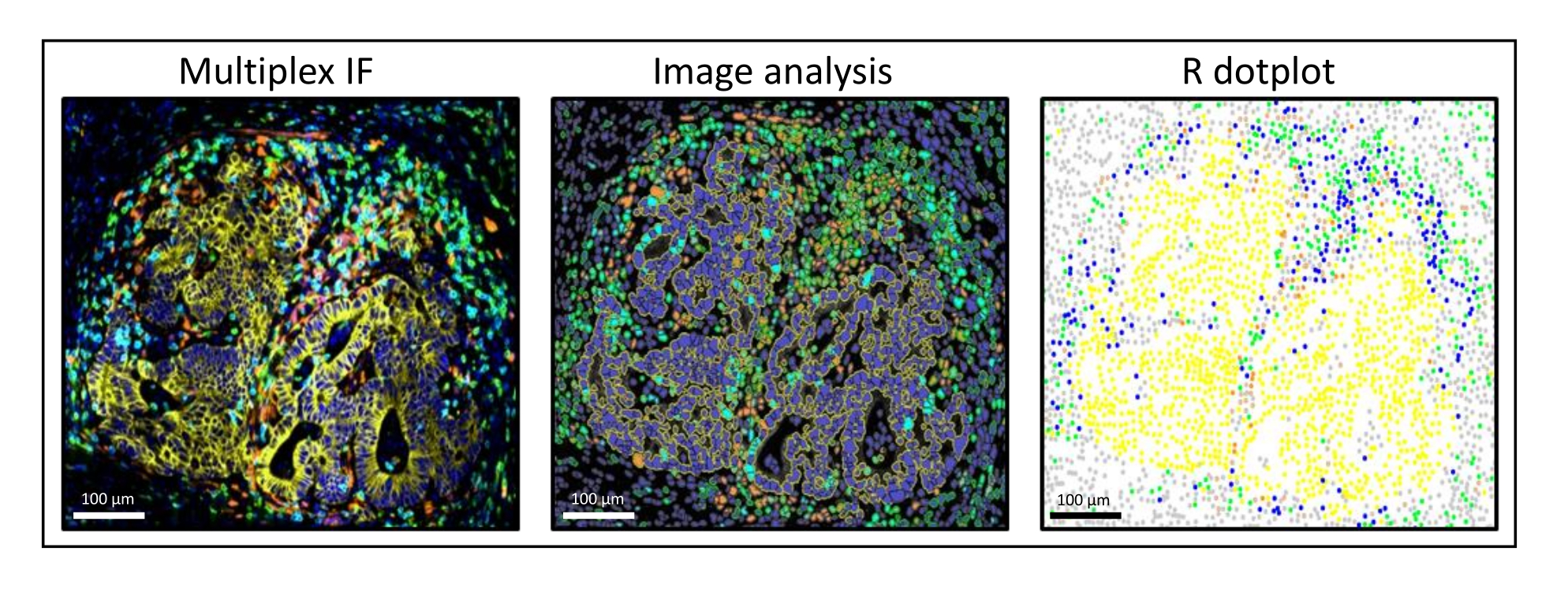
Abbildung 3: Bildanalyse und R-Punktplot-Rekonstitution einer Leberkolorektumkarzinommetastase. Auf der Multiplex-Färbung (links) ist das humane Pan-Zytokeratin gelb, CD3 grün, CD8 hellblau und IDO orange dargestellt. Im Punktdiagramm (rechts) sind humane Pan-Cytokeratin+-Zellen gelb, CD3+CD8−-Zellen grün, CD3+CD8+-Zellen blau und IDO+-Zellen orange dargestellt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

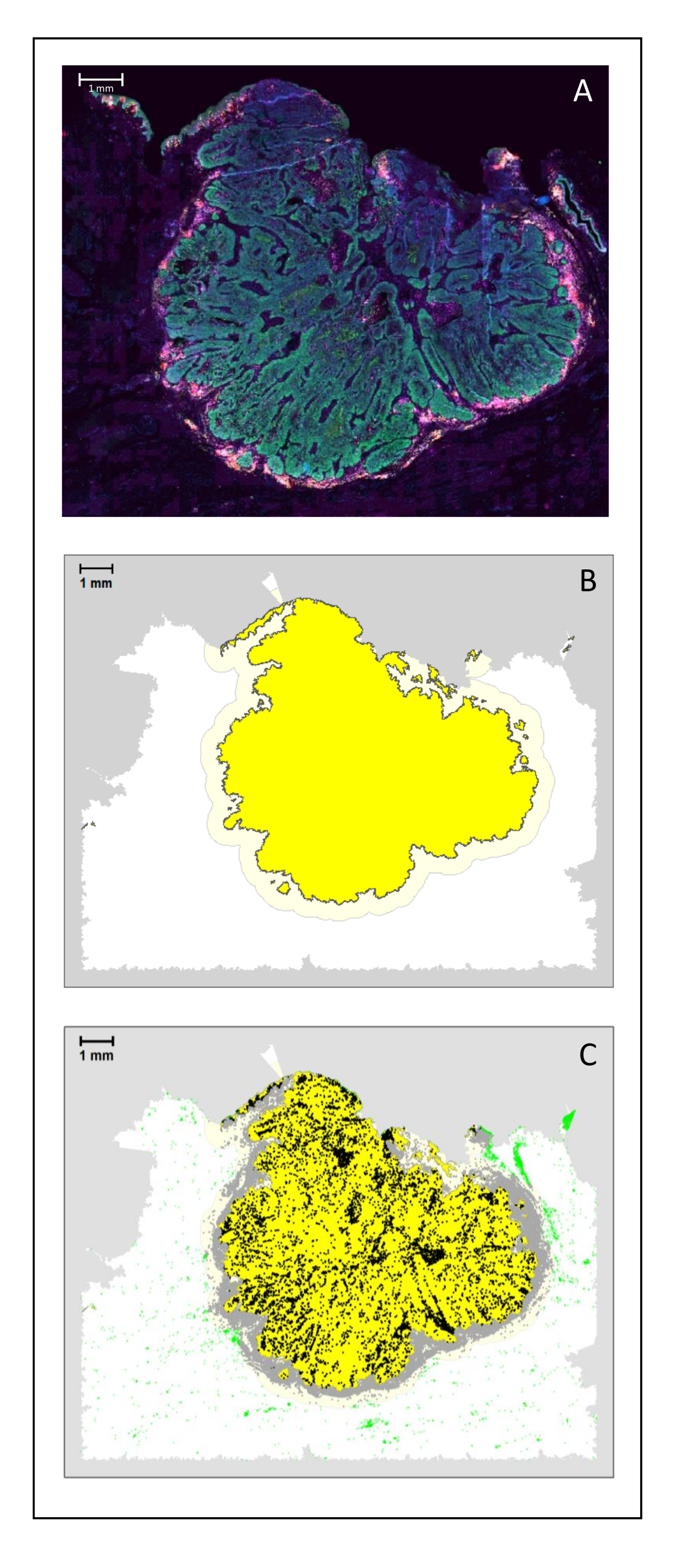
Abbildung 4: Analyse eines chirurgischen Schnitts eines HNSCC. (A) Ein chirurgischer Schnitt eines HNSCC. Krebszellen sind grün sichtbar. Peritumorale Zellen werden um die Tumorinseln herum sichtbar gemacht (CD3 in gelb und CD8 in lila). (B) Das Zentrum des Tumors (gelb mit schwarzem Rand) wird bioinformatisch durch den k-Nearest-Neighbor-Algorithmus berechnet, basierend auf dem Abstand zwischen den Tumorinseln von einem einzelnen Bereich. Um diesen Bereich herum wird ein invasiver Rand (hellgelb mit grauem Rand) auf einer beliebigen 500-μm-Basis berechnet. (C) Invasive T-Zellen sind mit schwarzen Punkten in der Mitte des Tumors und grauen Punkten am invasiven Rand hervorgehoben. Andere T-Zellen sind in hellgrünen Punkten hervorgehoben. Maßstabsleiste: 1 mm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

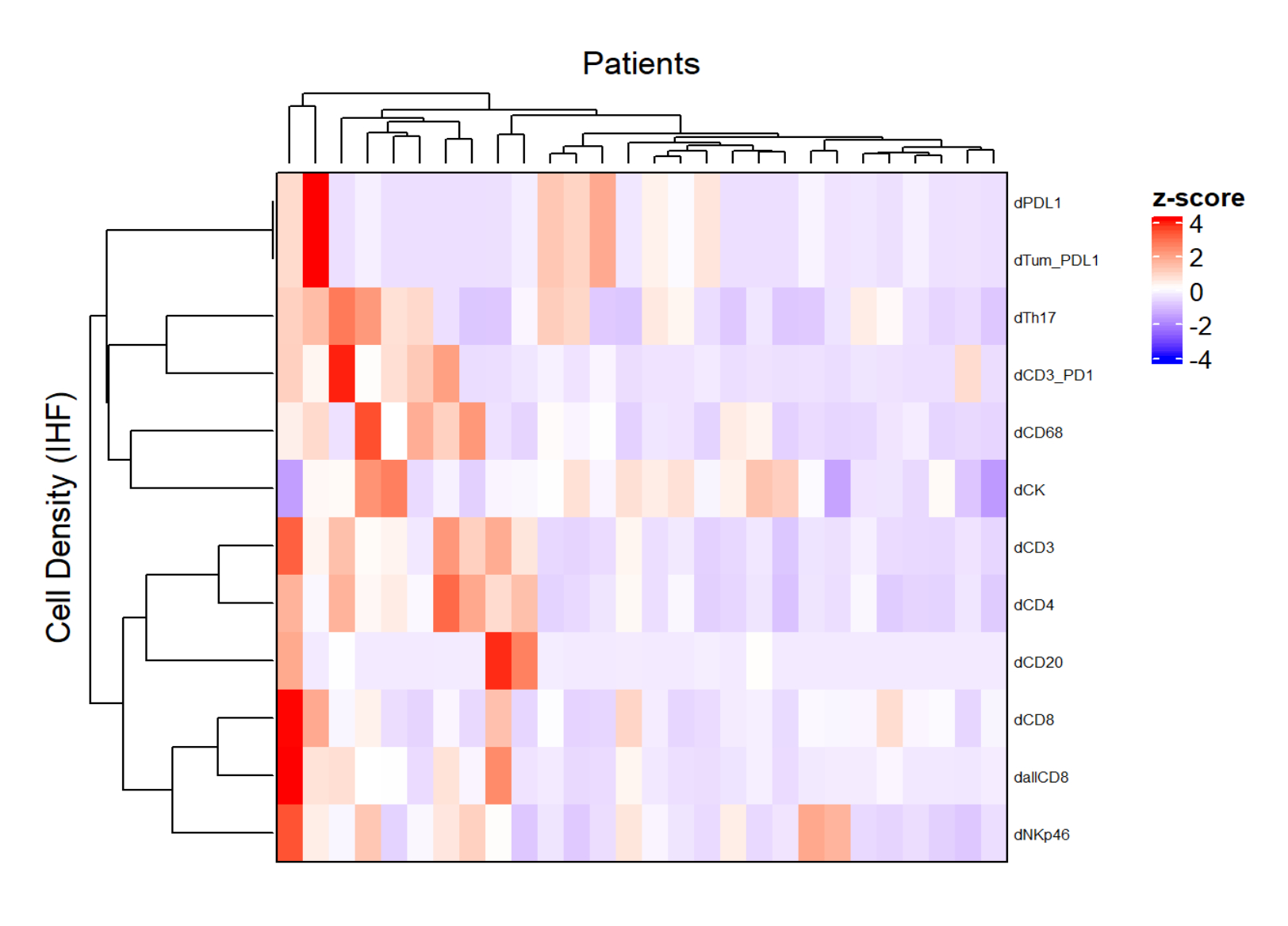
Abbildung 5: Heatmap der Dichte verschiedener Zelltypen von lokal fortgeschrittenen Rektumkarzinombiopsien. Die Heatmap wurde unter Verwendung von unüberwachtem Clustering der Dichten verschiedener Zelltypen aus verschiedenen Multiplex-Panels mit dem ComplexHeatmap-Paket erstellt. Für die Normalisierung wurden Skalierung und Zentrierung verwendet. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

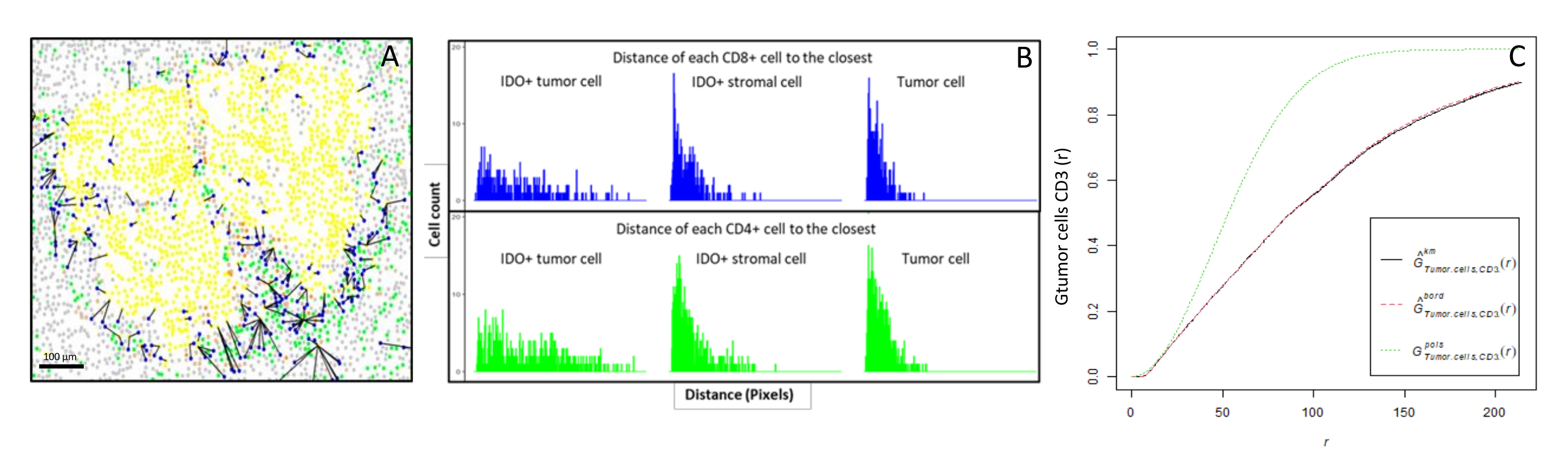
Abbildung 6: Abstände der CD4+- und CD8+-Zellen zu jeder IDO+- oder Tumorzelle. Humane Pan-Zytokeratin+-Zellen sind gelb, CD3+CD8−-Zellen grün, CD3+CD8+-Zellen blau und IDO+-Zellen orange. (A) Der geringste Abstand zwischen Tumorzellen und jeder CD8+ T-Zelle. (B) Barplots der Abstände zwischen IDO+-Zellen und jeder CD8+ T-Zelle (blau) oder CD4+ T-Zelle (grün). (C) Beispiel einer Probe, die mit der G-Kreuz-Funktion analysiert wurde. Die y-Achse zeigt die Wahrscheinlichkeit, dass eine Tumorzelle in einem Radius von 0-200 μm um die Tumorzelle auf einen CD3+-Lymphozyten trifft. Es werden drei Kurven gezeigt; Die theoretische Kurve ist grün gepunktet (Poisson-Verteilung), die korrigierte empirische Kurve mit km-Korrektur schwarz und die korrigierte empirische Kurve mit Randkorrektur rot gepunktet. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 7: Illustration eines Quadratcounts. Die Grenzberechnung und die Quadratanzahl wurden mit dem Paket spatstats durchgeführt. Die am stärksten infiltrierten Quadrate (Hotspots) können für nachgelagerte Statistiken verwendet werden. CD4 ist grün, CD8 rot und Tumorzellen gelb. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

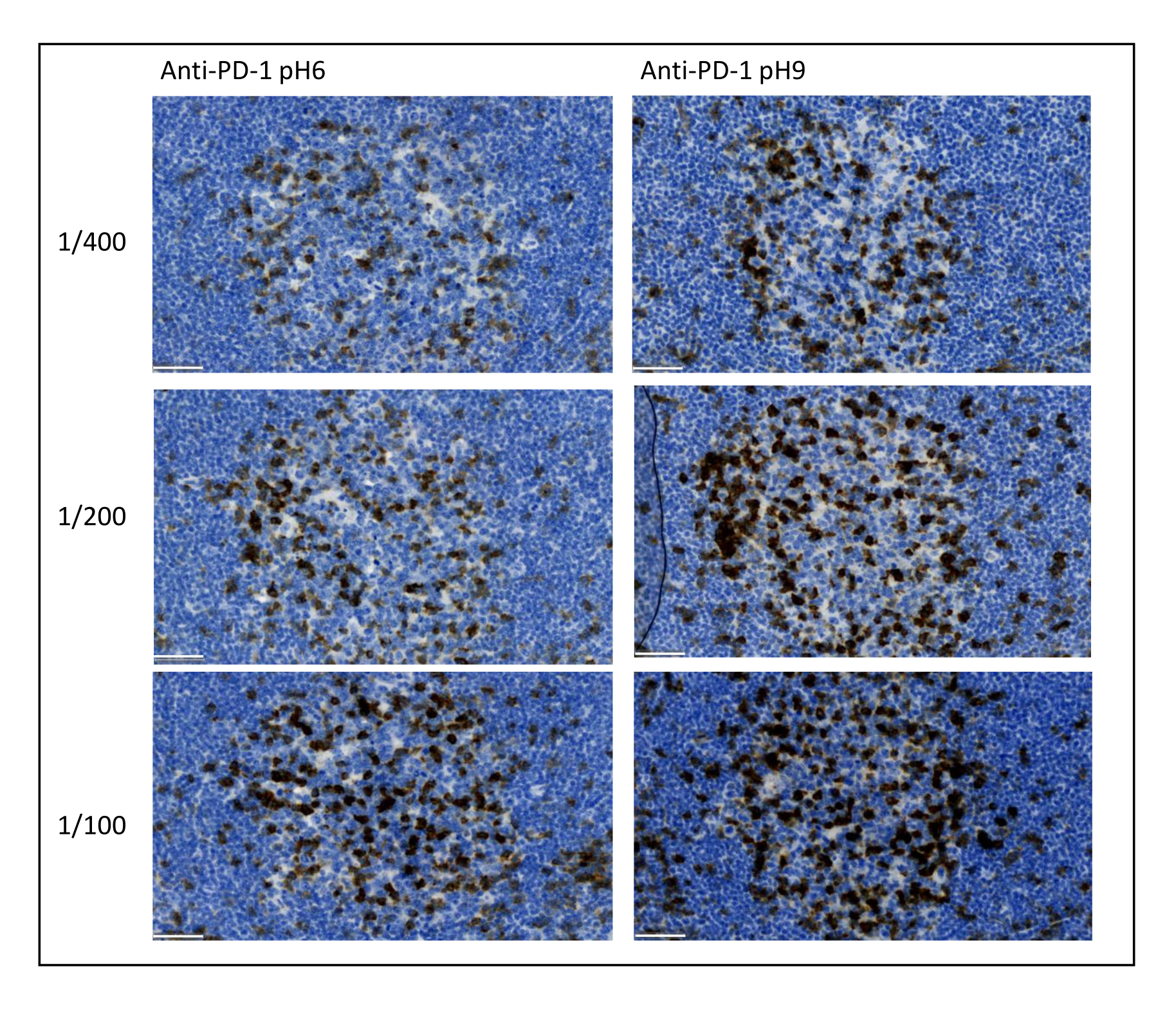
Abbildung 8: Antikörperverdünnung und Optimierung des Antigenabrufs. Chromogener Nachweis von PD-1 unter Verwendung von drei verschiedenen Verdünnungen und zwei verschiedenen Antigen-Rückgewinnungslösungen des primären Antikörpers (Citrat pH 6 und EDTA pH 9). Maßstabsleiste: 50 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
| Primärer Antikörper | Verdünnung | Antigen-Retrieval | Sekundärer Antikörper | Fluorochrom | Position |
| PD-1 | 1/100 | EDTA (pH 9) | Anti-Kaninchen | AF647 | 1 |
| PD-L1 | 1/1000 | EDTA (pH 9) | Anti-Kaninchen | AF488 | 2 |
| ROR-γ | 1/200 | EDTA (pH 9) | Anti-Maus | ATT0-425 | 3 |
| CD3 | 1/100 | Citrat (pH 6) | Anti-Kaninchen | AF555 | 4 |
| hPanCK | 1/50 | Citrat (pH 6) | Anti-Maus gekoppelt mit AF750 | 5 | |
Tabelle 1: Beispiel für ein optimiertes Multiplex-Panel. Abkürzungen: PD-1 = programmiertes Zelltodprotein 1; PD-L1 = Programmierter Todesligand 1; ROR-γ = RAR-verwandter Orphan-Rezeptor-Gamma; CD3 = Cluster der Differenzierung 3; hPanCK = humanes Pan-Zytokeratin; AF = AlexaFluor; EDTA = Ethylendiamintetraessigsäure. CD3 wird zum Nachweis von T-Lymphozyten verwendet; PD-1 wird verwendet, um erschöpfte Lymphozyten zu erkennen; ROR-γ wird verwendet, um Th-17 zu detektieren; und hPanCK wird zum Nachweis von Tumorzellen eingesetzt. Die Positionsspalte gibt die Reihenfolge an, in der der sequentielle Multiplex ausgeführt werden muss.
Diskussion
Die wichtigsten Parameter, die zur Optimierung der Multiplex-Färbung berücksichtigt werden müssen, sind die Verdünnung, die Spezifität und die Antigengewinnung, die für jeden Primärantikörper verwendet werden. Vor Beginn eines Multiplex-Protokolls muss die optimale Verdünnung und optimale Epitopgewinnung (pH 6 oder pH 9) jedes Primärantikörpers mittels chromogener Färbung (DAB) getestet werden. Wir empfehlen, drei Verdünnungen für jeden Antigen-Rückgewinnungspuffer zu testen: die Verdünnung, die normalerweise von der Marke angegeben wird, die den Antikörper kommerzialisiert, die gleiche Verdünnung zweifach geteilt und die gleiche Verdünnung zweifach multipliziert (Abbildung 8). Die Wahl der richtigen Verdünnung ist ein sehr wichtiger Schritt, um die Spezifität der Antikörper zu überprüfen und das Signal-Rausch-Verhältnis (SNR) der Färbung zu optimieren. Nach der Wahl der richtigen Verdünnung im DAB sollte die gleiche Verdünnung für jeden Primärantikörper mit Uniplex TSA getestet werden. Sobald die Verdünnung und der Epitop-Rückgewinnungspuffer für jede Antigenfärbung ausgewählt sind, ist es auch wichtig, die Sequenz des Multiplex korrekt einzustellen. Insbesondere sind einige Antigene an der ersten Position besser gefärbt und andere an der letzten Position. Wir empfehlen, die Multiplex-Markierung mit allen möglichen Ordnungspermutationen zu testen, um auszuwählen, welche Antigenfärbung zuerst, an zweiter Stelle usw. erfolgen soll. Dies ist auch ein sehr wichtiger Schritt, da einige fragile Antigene nach mehreren Runden der Epitopgewinnung abgebaut werden können und einige Antigene nach mehreren Runden der Epitopgewinnung besser gefärbt sind. Zum Beispiel ist das SNR in der letzten Position für CD3 und in der ersten Position für die PD-1-Färbung immer höher. Darüber hinaus kann die Färbung mehrerer kolokalisierter Antigene durch einen Umbrella-Effekt (die Sättigung von Tyramid-reaktiven Zentren) behindert werden. Dies kann durch eine Verringerung der Tyramidkonzentration abgeschwächt werden. Wenn die Expression eines Antigens durch die Expression eines anderen Antigens bedingt ist (CD8 kommt nur auf CD3-exprimierenden T-Zellen vor), empfehlen wir, das Antigen mit der breitesten Expression (in diesem Fall CD3) nach dem anderen zu färben. Schließlich ist auch die Auswahl des richtigen Fluorochroms für jede Antigenfärbung entsprechend den Besonderheiten des Scanners ein wichtiger Schritt, um Kreuzdetektionen zu vermeiden.
Die Hauptvorteile dieser Technik sind die Verstärkung und das erzielte Signal-Rausch-Verhältnis. Diese Technik hat jedoch eine Einschränkung, nämlich dass die Färbung sequenziell erfolgt und die Fluorochrome kovalent an das Gewebe gebunden sind. Nichtsdestotrotz ist es nach Durchführung aller Verstärkerrunden des Tyramid-Signals auch möglich, eine letzte Färbung mit einem sekundären Antikörper hinzuzufügen, der direkt mit einem Fluorochrom gekoppelt ist (kein TSA). In einigen Panels haben wir diese Methode verwendet, um Färbungen im 750-Kanal hinzuzufügen. Dies war notwendig, da zu diesem Zeitpunkt noch kein Tyramid-AF750 im Handel erhältlich war. Es ist zu beachten, dass die Expositionszeit (während des Scans) des mit AF750 gefärbten Antigens viel länger ist als bei den anderen mit TSA gefärbten Antigene. In diesem Fall empfehlen wir, ein stark exprimiertes Protein wie Zytokeratin zu färben oder die Konzentration des primären Antikörpers zu erhöhen. Auf diese Weise ist es möglich, je nach Fluoreszenzscanner maximal fünf bis sechs Antigene pro Objektträger in einer Charge zu färben.
Im Gegensatz dazu verwenden mehrere kommerziell erhältliche Techniken die serielle Färbung mit mehreren Runden der Färbung, des Scannens und des Striptens oder des Photobleachings, um die Anzahl der Antigene zu verbessern, die auf einem einzigen Gewebeschnitt gefärbt werden können. Diese Techniken sind jedoch oft zeitaufwändig, teuer, haben keine Signalverstärkung, erfordern fortgeschrittene Rechenschritte, um die seriellen Scans korrekt zusammenzuführen, und können unserer Erfahrung nach aufgrund der zahlreichen Verfahrensschritte irreversible Gewebeschäden induzieren. Nichtsdestotrotz wurde berichtet, dass mit dieser Methode bis zu 30 Antigene auf einem einzigen Gewebe gefärbt werden konnten14.
Zusammenfassend lässt sich sagen, dass unsere Methode eine robuste, reproduzierbare, einfach anzuwendende und kostengünstige Immunhistofluoreszenztechnik ist, die in jedem Labor mit einem Fluoreszenz-Objektträgerscanner eingesetzt werden kann. Jeder kommerzialisierte Primärantikörper, der für IHC geeignet ist, kann verwendet werden, und die Panels sind nicht spezifisch für kommerzielle Kits. Die Bildanalyse kann mit verschiedenen Programmen durchgeführt werden, einschließlich Open-Source-Programmen wie QuPath und R. Wir glauben jedoch, dass diese Methode in Zukunft sogar für große Antigen-Panels verbessert werden könnte, da sie die serielle Färbung/das Scannen desselben Objektträgers mit verschiedenen Antigen-Panels und mit dem Vorteil der Signalverstärkung ermöglicht.
Offenlegungen
Die Autoren haben keine Interessenkonflikte zu erklären.
Danksagungen
Die Autoren bedanken sich bei Dr. Derouane F. für ihre Hilfe und Unterstützung. Nicolas Huyghe ist wissenschaftlicher Mitarbeiter, der durch ein Stipendium des belgischen Nationalen Fonds für wissenschaftliche Forschung (Télévie/FNRS 7460918F) gefördert wird.
Materialien
| Name | Company | Catalog Number | Comments |
| anti-CD3 primary antibody | Abcam | ab16669 | rabbit monocolonal |
| anti-CD8 primary antibody | DAKO | M710301 | mouse monoclonal |
| anti-hPanCK primary antibody | DAKO | M3515 | mouse monoclonal |
| anti-PD-1 primary antibody | Cell Signalling | D4W2J | rabbit monocolonal |
| anti-PD-L1 primary antibody | Cell Signalling | 13684 | rabbit monocolonal |
| anti-RORC primary antibody | Sigma | MABF81 | mouse monoclonal |
| ATTO-425 | ATTOtec | ||
| Axioscan Z1 | Zeiss | Light source: Colibri 7 (385, 430, 475, 555, 590, 630, 735 nm) Filtersets: Excitation 379/34 – beam splitter 409 – emission 440/40; Excitation 438/24 – beam splitter 458 – emission 483/32; Excitation 490/20 – beam splitter 505 – emission 525/20; Excitation 546/10 – beam splitter 556 – emission 572/23; Excitation 592/21 – beam splitter 610 – emission 630/30; Excitation 635/18 – beam splitter 652 – emission 680/42; Excitation 735/40 – beam splitter QBS 405 + 493 + 611 + 762 - emission QBP 425/30 + 524/51 + 634/38 + 785/38; Objective: Plan-Apochromat 20x/0.8; Camera : Orca Flash 4.0 V3 | |
| Borosilicate Cover Glass | VWR | 631-0146 | |
| Envision+ anti-mouse | DAKO | K4001 | |
| Envision+ anti-rabbit | DAKO | K4003 | |
| Fluorescence mounting medium | DAKO | S3023 | |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 750 | ThermoFischer | A-21037 | |
| HALO software | Indicalabs | ||
| Hoescht | Sigma | 14533 | |
| Superfrost plus microscope slides | Fisherscientific/Epredia | 10149870 | |
| Tyramide-AF488 | ThermoFischer | B40953 | |
| Tyramide-AF555 | ThermoFischer | B04955 | |
| Tyramide-AF647 | ThermoFischer | B04958 |
Referenzen
- Ge, P., et al. Profiles of immune cell infiltration and immune-related genes in the tumor microenvironment of colorectal cancer. Biomedicine & Pharmacotherapy. 118, 109228 (2019).
- Fridman, W. H. The immune microenvironment as a guide for cancer therapies. Oncoimmunology. 1 (3), 261-262 (2012).
- Fridman, W. H., Pages, F., Sautes-Fridman, C., Galon, J. The immune contexture in human tumours: Impact on clinical outcome. in Nature Reviews. Cancer. 12 (4), 298-306 (2012).
- Hanahan, D., Weinberg, R. A. Hallmarks of cancer: The next generation. Cell. 144 (5), 646-674 (2011).
- Calu, V., et al. Key biomarkers within the colorectal cancer related inflammatory microenvironment. Scientific Reports. 11 (1), 7940 (2021).
- Havel, J. J., Chowell, D., Chan, T. A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nature Reviews. Cancer. 19 (3), 133-150 (2019).
- Pages, F., et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 391 (10135), 2128-2139 (2018).
- Mlecnik, B., et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 44 (3), 698-711 (2016).
- Gu, Z., Eils, R., Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 32 (18), 2847-2849 (2016).
- Baddeley, A., Rubak, E., Turner, R. . Spatial Point Patterns: Methodology and Applications with R. , (2022).
- Barua, S., et al. Spatial interaction of tumor cells and regulatory T cells correlates with survival in non-small cell lung cancer. Lung Cancer. 117, 73-79 (2018).
- Parra, E. R. Methods to determine and analyze the cellular spatial distribution extracted from multiplex immunofluorescence data to understand the tumor microenvironment. Frontiers in Molecular Biosciences. 8, 668340 (2021).
- El Sissy, C., et al. A diagnostic biopsy-adapted immunoscore predicts response to neoadjuvant treatment and selects patients with rectal cancer eligible for a watch-and-wait strategy. Clinical Cancer Research. 26 (19), 5198-5207 (2020).
- Bolognesi, M. M., et al. Multiplex staining by sequential immunostaining and antibody removal on routine tissue sections. The Journal of Histochemistry and Cytochemistry. 65 (8), 431-444 (2017).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten