Ein einfaches, schnelles und teilautomatisiertes Protokoll für die Isolierung von Einzelkernen aus gefrorenem Säugetiergewebe für die Einzelkernsequenzierung
In diesem Artikel
Zusammenfassung
Die Studie beschreibt ein einfaches, schnelles und teilautomatisiertes Protokoll zur Isolierung hochwertiger Zellkerne aus gefrorenem Säugetiergewebe für die nachgeschaltete Einzelkern-RNA-Sequenzierung.
Zusammenfassung
Die Einzelzell- und Einzelkern-RNA-Sequenzierung ist aufgrund der Fülle an transkriptomischen Informationen, die sie liefern, zu gängigen Laboranwendungen geworden. Insbesondere die Einzelkern-RNA-Sequenzierung ist nützlich, um die Genexpression in schwer zu dissoziierenden Geweben zu untersuchen. Darüber hinaus ist dieser Ansatz auch mit gefrorenem (Archiv-)Material kompatibel. Hier beschreiben wir ein Protokoll zur Isolierung hochwertiger Einzelkerne aus gefrorenem Säugetiergewebe für die nachgeschaltete Einzelkern-RNA-Sequenzierung in teilautomatisierter Weise unter Verwendung kommerziell erhältlicher Instrumente und Reagenzien. Konkret wird ein Roboterdissoziator verwendet, um die Gewebehomogenisierung zu automatisieren und zu standardisieren, gefolgt von einem optimierten chemischen Gradienten zur Filterung der Kerne. Schließlich zählen wir die Zellkerne genau und automatisch mit einem automatisierten Fluoreszenzzellzähler. Die Leistung dieses Protokolls wird am Gehirn von Mäusen, Rattennieren und Cynomolgus-Leber- und Milzgewebe demonstriert. Dieses Protokoll ist unkompliziert, schnell und leicht an verschiedene Säugetiergewebe anpassbar, ohne dass eine umfangreiche Optimierung erforderlich ist, und bietet qualitativ hochwertige Kerne für die nachgeschaltete RNA-Sequenzierung einzelner Kerne.
Einleitung
Die Einzelzell- (sc) und Einzelkern-RNA-Sequenzierung (sn) sind aufgrund der höheren Auflösung der Genexpression im Vergleich zur Massen-RNA-Sequenzierung zu häufig verwendeten Protokollen in der Molekular- und Zellbiologie geworden. Die Isolierung von qualitativ hochwertigen Einzelzell- und Einzelkernpräparaten aus festem Gewebe stellt jedoch nach wie vor eine Herausforderung dar und ist oft der geschwindigkeitsbestimmende Schritt in sc/sn-RNAseq-Experimenten. In der Tat wurde eine Fülle von Protokollen entwickelt, die verschiedene chemische und mechanische Verfahren verwenden, um Zell-/Kernsuspensionen zu erhalten 1,2,3,4,5,6,7,8,9,10,11,12,13,14,15 . Darüber hinaus reichen die Strategien zur Reinigung solcher Präparate von Schmutz/Klumpen usw. von der Durchflusssortierung über die Filtration bis hin zum Waschen. Solche Protokolle sind oft manuell (was zu benutzerbezogener Variabilität führt), können zeitaufwändig sein (was zu einer reduzierten Zell-/Zellkernlebensfähigkeit führt) und/oder erfordern möglicherweise den Zugang zu einem Durchflusszytometer für die Zell-/Zellkernsortierung. Diese Studie konzentrierte sich auf die Entwicklung eines einfachen, schnellen und teilautomatisierten Protokolls zur Isolierung einzelner Kerne aus gefrorenem Säugetiergewebe für nachgeschaltete RNA-Sequenzierungsanwendungen. Wir haben uns speziell auf die Zellkernisolierung im Gegensatz zur Zellisolierung konzentriert, da sie mit der Verwendung von gefrorenem Gewebe kompatibel ist, die Probenentnahme/-verarbeitung praktischer macht und eine unvoreingenommene Chargenbildung von Proben ermöglicht, insbesondere in Zeitverlaufsexperimenten. Obwohl das nukleäre Transkriptom das zelluläre Transkriptom nicht vollständig widerspiegelt, haben mehrere Studien nun gezeigt, dass die RNA-Sequenzierungsdaten der einzelnen Kerne mit den RNA-Sequenzierungsdaten der einzelnen Zellen für die Identifizierung von Zelltypen vergleichbar sind, auch wenn die Anteile der Zelltypen variieren können 6,16,17,18,19.
Die Zellkernisolierung besteht aus mehreren Schritten: 1) mechanischer oder chemischer Aufschluss des Gewebes zur Freisetzung der Kerne, 2) Reinigung von Ablagerungen und Klumpen und 3) genaue Zählung der Kerne zur Vorbereitung für nachgeschaltete Anwendungen. In einer Reihe von Protokollen beinhaltet Schritt 1 häufig die Verwendung eines Dounce-Homogenisators, um das Gewebe 3,20 aufzubrechen. Alternativ können auch chemische Methoden eingesetzt werden, die jedoch oft für unterschiedliche Gewebe optimiert werden müssen 2,5,6. Wir haben die Erfahrung gemacht, dass ein manuelles Verfahren zur Gewebeauffüllung anfällig für bedienerassoziierte Variabilität ist, was zu einer unterschiedlichen Qualität und Ausbeute von Zellkernen führt. Um die technische Variabilität zu minimieren und ein konsistenteres und reproduzierbareres Protokoll zu erhalten, das gewebeübergreifend funktioniert, wurde ein Protokoll entwickelt, das einen kommerziell erhältlichen robotergestützten Gewebedissoziator verwendet21. Obwohl der Pufferaustausch in der Regel das einfachste Mittel zum Waschen von Kernen ist, haben wir für Schritt 2 die Verwendung eines relativ kurzen Saccharosegradientenzentrifugationsschritts gewählt, um eine gründlichere Entfernung von Ablagerungen zu erreichen. Speziell für Hirngewebe verwenden wir einen Siliciumdioxid-Kolloid-Gradienten anstelle eines Saccharose-Gradienten, um eine effektivere Myelinentfernung zu erzielen. Schließlich ist für die Zählung die Verwendung eines Hämozytometers der Goldstandard für die Zählung und visuelle Inspektion der Kerne. In unserem Protokoll kann dieser Schritt unter Verwendung eines kommerziell erhältlichen automatisierten Fluoreszenzzellenzählers22 zuverlässig automatisiert werden. Dieses Protokoll wurde getestet und ist mit mehreren gefrorenen Säugetiergeweben, einschließlich Gehirn, Niere, Milz und Leber, von verschiedenen Säugetierarten (Ratte, Maus und nicht-menschlicher Primat) kompatibel und bietet qualitativ hochwertige Kerne für die nachgeschaltete RNA-Sequenzierung einzelner Kerne mit einer tröpfchenbasierten kommerziellen Plattform. Das Protokoll dauert etwa 75 Minuten von der Gewebevorbereitung bis zum Beginn des Einzelkern-RNA-Sequenzierungs-Workflows.
Protokoll
Alle Tierversuche wurden mit Genehmigung der kantonalen Veterinärbehörde Basel-Stadt unter strikter Einhaltung der schweizerischen Tierschutzbestimmungen oder mit Genehmigung des Institutionellen Tierpflege- und -verwendungsausschusses unter Einhaltung des deutschen Tierschutzgesetzes durchgeführt.
1. Gewebe- und Reagenz-/Instrumentenvorbereitung
- Reinigung und Instrumentenvorbereitung
- Reinigen Sie Arbeitsplatten und Pinzetten mit 70 % Ethanol und RNase-Dekontaminationslösung. Die Zentrifugen auf 4 °C vorkühlen.
- Die Kernisolationskartuschen im Kühlschrank bei 4 °C für mindestens 30 Minuten vorkühlen.
- Starten Sie den Roboter-Dissoziator und schalten Sie die Kühlung ein, indem Sie den Schieberegler oben rechts auf dem Bildschirm auf Kühlen stellen und darauf klicken, um die Kühlung zu starten, so dass der Schieberegler orange erscheint. Vergewissern Sie sich, dass die angebrachte NSR-Flasche (Nuclei Storage Reagenz) und die NIB-Flasche (Nuclei Isolation Buffer) noch genügend Flüssigkeit enthalten und ordnungsgemäß gekühlt sind.
- Bereiten Sie eine mit Trockeneis gefüllte Styroporschaumbox vor und kühlen Sie Petrischalen und Skalpellklingen auf Trockeneis vor.
- Vorbereitung der Puffer
- Bereiten Sie die 1,5 M Saccharose-Kissenlösung (SCS) wie in Tabelle 1 dargestellt vor. Verteilen Sie das SCS in 500-μl-Aliquots in 2-ml-DNase/RNase-Röhrchen, um vier 500-μl-SCS-Aliquots pro Probe zu erhalten. Bewahren Sie die Aliquots bis zur weiteren Verwendung auf Eis auf.
- Im Falle der Verarbeitung von Hirngewebe wird stattdessen eine 18%ige Siliciumdioxid-Kolloidlösung, wie in Tabelle 2 beschrieben, hergestellt, indem die Siliciumdioxid-Kolloid-Stammlösung in NSR verdünnt und RNase-Inhibitor hinzugefügt wird. Bereiten Sie 3 ml 18%ige Siliciumdioxid-Kolloidlösung pro Probe vor und bewahren Sie sie auf Eis auf.
2. Gewebehomogenisierung und Zellkernisolierung
- Nehmen Sie die Probe aus dem Gefrierschrank bei -80 °C und legen Sie sie sofort auf Trockeneis.
- Schneiden Sie die Probe auf einer vorgekühlten Petrischale oder Metallplatte auf Trockeneis mit einem vorgekühlten Skalpell in ein 15-50 mg Stück (falls es nicht bereits die richtige Größe hat). Achten Sie darauf, die Probe in die richtige Richtung zu schneiden, damit die Probe immer noch repräsentativ für die interessierenden Organstrukturen ist.
HINWEIS: Bei diesem Protokoll sind 15-50 mg die optimale Probengröße für die Kernextraktion. Um nach der Aufreinigung eine gute Ausbeute zu erzielen, wird eine Probengröße von mindestens 25 mg empfohlen. Für kleinere Proben stehen spezielle Kartuschen zur Verfügung, die für die Verarbeitung kleiner Eingaben mit dem Roboterdissoziator optimiert sind. Kleine Eingangskern-Isolationskartuschen wurden verwendet, um Gewebeproben mit einem Gewicht von nur 4 mg mit ausreichender Ausbeute für die nachgeschaltete Einzelkern-RNA-Sequenzierung zu dissoziieren. Ein Beispiel für die Ausbeute von Zellkernen unter Verwendung der Low-Input-Kartusche aus Rattenlebergewebe ist in Tabelle 3 dargestellt. - Nehmen Sie die Kernisolationskartusche aus dem Kühlschrank, packen Sie sie aus, nehmen Sie die Mühle heraus und pipettieren Sie 15 μl RNase-Inhibitor (40 U/μl) auf den Boden der Kartusche.
HINWEIS: Während der Kernextraktion fügt der Roboterdissoziator der Kartusche NIB und NSR zu einem Gesamtvolumen von 3 ml hinzu (mit dem Protokoll zur Extraktion von Kernen mit geringem Volumen). Durch Zugabe von 15 μl RNase-Inhibitor (40 U/μl) zur Kartusche vor der Extraktion weist die Suspension die gewünschte RNAse-Inhibitorkonzentration von 0,2 U/μl auf. - Legen Sie die Gewebeprobe mit einer Pinzette auf den Boden der Kartusche. Platzieren Sie die Probe nicht genau in der Mitte der Kartusche, um eine optimale Aufbrucheffizienz zu erzielen.
- Wählen Sie Protokoll ausführen auf dem Gerät und klicken Sie auf die Option Kerne in der oberen linken Ecke.
- Wählen Sie im Menü das Protokoll Low Volume Nuclei Isolation aus und überprüfen Sie, ob die Unterbrechungsgeschwindigkeit auf schnell eingestellt ist, indem Sie auf Ändern klicken. Legen Sie den Tonabnehmer in das Instrument, indem Sie die Klappe öffnen und durch Anheben des roten Knopfes aus dem Tisch schieben.
- Setzen Sie die Kassette an der dafür vorgesehenen Stelle ein, drehen Sie die Kartuschenverriegelung und schieben Sie den Tisch ein, bis der rote Knopf einrastet. Schließen Sie die Tür und starten Sie die Kernextraktion auf dem Gerät, indem Sie auf Weiter klicken. Der Lauf dauert ca. 7 Minuten.
- Sobald der Lauf beendet ist, entfernen Sie den Tonabnehmer aus dem Instrument, indem Sie den roten Knopf anheben und den Tisch herausziehen. Legen Sie die Kartusche sofort auf Eis.
- Fahren Sie für alle Gewebe mit Ausnahme des Gehirns mit Schritt 3.1 fort. Für Gehirnproben fahren Sie direkt mit Schritt 3.2 fort.
3. Reinigung der Kerne
- Saccharose-Gradienten-Bereinigung
HINWEIS: Überspringen Sie diesen Schritt für Hirngewebe und fahren Sie direkt mit Schritt 3.2 fort. Alle Reinigungsschritte werden auf Eis durchgeführt, um den RNA-Abbau zu minimieren. Puffer und Röhrchen sowie die Zentrifugen müssen vorgekühlt werden. Alle Resuspensions- und Mischschritte werden nur durch sorgfältiges Pipettieren durchgeführt, da Wirbel die Qualität und Integrität der Kerne beeinträchtigen könnten.- Stechen Sie die runde Folie vorsichtig mit einer Pipettenspitze in die Dissoziatorkartusche.
HINWEIS: Nach der Dissoziation hat die resultierende Kernsuspension ein ungefähres Volumen von 2 ml. Um die Reinigung des Saccharosegradienten zu erleichtern, teilen Sie die Zellkernsuspension in zwei 900-μl-Aliquote, was zu einem Gesamtvolumen von 1,8 ml Kernsuspensionen führt, die während der Reinigung verwendet werden. - Das erste 900-μl-Aliquot der Kernsuspension wird aus der Kartusche entnommen und in ein 500-μl-SCS-Aliquot gegeben, das zuvor in einem 2-ml-Röhrchen zubereitet wurde. Durch Pipettieren gut mischen, bis die Mischung homogen ist.
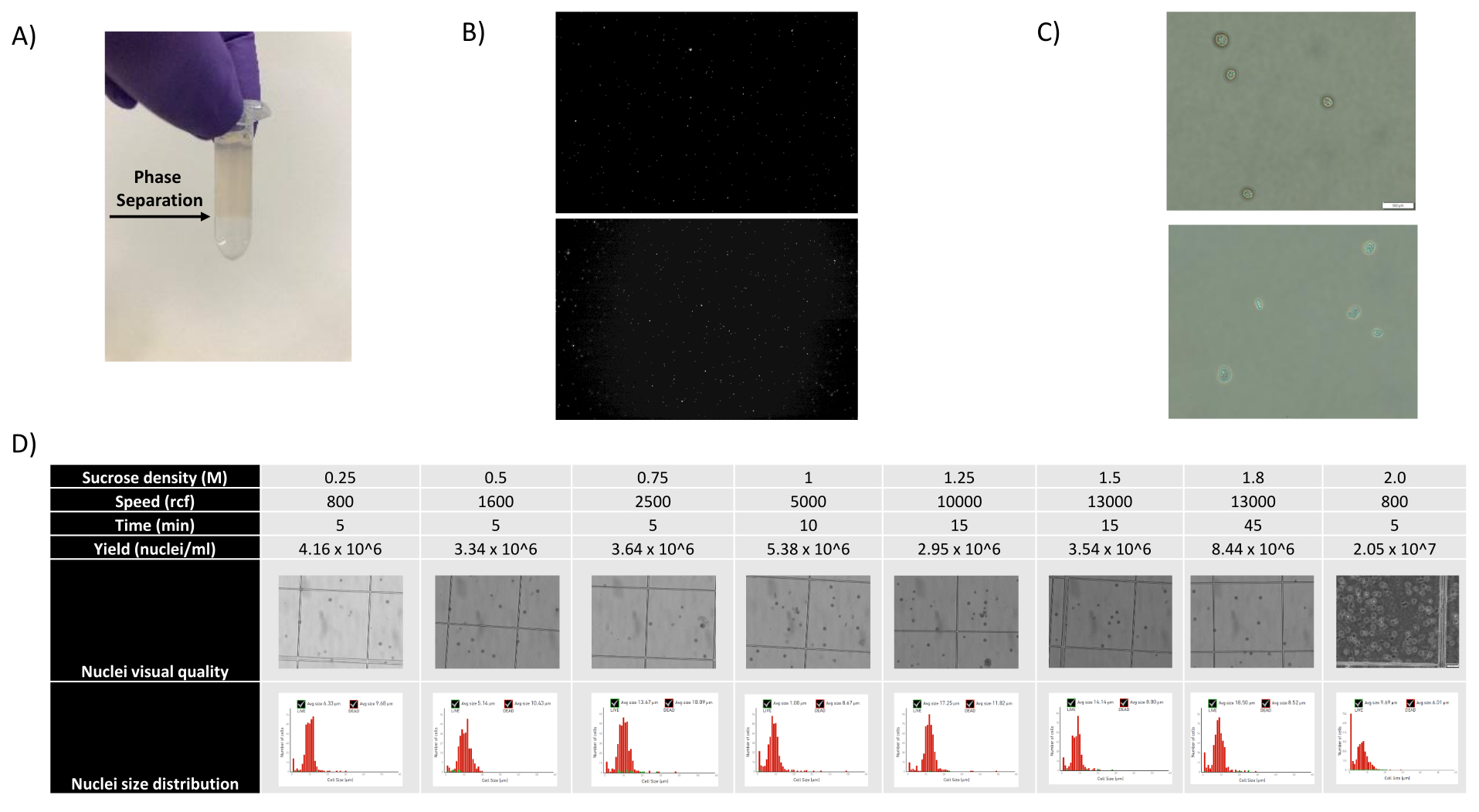
- Entfernen Sie das 1400-μl-Kernsuspensions-SCS-Gemisch und schichten Sie es vorsichtig auf ein neues 500-μl-SCS-Aliquot, indem Sie das Röhrchen schräg halten und das Gemisch tropfenweise hinzufügen, wodurch eine deutlich sichtbare Phasentrennung entsteht (siehe Abbildung 1A).
- Verschließen Sie das Röhrchen vorsichtig und legen Sie es wieder auf Eis, ohne die Phasentrennung zu stören.
- Wiederholen Sie die Schritte 3.1.2-3.1.4 mit dem zweiten 900-μl-Aliquot der Suspension und einem neuen SCS-Aliquot, um zwei 2-ml-Röhrchen pro Probe mit einer deutlich sichtbaren Phasentrennung für die Gradientenzentrifugation zu erhalten.
- Die Röhrchen vorsichtig in eine vorgekühlte Zentrifuge geben und bei 13.000 x g 15 min bei 4 °C schleudern.
- In der Zwischenzeit wird die in Tabelle 4 beschriebene NSR hergestellt, indem der RNase-Inhibitor zu einem Aliquot der NSR hinzugefügt wird. Bereiten Sie 1 ml NSR pro Probe vor.
HINWEIS: Zu diesem Zeitpunkt können die Einzelzell-Genexpressions-Reagenzgelkügelchen aus dem -80 °C-Gefrierschrank entnommen werden, so dass sie sich auf Raumtemperatur (RT) ausgleichen können, und das Template-Switch-Oligo kann in einem Puffer mit niedrigem TE-Wert resuspendiert werden. - Entfernen Sie nach der Zentrifugation den Überstand aus beiden Röhrchen, ohne das Pellet zu stören, und resuspendieren Sie das Pellet vorsichtig in 50 μl eiskaltem NSR gemäß der Empfehlung des Herstellers. Fassen Sie die beiden Pellets derselben Probe in einem neuen 1,5-ml-Röhrchen zusammen und fügen Sie 900 μl eiskaltes NSR zu einem Gesamtvolumen von 1 ml hinzu. Durch Pipettieren gut mischen.
- Die Probe wird bei 500 x g für 5 min bei 4 °C mit einer Rotorzentrifuge zentrifugiert.
HINWEIS: Es wird dringend empfohlen, einen Rotor mit schwingender Schaufel zu verwenden, um den Zellkernverlust zu minimieren, insbesondere wenn eine geringe Kernausbeute erwartet wird oder wenn mit kleinen Gewebemengen begonnen wird. - In der Zwischenzeit werden 500 μl pro Probe von 1x PBS (ohne Ca2+ und Mg2+) mit 0,04 % Rinderserumalbumin (BSA) und 0,2 U/μl RNase-Inhibitor hergestellt, wie in Tabelle 5 beschrieben.
- Entfernen Sie den Überstand vorsichtig, ohne das Pellet zu verwerfen, und resuspendieren Sie das Pellet in 100 μl PBS-Lösung, wie oben vorbereitet (1x PBS + 0,04% BSA + RNase-Inhibitor bei 0,2 U/μl).
HINWEIS: Bei kleinen Gewebeproben kann die Kernkonzentration niedrig sein, daher wird empfohlen, das Pellet in nur 50 μl PBS-Lösung zu resuspendieren, um ausreichend hohe Konzentrationen für die RNA-Sequenzierung einzelner Kerne zu gewährleisten. - Fahren Sie direkt mit Schritt 4 fort.
- Stechen Sie die runde Folie vorsichtig mit einer Pipettenspitze in die Dissoziatorkartusche.
- Reinigung von Silica-Kolloid-Gradienten
HINWEIS: Für Hirngewebe ist ein Silica-Kolloid-Gradient besser geeignet als ein Saccharose-Gradient, um Myelin und Trümmer aus der Kernsuspension zu entfernen. Alle Reinigungsschritte werden auf Eis durchgeführt, um den RNA-Abbau zu minimieren. Puffer und Röhrchen sowie die Zentrifugen müssen vorgekühlt werden. Alle Resuspensions- und Mischschritte werden nur durch sorgfältiges Pipettieren durchgeführt, da Wirbel die Qualität und Integrität der Kerne beeinträchtigen könnten.- Stechen Sie die runde Folie vorsichtig mit einer Pipettenspitze in die Dissoziatorkartusche.
- Entfernen Sie die Kernsuspension aus der Kartusche und geben Sie sie in ein 5-ml-Röhrchen.
- Bei 500 x g für 5 min bei 4 °C in einer vorgekühlten Zentrifuge zentrifugieren.
- Entfernen Sie den Überstand vorsichtig, ohne das Pellet zu stören, und resuspendieren Sie das Pellet in 1 ml eiskalter 18%iger Kieselsäure-Kolloidlösung.
- Fügen Sie weitere 2 ml 18%ige Siliciumdioxid-Kolloidlösung hinzu, um ein Gesamtvolumen von 3 ml zu erhalten, und mischen Sie es durch Pipettieren gut.
- Die Probe wird bei 700 x g für 5 min bei 4 °C in einem Rotor mit ausgeschalteter Bremse zentrifugiert.
- In der Zwischenzeit wird die in Tabelle 4 beschriebene NSR durch Zugabe des RNase-Inhibitors zu einem Aliquot der NSR hergestellt. Bereiten Sie 1 ml NSR pro Probe vor.
HINWEIS: Zu diesem Zeitpunkt können die Einzelzell-Genexpressions-Reagenzgelkügelchen aus dem -80 °C-Gefrierschrank entnommen werden, so dass sie sich zu RT äquilibrieren können und das Template-Switch-Oligo in einem niedrigen TE-Puffer resuspendiert werden kann. - Entnehmen Sie die Probe vorsichtig aus der Zentrifuge, ohne die darüber schwimmende Myelinschicht zu stören.
- Entfernen Sie zunächst die Myelinschicht von der Oberseite und entsorgen Sie sie. Entfernen Sie dann vorsichtig den gesamten Überstand, ohne das Pellet zu stören.
HINWEIS: Die Myelinschicht kann leicht entfernt werden, indem ein steriles, fusselfreies Tuch um eine 1-ml-Pipettenspitze gewickelt wird, um die Myelinschicht zusammen mit 1-2 ml des Überstands abzusaugen. - Resuspendieren Sie das Pellet in 1 ml eiskaltem NSR gemäß der Empfehlung des Herstellers.
- Die Probe wird bei 500 x g für 5 min bei 4 °C in einer Zentrifuge mit einem Ausschwingrotor zentrifugiert.
HINWEIS: Es wird dringend empfohlen, einen Rotor mit schwingender Schaufel zu verwenden, um den Zellkernverlust zu minimieren, insbesondere wenn eine geringe Kernausbeute erwartet wird oder wenn mit kleinen Gewebemengen begonnen wird. - In der Zwischenzeit werden 500 μl pro Probe von 1x PBS (ohne Ca2+ und Mg2+) mit 0,04 % Rinderserumalbumin (BSA) und 0,2 U/μl RNase-Inhibitor hergestellt, wie in Tabelle 5 beschrieben.
- Entfernen Sie den Überstand vorsichtig, ohne das Pellet zu stören, und resuspendieren Sie die Probe in 100 μl PBS-Lösung, wie oben zubereitet (1x PBS + 0,04% BSA + RNase-Inhibitor bei 0,2 U/μl).
HINWEIS: Bei kleinen Gewebeproben kann die Kernkonzentration niedrig sein, daher wird empfohlen, das Pellet in nur 50 μl PBS-Lösung zu resuspendieren, um ausreichend hohe Konzentrationen für die RNA-Sequenzierung einzelner Kerne zu gewährleisten.
4. Zählen
- Für jede zu zählende Probe werden 10 μl der Kernsuspension in 20 μl PBS-Lösung verdünnt, um eine Verdünnung von 1:3 zu erhalten.
- Zum Zählen 25 μl Propidiumiodid (PI)-Färbelösung in eine Mischvertiefung der fluoreszierenden Zählplatte geben. 25 μl der verdünnten Kernsuspension zugeben und durch Pipettieren gut mischen. Die 50 μl gefärbte Probe aus der Mischvertiefung in die Beschickungsvertiefung überführen.
- Legen Sie die Zählplatte auf den Zellenzähler und starten Sie die Zählung.
HINWEIS: Die Anzahl der Kerne wird aus dem roten Fluoreszenzkanal mit einer Belichtungszeit von 700 ms entnommen. Dieser Kanal wurde optimiert, um eine genaue Kernzählung zu erhalten, indem er mit der manuellen Zählung mit einer Neubauer-Kammer und der Trypanblau-Färbung unter dem Mikroskop verglichen wurde. Bei hohen Konzentrationen liegen die Kerne sehr dicht beieinander, was es für die Software schwierig macht, sie zu trennen. In diesem Fall wird empfohlen, die Probe in einer geeigneten Verdünnung nachzuzählen. Die Unversehrtheit der Zellkerne sowie die Sauberkeit können anhand des Hellfeldbildes oder unter dem Mikroskop beurteilt werden. - Verdünnen Sie die Proben mit PBS (1x PBS + 0,04% BSA + RNase-Inhibitor bei 0,2 U/μL) auf die gewünschte Konzentration für die Einzelkern-RNA-Sequenzierung.
HINWEIS: Konzentrationen zwischen 700 und 1.200 Kernen/μl gelten als optimal für die RNA-Sequenzierung einzelner Kerne. Niedrigere Zellkonzentrationen, wie z. B. 700 Zellkerne/μl, können zu einer geringeren Hintergrundkontamination durch Umgebungs-RNA führen.
5. Vorbereitung der Bibliothek
- Führen Sie die RNA-Sequenzierung einzelner Kerne mit den Einzelzell-Genexpressionsreagenzien unter Verwendung des Herstellerprotokolls durch, das auf eine Zellkernrückgewinnung von 8000-10.000 Kernen pro Probe abzielt.
6. Sequenzierung
- Sequenzieren Sie die Bibliotheken mit der gewünschten Sequenzierungstiefe mit Paired-End, Dual-Indexierung und den folgenden Sequenzierungsreads: Lesen 1: 28 Zyklen, i7 Index: 10 Zyklen, i5 Index: 10 Zyklen und Lesen 2: 90 Zyklen.
Repräsentative Ergebnisse
Die Leistungsfähigkeit und Vielseitigkeit dieses Protokolls wird durch die Durchführung von Einzelkern-RNA-Sequenzierungen an frischem, gefrorenem Hinterhauptsrindengewebe des Gehirns von drei B6-Mäusen, frischem, gefrorenem, quer geschnittenem Nierengewebe von drei Wistar-Ratten, archiviertem (11-jährigem) Leber- und Milzgewebe von drei Mauritius-Cynomolgus-Makaken demonstriert. Alle Tiere wurden nicht durchblutet.
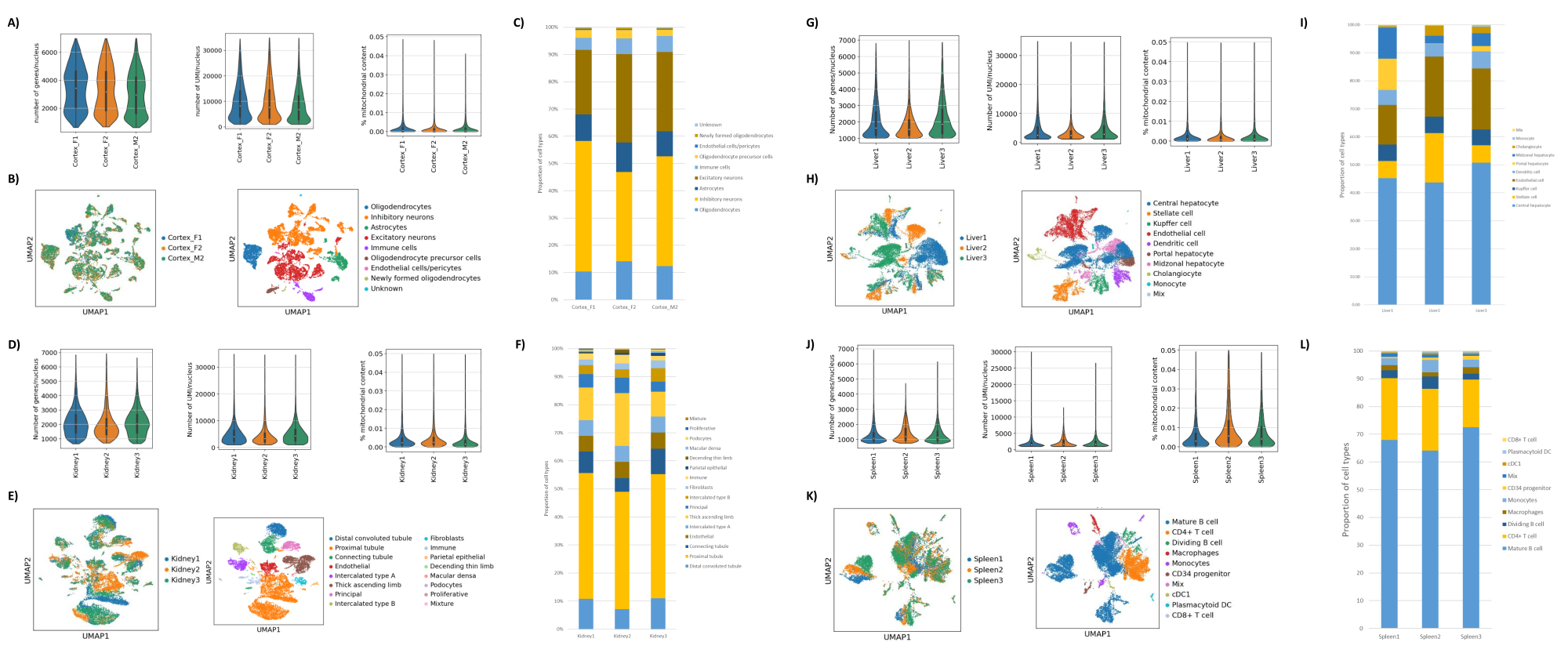
Wie in den Abbildungen 1B und C gezeigt, wurden Kerne von guter Qualität erhalten, die frei von Anzeichen von Blasenbildung, Trümmern und Verklumpung waren. Die auf Saccharosegradienten basierende Filtration wurde optimiert, um den Großteil der Ablagerungen zu entfernen, indem verschiedene Dichten, Schleudergeschwindigkeiten und -zeiten getestet und die nukleare Reinheit/Integrität unter einem Mikroskop sowie die Größenverteilung und Ausbeute der Kerne beurteilt wurden (Abbildung 1D). Dies ermöglichte es uns, eine Saccharosegradientendichte von 1,5 M zu wählen und eine kurze Schleuderzeit von 15 min zu verwenden. Um die Qualität der Kerne weiter zu beurteilen, wurden die Daten mit 10X Cell Ranger vorverarbeitet und eine weitere nachgelagerte Datenanalyse mit Besca23 durchgeführt. Zellkerne mit >5% Prozent Mitochondriengehalt (da diese dazu neigen, beschädigte/gestresste Kerne zu sein) wurden herausgefiltert, und Kerne mit 500-7.000 Genen (zur Minimierung leerer Tröpfchen und Multipletts) wurden zurückbehalten. Wir schlossen nur Gene ein, die in mindestens 30 Zellkernen vorhanden waren. Wir zielten auf 8.000 Kerne pro Hirnrindenprobe und 10.000 Kerne pro Nieren-, Leber- und Milzprobe ab. Nach der Filtration wurden 10.644 hochwertige Kerne aus den drei Hirnproben, 14.960 hochwertige Kerne aus den drei Nierenproben, 18.795 hochwertige Kerne aus den drei Leberproben und 13.882 hochwertige Kerne aus den drei Milzproben gewonnen. Die Abbildungen 2A, D, G, J zeigen Geigendiagramme, die die Verteilung der UMI-Zahlen, der Genzahlen und des Mitochondriengehalts in jeder Probe darstellen. Die mediane Anzahl der Zählungen über alle Hirnproben hinweg betrug 7.563 UMI/Zellkern und 3.208 Gene/Zellkern. Die mediane Anzahl der Zählungen über alle Nierenproben betrug 3.841 UMI/Zellkern und 1.915 Gene/Zellkern. Die mediane Anzahl der Zählungen über alle Leberproben betrug 2.649 UMI/Zellkerne und 1.676 Gene/Zellkerne. Die mediane Anzahl der Zählungen über alle Milzproben betrug 1.609 UMI/Zellkerne und 1.138 Gene/Kerne. Anschließend generierten wir Cluster mit hochvariablen Genen und annotierten sie mit bekannten Markergenen 17,24,25,26. Wie in Abbildung 2B, E, H, K zu sehen ist, konnten wir die erwarteten Zelltypen aus jedem Gewebe identifizieren. Wie in Abbildung 2B, E, H, K zu sehen ist, trugen alle Tiere zu allen Clustern bei, was auf eine insgesamt geringe technische Variabilität hindeutet, die durch das Protokoll eingeführt wurde. Darüber hinaus waren die zellulären Anteile in allen drei Proben pro Gewebetyp vergleichbar, ebenso wie die UMI und die Genzahlen (Abbildung 2A,C,D,F,G,I,J,L). Eine bemerkenswerte Ausnahme ist die Leber, wo sich die Hepatozytenpopulationen unter den drei Leberproben in Anteilen und Profil unterschieden. Dies ist höchstwahrscheinlich auf biologische Unterschiede zwischen den Tieren zurückzuführen (Geschlecht, Alter, Stoffwechselstatus).

Abbildung 1: Beurteilung der Kernqualität und Saccharosegradientenoptimierung. (A) Die erwartete Phasentrennung während der Saccharosegradientenzentrifugation ist mit einem Pfeil dargestellt. (B) Repräsentative Fluoreszenzbilder von Propidiumiodid-gefärbten Nukleen der Rattenniere (oben) und Cynomolgus-Milz (unten), die mit dem Protokoll aufgenommen wurden. (C) Repräsentative hellfeldmikroskopische Aufnahmen von Zellkernen, die aus der Leber der Maus (oben) und dem Mäusegehirn (unten) isoliert wurden, Maßstab 500 μm. Man beachte die regelmäßig glatte Oberfläche der Kerne, die auf eine gute Kernqualität hinweist. (D) Saccharose-Gradienten-Optimierung. Es wurden verschiedene Saccharosedichten, Schleudergeschwindigkeiten und Schleuderzeiten getestet. Hellfeldmikroskopische Bilder von Kernen, Kerngrößenverteilung und Kernausbeute werden für jede Bedingung gezeigt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Repräsentative Daten von snRNAseq auf dem okzipitalen Kortex des Mäusegehirns, der Rattenniere (Kortex und Medulla) und der Leber und Milz des Cynomolgus-Makaken. (A) Violindiagramme, die die Verteilung von Genen/Zellkern, UMIs/Zellkern und den prozentualen Mitochondriengehalt pro Gehirnprobe zeigen. (B) Linkes Bild: UMAP-Diagramm, das den Beitrag jeder Probe zu den im Gehirn identifizierten Clustern zeigt. Rechtes Bild: UMAP zeigt die Identitäten der Cluster, die auf der Grundlage von Markergenen im Hirngewebe annotiert sind. (C) Zelluläre Anteile, die in den 3 Gehirnproben beobachtet wurden. (D) Violindiagramme, die die Verteilung von Genen/Zellkern, UMIs/Zellkern und den prozentualen Mitochondriengehalt pro Nierenprobe zeigen. (E) Linkes Bild: UMAP-Diagramm, das den Beitrag jeder Probe zu den in der Niere identifizierten Clustern zeigt. Rechtes Bild: UMAP zeigt die Identitäten der Cluster, die auf der Grundlage von Markergenen im Nierengewebe annotiert sind. (F) Zelluläre Anteile, die in den 3 Nierenproben beobachtet wurden. (G) Violindiagramme, die die Verteilung von Genen/Zellkern, UMIs/Zellkern und den prozentualen Mitochondriengehalt pro Leberprobe zeigen. (H) Linkes Bild: UMAP-Diagramm, das den Beitrag jeder Probe zu den in der Leber identifizierten Clustern zeigt. Rechtes Bild: UMAP zeigt die Identitäten der Cluster, die auf der Grundlage von Markergenen im Lebergewebe annotiert sind. (I) Zelluläre Anteile, die in den 3 Leberproben beobachtet wurden. (J) Violindiagramme, die die Verteilung von Genen/Zellkern, UMIs/Zellkern und den prozentualen Mitochondriengehalt pro Milzprobe zeigen. (K) Linkes Bild: UMAP-Diagramm, das den Beitrag jeder Probe zu den in der Milz identifizierten Clustern zeigt. Rechts im Bild: UMAP zeigt die Identitäten der Cluster, die auf der Grundlage von Markergenen im Milzgewebe annotiert sind. (L) Zelluläre Anteile, die in den 3 Milzproben beobachtet wurden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
| Komponenten | Bestandskonzentration | Volumen pro Probe | Endkonzentration |
| Saccharose-Kissen-Lösung | 2 Mio | 1500 μl | 1,5 Mio. |
| Saccharose-Kissen-Puffer | - | 500 μl | - |
| Dithiothreitol (DTT) | 1 Mio. | 2 μl | 1 mM |
| RNASE-Inhibitor | 40 U/μL | 10 μl | 0,2 U/μL |
Tabelle 1: Herstellung von 1,5 M Saccharosekissenlösung (SCS). Diese Lösung wird für die Saccharose-Gradientenzentrifugation während der Reinigung in Schritt 3.1 verwendet und sollte jedes Mal frisch zubereitet werden, bevor das Protokoll gestartet wird. Halten Sie das SCS während des Protokolls immer auf Eis. Die in dieser Tabelle erwähnten Lösungen sind in der Materialtabelle aufgeführt.
| Komponenten | Bestandskonzentration | Volumen pro Probe | Endkonzentration |
| Silica-Kolloid-Stammlösung | 90% | 600 μl | 18% |
| Reagenz zur Lagerung von Zellkernen (S2-Genomik) | - | 2400 μl | - |
| RNASE-Inhibitor | 40 U/μL | 15 μl | 0,2 U/μL |
Tabelle 2: Herstellung einer 18%igen Siliciumdioxid-Kolloidlösung. Diese Lösung wird für die Siliciumdioxid-Kolloid-Gradientenzentrifugation während der Reinigung in Schritt 3.2 verwendet und sollte jedes Mal frisch zubereitet werden, bevor mit dem Protokoll begonnen wird. Bewahren Sie die 18%ige Silica-Kolloidlösung während des Protokolls immer auf Eis auf.
| Gewebe | Probengewicht | Patrone | Ertrag |
| Rattenleber | 25 mg | Kern-Isolationskartusche | 65.000 Zellkerne pro mg Gewebe |
| Rattenleber | 4 mg | Isolationskassette für kleine Eingangskerne | 32.000 Zellkerne pro mg Gewebe |
Tabelle 3: Kernausbeute aus der Nuklei-Isolationskartusche mit niedrigem Input im Vergleich zur Kern-Isolationskartusche nach der Saccharose-Gradienten-Reinigung.
| Komponenten | Bestandskonzentration | Volumen pro Probe | Endkonzentration |
| Reagenz für die Lagerung von Kernen | - | 1000 μl | - |
| RNASE-Inhibitor | 40 U/μL | 5 μl | 0,2 U/μL |
Tabelle 4: Herstellung von Zellkern-Speicherreagenz (NSR). Diese Lösung wird während der Kernisolierung in den Schritten 3-5 sowie während der Bereinigung in Schritt 3.1.8 verwendet. Es kann bei 4 °C bis zu 4 Monate gelagert werden. Bereiten Sie ein frisches Aliquot mit RNase-Inhibitor während des Zentrifugationsschritts in der Aufreinigungsstufe 6 vor. Die in dieser Tabelle erwähnten Lösungen sind in der Materialtabelle aufgeführt.
| 1x PBS + 0,04% BSA-Stammlösung | |||
| Komponenten | Bestandskonzentration | Volumen für Lagerbestand | Endkonzentration |
| PBS (kein Ca 2+, kein Mg2+) | 1-fach | 30 ml | - |
| Rinderserumalbumin (BSA) | 30% | 40 μl | 0.04% |
| 1x PBS + 0,04% BSA + 0,2 U/μL RNAse-Inhibitor | |||
| Komponenten | Bestandskonzentration | Volumen pro Probe | Endkonzentration |
| 1x PBS + 0,04% BSA Aktienlösung | - | 500 μl | - |
| RNASE-Inhibitor | 40 U/μL | 2,5 μl | 0,2 U/μL |
Tabelle 5: Herstellung von PBS + 0,04% BSA. Diese Lösung wird am Ende der Aufreinigung in Schritt 3.1.10 und nach der Zählung verwendet, um die Kernsuspension auf die gewünschte Konzentration für die 10-fache Einzelkern-RNA-Sequenzierung zu verdünnen (Zählschritt 4.4). Die Stammlösung kann bei 4 °C bis zu 1 Monat gelagert werden. Bereiten Sie ein frisches Aliquot mit RNase-Inhibitor während des Zentrifugationsschritts in der Aufreinigungsstufe 6 vor.
Diskussion
Wir haben ein vielseitiges und teilautomatisiertes Protokoll entwickelt, um qualitativ hochwertige Einzelkerne aus gefrorenem Säugetiergewebe zu gewinnen, und haben das Protokoll an Mäusegehirn, Rattennieren und Cynomolgus-Leber- und Milzgewebe demonstriert.
Wenn wir die Leistung dieses Protokolls mit der anderer veröffentlichter Protokolle für die Einzelkern-RNA-Sequenzierung in Gehirn-, Nieren-, Milz- und Lebergewebe vergleichen 6,7,20,24,25,26, stellen wir fest, dass wir in der Lage sind, eine ähnliche Anzahl von Genen und UMI-Zahlen pro Zellkern zu detektieren und die erwarteten Zelltypen zu gewinnen. Im Vergleich zu bestehenden Methoden bietet dieses Protokoll mehrere Vorteile. Erstens automatisiert das Protokoll in dieser Studie die Gewebehomogenisierung und Isolierung einzelner Kerne. Dies wird durch die Verwendung eines robotergestützten Gewebedisruptors21 erreicht. In den meisten Protokollen wird das Gewebe mit einem Dounce-Homogenisator homogenisiert, um einzelne Kerne freizusetzen 3,20. Wir haben jedoch festgestellt, dass dieser manuelle Schritt zu experimentellen Schwankungen in der Ausbeute und Integrität der Kerne führen kann, abhängig von der Höhe der Kraft, die während der Homogenisierung ausgeübt wird, was die Reproduzierbarkeit der Experimente beeinträchtigt. Hier wurden durch den Einsatz eines automatisierten Gewebeschleifers mit festen Einstellungen experimentell eine gute Kernqualität und Ausbeute mit größerer Konsistenz erzielt. Darüber hinaus reduziert die Automatisierung dieses Schritts auch die praktische Zeit des Protokolls (der Schritt des Gewebeaufschlusses dauert etwa 7 Minuten), so dass sich der Benutzer auf die nachfolgenden Schritte vorbereiten kann. Zweitens ist das in dieser Studie beschriebene Protokoll vielseitig, d.h. es ist mit verschiedenen Geweben verschiedener Spezies kompatibel. Dies ermöglicht es uns, langwierige Protokolloptimierungen zu vermeiden, z. B. zur Identifizierung von Homogenisierungspuffern/Detergenzien für verschiedene Gewebe 2,5,6. Drittens ist dieses Protokoll nicht auf den Zugang zu einem Durchflusssortierer angewiesen, um saubere Kerne zu erhalten, wodurch es für Laboratorien, die nicht über die erforderliche Ausrüstung/das erforderliche Fachwissen für die Durchflusssortierung verfügen, leichter zugänglich ist. Stattdessen haben wir die auf Saccharose basierende Gradientenfiltration optimiert, um den größten Teil des Schmutzes zu entfernen. Insbesondere für Hirngewebe wird jedoch die Verwendung eines Siliciumdioxid-Kolloid-Gradienten anstelle eines Saccharose-Gradienten empfohlen, um die Myelinentfernung effizienter zu gestalten. Wir haben auch festgestellt, dass die Verwendung eines Rotors mit schwingender Schaufel am Ende des Gradientenzentrifugationsschritts der Saccharose/Siliciumdioxid-Kolloid-Gradientenzentrifugation den Kernverlust minimiert. Daher ist der Einsatz eines solchen Rotors sehr zu empfehlen. Viertens wird nach dem Testen mehrerer Verfahren zum Zählen von Kernen (manuelles Zählen unter dem Mikroskop, Verwendung mehrerer automatisierter Zähler) die Verwendung eines automatisierten Fluoreszenzzellenzählers22 empfohlen. Die Verwendung eines DNA-interkalierenden Farbstoffs, wie z. B. Propidiumiodid, erhöht die Genauigkeit der Zellkernzählung. Fünftens dauert dieses Protokoll etwa 75 Minuten vom Start bis zum Laden des Mikrofluidik-Chips. Dadurch wird sichergestellt, dass die Integrität der Kerne bei der Verarbeitung mehrerer Proben hoch bleibt. Schließlich haben wir festgestellt, dass das Protokoll auch mit OCT-eingebettetem Gewebe (Optimal Cutting Temperature Compound) kompatibel ist. Bei Verwendung eines solchen Materials kann das Gewebe vor der Homogenisierung mit einem Skalpell aus dem OCT-Block entfernt werden.
Eine häufige Herausforderung bei Einzelkern-RNA-Sequenzierungsdatensätzen ist das Vorhandensein von Umgebungs-RNA, die sowohl nicht-nukleär (z. B. mitochondrial) als auch nukleär sein kann27,28. In unserem Protokoll ist die mitochondriale RNA (ein Proxy für nicht-nukleäre Umgebungs-RNA) bereits vor der Filterung niedrig (0,1-1,6 % für die gezeigten Gewebe). Ähnlich wie bei anderen Protokollen und Datensätzen ist die RNA-Kontamination durch hochexprimierte Gene in den Zellkernen zahlreicher Zelltypen (z. B. Hepatozyten in der Leber, Neuronen im Gehirn usw.) jedoch immer noch vorhanden27. Es gibt mehrere bioinformatische Werkzeuge, wie z. B. CellBender, SoupX usw., die eine solche RNA-Kontamination vor der Kernannotation entfernen können 29,30,31. Eine weitere Einschränkung dieses Protokolls besteht darin, dass der Durchsatz dieses Schritts immer noch begrenzt ist, obwohl der Gewebeaufschluss und die Zellkernisolierung automatisiert sind, da jeweils nur eine Probe verarbeitet werden kann. Da dieser Schritt jedoch nur ca. 7 min pro Stück Gewebe dauert, können immer noch mehrere Proben in einem Batch verarbeitet werden. Wir verarbeiten in der Regel vier Proben pro Charge, haben aber auch bis zu sechs Proben pro Charge mit guten Ergebnissen durchgeführt. Jüngste Verbesserungen des Roboterdissoziators, der die parallele Verarbeitung von zwei Proben gleichzeitig ermöglicht, ermöglichen die Verarbeitung von 8-12 Proben pro Charge, was mit dem Durchsatz des Mikrofluidik-Chips kompatibel ist, der für die Verkapselung einzelner Kerne verwendet wird.
Obwohl wir die durch dieses Protokoll isolierten Kerne nicht für andere Downstream-Anwendungen wie ATAC-seq oder snRNAseq auf anderen Plattformen verwendet haben, glauben wir, dass unser Protokoll aufgrund der Qualität der Daten, die mit den hier verwendeten Genexpressionsreagenzien gewonnen wurden, mit weiteren Downstream-Anwendungen kompatibel sein sollte. Zukünftige Arbeiten werden jedoch beinhalten, dieses Protokoll mit anderen nachgelagerten Anwendungen wie ATAC-seq zu testen.
Zusammenfassend lässt sich sagen, dass wir ein schnelles, einfaches und teilautomatisiertes Kernisolationsprotokoll für die nachgeschaltete Einzelkern-RNA-Sequenzierung entwickelt haben, das sich als kompatibel mit verschiedenen Arten von gefrorenem Säugetiergewebe erwiesen hat.
Offenlegungen
Alle Autoren sind/waren während der Durchführung der Studie Mitarbeiter von F. Hoffmann-La Roche.
Danksagungen
Die Autoren danken Filip Bochner, Marion Richardson, Petra Staeuble und Matthias Selhausen für die Bereitstellung der tierischen Gewebe, die in diesem Manuskript analysiert wurden. Wir danken auch Petra Schwalie, Klas Hatje, Roland Schmucki und Martin Ebeling für ihre bioinformatische Unterstützung.
Materialien
| Name | Company | Catalog Number | Comments |
| 1 M DTT | Thermo Fisher Scientific | P2325 | |
| 10% Tween 20 | Bio-Rad | 1662404 | |
| 10x Magnetic Separator | 10x genomics | PN-120250 | |
| 10x Vortex Adapter | 10x genomics | PN-120251 | |
| 1x DPBS (10x), no calcium, no magnesium | Thermo Fisher Scientific | 14190144 | stored at 4°C |
| 30% Bovine Serum Albumin | Sigma-Aldrich | A9576_50ML | |
| 400 mM Tris-HCl, pH 8.0 | Thermo Fisher Scientific | 15568025 | |
| 40U/μl RNaseOUT Recombinant Ribonuclease Inhibitor | Thermo Fisher Scientific | 10777019 | Stored at -20 °C |
| Agilent High Sensitivity DNA Kit | Agilent | 5067-4626 | |
| Cellaca MX High-throughput Automated Cell Counter | Nexcelom Bioscience | CELMXSYSF2 | Automated fluorescent cell counter |
| Chromium Next GEM Chip G Single Cell Kit, 16 rxns | 10x genomics | PN-1000127 | Single cell gene expression reagent, stored at room temperature |
| Chromium Next GEM Secondary Holder | 10x genomics | PN-1000195 | |
| Chromium Next GEM Single Cell 3' Gel Bead Kit v3.1, 4 rxns | 10x genomics | PN-1000129 | Single cell gene expression reagent, stored at -80 °C |
| Chromium Next GEM Single Cell 3' GEM, Library & Gel Bead Kit v3.1, 4 rxns | 10x genomics | PN-1000128 | Single cell gene expression reagent |
| Chromium Next GEM Single Cell 3' Library Kit v3.1 4 rxns | 10x genomics | PN-1000158 | Single cell gene expression reagent, stored at -20 °C |
| Chromium Next GEM Single Cell 3'GEM Kit v3.1 4 rxns | 10x genomics | PN-1000130 | Single cell gene expression reagent, stored at -20 °C |
| Divided Polystyrene Reservoirs | VWR | 41428-958 | |
| DNA LoBind Tubes 1.5ml Eppendorf | Sigma-Aldrich | EP0030108051 | |
| DNA LoBind Tubes 2ml Eppendorf | Sigma-Aldrich | EP0030108078 | |
| Dry ice | - | - | |
| Dynabeads MyOne SILANE | 10x genomics | PN-2000048 | Single cell gene expression reagent, stored at 4 °C |
| Ethanol Pure | Sigma-Aldrich | E7023 | |
| Glycerin (Glycerol), 50% (v/v) | Ricca Chemical Company | 3290-16 | |
| Heatblock | |||
| High-Throughput Nexcelom Counting Plates | Nexcelom Bioscience | CHM24-A100-001 | Cell counter counting plate |
| Low TE Buffer (10 mM Tris-HCl pH 8.0, 0.1 mM EDTA) | Thermo Fisher Scientific | 12090015 | |
| Mini Centrifuge | - | - | |
| NovaSeq 6000 SP Reagent Kit v1.5 (100 cycles) | Illumina | 2002840 | |
| Nuclei Isolation Buffer | S2 Genomics | 100-063-396 | Stored at 4 °C |
| Nuclei Isolation Cartridge | S2 Genomics | 100-063-287 | Precooled at 4 °C before use |
| Nuclei PURE 2 M Sucrose Cushion Solution | Sigma-Aldrich | NUC201-1KT | Sucrose cushion solution |
| Nuclei PURE Sucrose Cushion Buffer | Sigma-Aldrich | NUC201-1KT | |
| Nuclei Storage Reagent | S2 Genomics | 100-063-405 | Stored at 4 °C |
| PCR Tubes 0.2 ml 8-tube strips | Eppendorf | 30124359 | |
| Percoll | GE Healthcare | 17-0891-02 | Silica colloid solution |
| PhiX Control v3 | Illumina | FC-110-3001 | |
| Qiagen Buffer EB | Qiagen | 19086 | |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Q32854 | |
| Refrigerated Centrifuge (Eppendorf 5804R) | Eppendorf | 5805000010 | |
| Refrigerated Centrifuge with Swinging-Bucket Rotor (Eppendorf 5810R) | Eppendorf | 5811000015 | |
| RNAseZap | Ambion | AM9780 | RNAse decontamination solution |
| Round cell culture petri dish | SPL | 330005 | |
| Scalpel disposable | Aesculap AG | BA210 | pre-cooled on dry ice before use |
| Single Index Kit T Set A, 96 rxns | 10x genomics | PN-1000213 | Single cell gene expression reagent, stored at -20 °C |
| Singulator 100 System | S2 Genomics | - | Commercially available robotic tissue dissociator |
| Sodium Hydroxide 1M | Sigma-Aldrich | 72068 | |
| SPRIselect Reagent Kit | Beckman Coulter | b23318 | |
| Sterile tweezers | - | - | |
| UltraPure DNase/RNase-Free Distilled Water | Thermo Fisher Scientific | 10977049 | |
| ViaStain PI Staining Solution | Nexcelom Bioscience | CS1-0109-5mL | Propidium iodide staining solution |
| Vortex Mixer+A2:D44 | VWR | - |
Referenzen
- Burja, B., et al. An Optimized Tissue Dissociation Protocol for Single-Cell RNA Sequencing Analysis of Fresh and Cultured Human Skin Biopsies. Front Cell Dev Biol. 10, 872688 (2022).
- Kimbley, L. M., et al. Comparison of optimized methodologies for isolating nuclei from esophageal tissue. Biotechniques. 72 (3), 104-109 (2022).
- Maitra, M., et al. Extraction of nuclei from archived postmortem tissues for single-nucleus sequencing applications. Nature Protocols. 16 (6), 2788-2801 (2021).
- Nadelmann, E. R., et al. Isolation of nuclei from mammalian cells and tissues for single-nucleus molecular profiling. Current Protocols. 1 (5), e132 (2021).
- Rousselle, T. V., et al. An optimized protocol for single nuclei isolation from clinical biopsies for RNA-seq. Scientific Reports. 12, 9851 (2022).
- Slyper, M., et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nature Medicine. 26 (5), 792-802 (2020).
- Leiz, J., et al. Nuclei isolation from adult mouse kidney for single-nucleus RNA-sequencing. Journal of Visualized Experiments: JoVE. (175), 62901 (2021).
- Alvarez, M., et al. Isolation of nuclei from human snap-frozen liver tissue for single-nucleus RNA sequencing. Bio-Protocol. 13 (3), e4601 (2023).
- Ayhan, F., Douglas, C., Lega, B. C., Konopka, G. Nuclei isolation from surgically resected human hippocampus. STAR Protocols. 2 (4), 100844 (2021).
- Joshi, N., Misharin, A. Single-nucleus isolation from frozen human lung tissue for single-nucleus RNA-seq. Protocols.io. , (2019).
- Martelotto, L. G., Luciano Martelotto, L. 'Frankenstein' protocol for nuclei isolation from fresh and frozen tissue for snRNAseq. Protocols.io. , (2020).
- Masilionis, I., Chaudhary, O., Chaligne, R., Mazutis, L. Nuclei extraction for single-cell RNAseq from frozen tissue using Singulator™ 100. Protocols.io. , (2022).
- Matson, K. J. E., et al. Isolation of adult spinal cord nuclei for massively parallel single-nucleus RNA sequencing. Journal of Visualized Experiments: JoVE. (140), 58413 (2018).
- Mendelev, N., et al. Multi-omics profiling of single nuclei from frozen archived postmortem human pituitary tissue. STAR Protocols. 3 (2), 101446 (2022).
- Soule, T. G., et al. A protocol for single nucleus RNA-seq from frozen skeletal muscle. Life Science Alliance. 6 (5), e202201806 (2023).
- Bakken, T. E., et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS One. 13 (12), e0209648 (2018).
- Ding, J., et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nature Biotechnology. 38, 737-746 (2020).
- Hu, P., et al. Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes & Development. 32 (19-20), 1344-1357 (2018).
- Lake, B. B., et al. A comparative strategy for single-nucleus and single-cell transcriptomes confirms accuracy in predicted cell-type expression from nuclear RNA. Scientific Reports. 7, 6031 (2017).
- Narayanan, A., et al. Nuclei Isolation from Fresh Frozen Brain Tumors for Single-Nucleus RNA-seq and ATAC-seq. Journal of Visualized Experiments: JoVE. (162), 61542 (2020).
- Jovanovich, S., et al. . Automated processing of solid tissues into single cells or nuclei for genomics and cell biology applications with the Singulator™ 100 and 200 systems. , (2022).
- Bell, J., et al. Characterization of a novel high-throughput, high-speed and high-precision plate-based image cytometric cell counting method. Cell & Gene Therapy Insights. 7 (4), 427-447 (2021).
- Madler, S. C., et al. Besca, a single-cell transcriptomics analysis toolkit to accelerate translational research. NAR Genomics and Bioinformatics. 3 (4), lqab102 (2021).
- Wu, H., et al. Mapping the single-cell transcriptomic response of murine diabetic kidney disease to therapies. Cell Metabolism. 34 (7), 1064-1078 (2022).
- Han, L., et al. Cell transcriptomic atlas of the non-human primate Macaca fascicularis. Nature. 604 (7907), 723-731 (2022).
- Madissoon, E., et al. scRNA-seq assessment of the human lung, spleen, and esophagus tissue stability after cold preservation. Genome Biology. 21 (1), 1 (2019).
- Caglayan, E., Liu, Y., Konopka, G. Neuronal ambient RNA contamination causes misinterpreted and masked cell types in brain single-nuclei datasets. Neuron. 110 (24), 4043-4056 (2022).
- Luecken, M. D., Theis, F. J. Current best practices in single-cell RNA-seq analysis: a tutorial. Molecular Systems Biology. 15 (6), e8746 (2019).
- Fleming, S. J., et al. Unsupervised removal of systematic background noise from droplet-based single-cell experiments using CellBender. bioRxiv. , (2022).
- Yang, S., et al. Decontamination of ambient RNA in single-cell RNA-seq with DecontX. Genome Biology. 21 (1), 57 (2020).
- Young, M. D., Behjati, S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience. 9 (12), giaa151 (2020).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten