Mikrofluidischer Ansatz zur Auflösung der gleichzeitigen und sequentiellen Zytokinsekretion einzelner polyfunktioneller Zellen
* Diese Autoren haben gleichermaßen beigetragen
In diesem Artikel
Zusammenfassung
Das Protokoll beschreibt eine fortschrittliche mikrofluidische Plattform zur quantitativen Messung der Zytokinsekretionsdynamik einzelner mononukleärer Zellen des menschlichen peripheren Blutes. Die Plattform misst bis zu drei Zytokine parallel (IL-6, TNFα und IL-1β) für jede einzelne Zelle, die beispielsweise mit Lipopolysaccharid stimuliert wird.
Zusammenfassung
Infektionen, Autoimmunerkrankungen, erwünschte und unerwünschte immunologische Reaktionen auf die Behandlung können in vivo zu einer komplexen und dynamischen Zytokinantwort führen. An dieser Reaktion sind zahlreiche Immunzellen beteiligt, die verschiedene Zytokine ausschütten, um die Immunreaktion zu orchestrieren. Die Sekretionsdynamik, die Mengen und das gleichzeitige Auftreten der verschiedenen Zytokine durch verschiedene Zellsubtypen sind jedoch aufgrund des Mangels an geeigneten Instrumenten zu ihrer Untersuchung nach wie vor unzureichend verstanden. Hier beschreiben wir ein Protokoll unter Verwendung einer mikrofluidischen Tröpfchenplattform, das die zeitaufgelöste quantitative Messung der Sekretionsdynamik für mehrere Zytokine parallel auf Einzelzellebene ermöglicht. Dies wird ermöglicht durch die Verkapselung einzelner Zellen in mikrofluidische Tröpfchen zusammen mit einem gemultiplexten Immunoassay zur parallelen Quantifizierung von Zytokinkonzentrationen, deren Immobilisierung für die dynamische Fluoreszenzbildgebung und die Analyse der jeweiligen Bilder zur Ableitung von sezernierten Mengen und Dynamiken. Das Protokoll beschreibt die Präparation funktionalisierter magnetischer Nanopartikel, Kalibrierungsexperimente, die Zellpräparation und die Verkapselung der Zellen und Nanopartikel in Tröpfchen für die Fluoreszenzbildgebung und die anschließende Bild- und Datenanalyse am Beispiel von Lipopolysaccharid-stimulierten mononukleären Zellen des peripheren Blutes. Die vorgestellte Plattform identifizierte ein ausgeprägtes Zytokinsekretionsverhalten für einzelne und co-sezernierende Zellen und charakterisierte die erwartete phänotypische Heterogenität in der gemessenen Zellprobe. Darüber hinaus ermöglicht der modulare Charakter des Assays seine Anpassung und Anwendung zur Untersuchung einer Vielzahl von Proteinen, Zytokinen und Zellproben, was möglicherweise zu einem tieferen Verständnis des Zusammenspiels zwischen verschiedenen Immunzelltypen und der Rolle der verschiedenen dynamisch sezernierten Zytokine bei der Gestaltung der streng regulierten Immunantwort führt. Diese neuen Erkenntnisse könnten besonders interessant für die Erforschung von Immundysregulationen oder für die Identifizierung von Zielpopulationen in der Therapie- und Medikamentenentwicklung sein.
Einleitung
Infektionen verursachen oft komplexe Wirtsreaktionen, an denen das angeborene und das adaptive Immunsystem beteiligt sind 1,2. Bei einer Infektion oder Erkennung von Infektionserregern können Wirtszellen eine Vielzahl von Chemo- und Zytokinen produzieren, bei denen es sich um kleine Proteine handelt, die als kritische Kommunikatoren bekannt sind und das Immunsystem modulieren3. Entzündungsfördernde Zytokine werden früh nach der Infektion freigesetzt, um die Immunantwort einzuleiten, gefolgt von entzündungshemmenden Zytokinen, die entscheidend sind, um Gewebeschäden und nachfolgende chronische oder autoinflammatorische Erkrankungen zu verhindern. Dieses Gleichgewicht zwischen Bedrohungseliminierung und Gewebeschutz manifestiert sich in einem breiten Repertoire an Zytokinen, die während der Infektion unterschiedliche Funktionen ausüben und eine Feinabstimmung der Reaktion ermöglichen 4,5. Innerhalb dieser Mischung können einzigartige Signaturen beobachtet werden, die vom Erreger und den von ihm induzierten Signalen, der Gewebelage und den Immunzellen, von denen sie stammen, abhängen. Die Freisetzung von Zytokinen scheint jedoch auch ein multifunktionaler biologischer Prozess zu sein, der für jede Zellpopulation einzigartig ist und sich in der Sekretionsdynamik und der individuellen Reaktion unterscheidet. Diese Heterogenität wird in der Literatur seit vielen Jahren beschrieben, z. B. bei T-Zell-Subpopulationen 6,7, wo Untersuchungen zu autoinflammatorischen Erkrankungen und schweren COVID-19-Infektionen eine große funktionelle Vielfalt an Entzündungsmarkern innerhalb und zwischen Patienten zeigten 8,9. In jüngster Zeit hat das Aufkommen der Einzelzellsequenzierung die hohe Plastizität und Wechselwirkung zwischen Subpopulationen innerhalb von Immunmikroumgebungen hervorgehoben, die zuvor nicht offensichtlich waren, was darauf hindeutet, dass Einzelzellmethoden notwendig sind, um diese Heterogenität zu erfassen10,11. Während neuartige Methoden zur Analyse des Transkriptoms entwickelt werden, bleibt die phänotypische Analyse eine Herausforderung, da sie gleichzeitige, quantitative und zeitaufgelöste Messungen der Proteinsekretion auf Einzelzellebene erfordert. Solche Messungen ermöglichen es uns, sezernierende Zellidentitäten, -dynamiken und -sekretionsmuster (langsam/schnell, früh/spät, simultan/sequentiell) für ein Repertoire oder eine Gruppe von Zytokinen zu untersuchen. Durch die quantitative und zeitliche Auflösung der Dynamik der Zytokinfreisetzung während einer Immunantwort könnten die daraus resultierenden Erkenntnisse ein Verständnis des zellulären Ensembles und der induzierten Reaktion ermöglichen.
In Standardprotokollen werden Zytokine in der Regel im Überstand von Zellsuspensionen und Serum unter Verwendung des Enzyme-Linked Immunosorbent Assay (ELISA) nachgewiesen, was zu Massensekretionsmengen führt. Massenmessungen ermöglichen keine Quantifizierung der von jeder Zelle produzierten Zytokinmengen, ein Problem, das besonders bei heterogenen Zellproben deutlich wird. Alternative Methoden wie die intrazelluläre Zytokinfärbung, der Enzyme-Linked Immunospot (ELISpot)-Assay oder mikrogravierte Assays (z. B. Isoplexis) weisen Zytokine nach, die von einzelnen Zellen exprimiert werden, liefern aber nur Endpunktmessungen12,13. Das bedeutet, dass die Sekretionsdynamik und Veränderungen, die im zellulären Sekretionsmuster während der Inkubationszeit auftreten können, ignoriert werden. Darüber hinaus können bei Endpunktmessungen nicht zwischen gleichzeitiger und sequentieller Zytokinsekretion unterschieden werden, so dass das wahre Ausmaß der gleichzeitigen Polyfunktionalität von Immunzellen in der Zytokinsekretion mit diesen Methoden unklar bleibt.
Eine Einzelzellauflösung kann mit Hilfe der Tröpfchenmikrofluidik erreicht werden, um Pikoliter-große physikalische Kompartimente zu erzeugen und zu verarbeiten, um Immunzellen auf ihre einzigartigen Zytokinsekretionsphänotypen auf Einzelzellebene zu untersuchen14,15. Diese Kompartimente bestehen aus Wasser-in-Öl-Emulsionen und können mit Hilfe von Mikrofluidik-Chips16,17 erzeugt werden. In der Tat haben tröpfchenbasierte mikrofluidische Assays eine extreme Vielseitigkeit bewiesen, wenn es darum geht, die Analyse verschiedener biologischer Proben und Repertoires auf Einzelzellebene und ihre Integration in vorgelagerte (Zell- und Reagenzienverarbeitung) und nachgelagerte Prozesse (Einzelzellsortierung, Proteomik oder Sequenzierung) zu ermöglichen18,19,20,21,22. Insbesondere ermöglichen Aufbauten, die eine Tröpfchenimmobilisierung ermöglichen, die Messung einer Einzelzellfunktionalität über die Zeit, was für die Analyse der Proteinsekretion wertvoll ist18. Darüber hinaus ermöglicht die Integration von gemultiplexten, quantitativen Assays zusätzliche Untersuchungen in bisher unzugänglichen Dimensionen, in Prozessen wie der Co-Sekretion und der Identifizierung polyfunktioneller Immunzellen23,24.
In diesem Protokoll beschreiben wir einen immobilisierten tröpfchenbasierten Einzelzell-Workflow, um die Sekretion von bis zu drei Zytokinen parallel aus einzelnen Zellen zu detektieren, zu quantifizieren und zeitlich zu messen17,23. Die Technologie bietet die Möglichkeit, die Zytokinreaktionen von über 20.000 Zellen parallel zu überwachen.
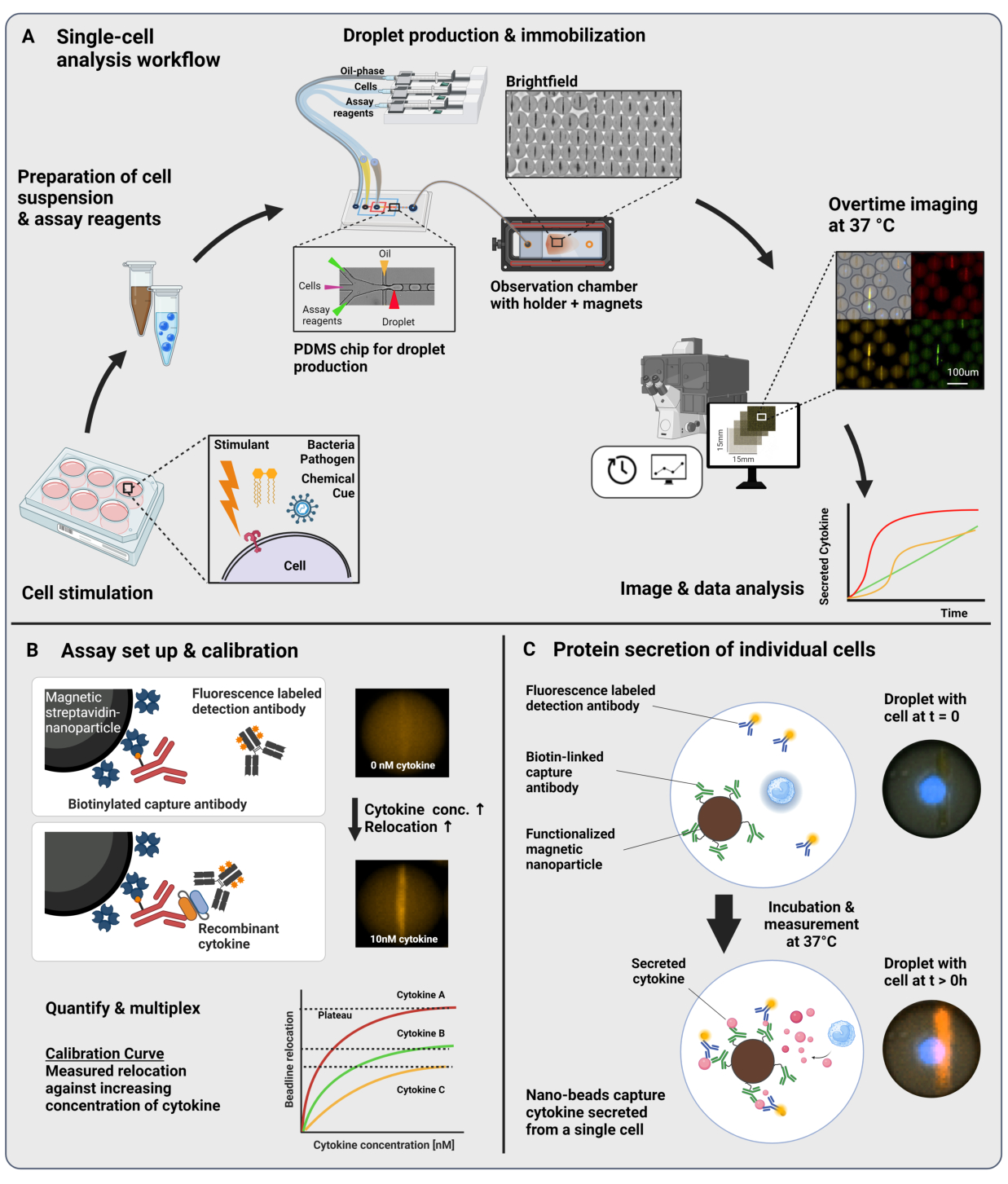
Der vorgestellte Arbeitsablauf besteht aus der mikrofluidischen Verkapselung einzelner Immunzellen und funktionalisierter Nanopartikel in 60 pL Wasser-in-Öl-Tröpfchen. Die Immobilisierung von >100.000 Tröpfchen in einer Beobachtungskammer und die zeitaufgelöste Fluoreszenzmikroskopie ermöglichen die Messung der Dynamik der Zytokinsekretion in jedem Tröpfchen und jedem Zytokin (Abbildung 1A). Für jede einzelne Zelle innerhalb eines Tröpfchens wird die Zytokinsekretion durch einen Sandwich-Immunoassay gemessen, bei dem magnetische Nanopartikel, die mit einem spezifischen Fängerantikörper funktionalisiert sind, das sezernierte Zytokin binden, was zur anschließenden Verlagerung und Bindung von fluoreszenzmarkierten Detektionsantikörpern führt (Abbildung 1B, C). Durch Ausrichten der magnetischen Nanopartikel wird eine Beadline gebildet, an die die Fluoreszenzverlagerung in Gegenwart von Zytokinen quantifiziert werden kann. Hier wird die Fluoreszenzverlagerung definiert als die durchschnittliche Fluoreszenzintensität auf der Perlenlinie dividiert durch die durchschnittliche Fluoreszenzintensität des verbleibenden Tröpfchens. Dieser Assay kann für mehrere Zytokine gemultiplext werden, indem unterschiedlich funktionalisierte Nanopartikelchargen und entsprechende Detektionsantikörper, die in verschiedenen Fluoreszenzkanälenmarkiert sind, gemischt werden, was zu spezifischen Fluoreszenzverlagerungen in den verschiedenen Kanälen führt. Mit Hilfe eines angepassten Analyseskripts können Fluoreszenzverlagerungswerte extrahiert und die Bilder in dynamische Sekretionsprofile für jede einzelne Zelle und jedes Zytokin umgewandelt werden. Daher liefern die resultierenden Datensätze zahlreiche Messwerte, wie z.B. die quantitative Sekretionsmessung über die Zeit, die Identifizierung von co-sezernierenden Subpopulationen und die Verteilungen der Zellen nach sezernierten Mengen, Raten und Kombinationen von Zytokinen.

Abbildung 1: Arbeitsablauf und Assay-Prinzip. (A) Überblick über den Arbeitsablauf zur Analyse von Zytokin-sezernierenden Zellen nach Stimulation. Einzellige Suspensionen und magnetische Nanopartikel werden hergestellt und in Öl/Wasser-Emulsionen (Tröpfchen) mit einem Volumen von 60 pL verkapselt. Tröpfchen werden immobilisiert und Nanopartikel in einem Magnetfeld ausgerichtet, bevor die Messung alle 30 Minuten für bis zu 4 Stunden erfolgt. Schließlich werden die Bilder analysiert und die Parameter für jedes Tröpfchen, jeden Zeitpunkt und jeden Fluoreszenzkanal extrahiert. Diese Zahl wurde von17 geändert. (B) Prinzip des Tröpfchen-Sandwich-Bioassays. Funktionalisierte Nanopartikel binden die sezernierten Zytokine, was dazu führt, dass fluoreszenzmarkierte Detektionsantikörper anschließend auf die Nanopartikel umgelagert werden. Diese Verschiebung der Fluoreszenz wird quantifiziert und durch Kalibrierungsexperimente validiert, die mit rekombinanten Zytokinen durchgeführt werden. Das Mischen verschiedener funktionalisierter Nanopartikel ermöglicht die Multiplexmessung von bis zu drei Zytokinen gleichzeitig. (C) In zellbasierten Experimenten werden Tröpfchen über die Messzeit verfolgt und sezernierende Zellen durch eine zeitliche Zunahme der Fluoreszenzverlagerung auf die Nanopartikel identifiziert. Die Schaltpläne sind nicht skalierbar. Mit BioRender.com erstellte Abbildung. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Protokoll
Alle Experimente wurden unter der Ethikvereinbarung EK202-N-56 durchgeführt und von der Ethikkommission der ETH Zürich genehmigt. Die Handhabung der menschlichen Zellen erfolgte in einem Laminar-Flow-Schrank, der sich in einem Labor der Biosicherheitsstufe 2 befand.
HINWEIS: In den folgenden Abschnitten wird das Protokoll zur Messung der zeitaufgelösten Zytokinsekretion auf Einzelzellebene beschrieben. Das hier skizzierte Verfahren wird auf die Stimulation von mononukleären Zellen des peripheren Blutes (PBMC) mit Lipopolysaccharid (LPS) und die parallele Messung der Zytokine IL-6, TNFα und IL-1β angewendet. Bei Bedarf kann das Protokoll jedoch an andere Zelltypen, Stimulanzien und Zytokine angepasst werden.
1. Herstellung der Beobachtungskammer
HINWEIS: Um eine Bewegung der Tröpfchen während der Bildgebung zu vermeiden, wird eine Beobachtungskammer mit einer Höhe vorbereitet, die etwa 10 % kleiner ist als der Tröpfchendurchmesser.
- Vorbereitung des Schneidens des doppelseitigen Klebebandes und des oberen Glasobjektträgers
- Zeichnen oder laden Sie das gewünschte Design des Kammerausschnitts auf der Registerkarte Design der Schneidsoftware. Die hier verwendeten Dimensionen finden Sie in Abbildung 2E.
- Befestigen Sie das doppelseitige Klebeband mit einer Dicke von 32 μm mit Klebeband auf der selbstklebenden Schneidematte und legen Sie die Schneidematte in den Schneideautomaten.
- Schneiden Sie das Kammerdesign aus dem Klebeband heraus und achten Sie dabei darauf, die langen Kanten der Kammer in Schritt 1.3 in die gleiche Richtung zu schneiden, um das Ablösen zu erleichtern.
- Lagern Sie die Bandausschnitte für eine langfristige Lagerung bei Raumtemperatur. Für die kurzfristige Lagerung lagern Sie sie bei -20 °C und nehmen sie erst kurz vor Schritt 1.3 heraus. für eine einfachere Handhabung.
- Bohren Sie zwei Löcher mit einem Durchmesser von ca. 1 mm in die Mitte eines Standard-Objektträgers (76 mm x 26 mm x 1 mm), wobei der Abstand zwischen den beiden Löchern etwa 3,5 cm beträgt.
- Reinigung und Plasmaaktivierung von Objektträgern
- Reinigen Sie einen Objektträger mit Löchern und einen ohne Löcher mit Seife. Gut mit destilliertem Wasser abspülen und mit fusselfreien Präzisionstüchern trocknen.
- Legen Sie die Objektträger in einen Plasmareiniger und behandeln Sie die oberen Oberflächen 10 Minuten lang bei 55 W. Entfernen Sie die Objektträger und fahren Sie mit Schritt 1.3 fort.
- Montage der Kammer
- Stellen Sie den Objektträger mit Löchern mit der plasmaaktivierten Seite nach oben auf eine saubere Oberfläche, ohne die aktivierte Oberfläche zu berühren.
- Entfernen Sie die Schutzschicht von einer Seite des doppelseitigen Klebebandes in gleicher Schnittrichtung. Richten Sie den Bandausschnitt ohne Berührung an den Kanten des Glasschiebers und den Bohrlöchern aus und bringen Sie das Klebeband langsam von der kurzen Kante aus in Kontakt mit dem Glasträger.
HINWEIS: Achten Sie darauf, keine Dehnungen oder Falten im Band zu erzeugen, da dies zu falschen Kammerhöhen führt. Da dieser Schritt fehleranfällig ist und etwas praktische Erfahrung erfordert, empfiehlt es sich, mehrere Objektträger parallel zu präparieren. - Entfernen Sie die zweite Schutzschicht vom Klebeband, wieder in Schnittrichtung, und platzieren Sie den zweiten Glasobjektträger ohne Löcher mit der aktivierten Fläche nach unten. Drücken Sie die gesamte Oberfläche der beiden Objektträger zusammen, indem Sie ein flaches Brett darauf legen und mit Oberkörperkraft ca. 10 s lang nach unten drücken.
- Nachdem du die beiden Glasschienen zusammengebaut hast, drehst du die Kammer so, dass die beiden Löcher zu dir zeigen. Klebe die Nanoports auf die beiden Löcher, indem du eine kleine Menge UV-härtenden Kleber in den Ring unter der Öffnung gibst und die Öffnung auf das Loch im Objektträger legst. Fügen Sie einen Ring aus UV-härtendem Kleber um den Port hinzu und härten Sie den Kleber mit einer UV-Lampe aus. Die Kammer sollte nun wie in Abbildung 2E dargestellt aussehen. Fahren Sie sofort mit Schritt 1.4 fort.
HINWEIS: UV-Licht kann Augen und Haut schädigen. Tragen Sie geeignete Schutzausrüstung.
- Fluorophile Beschichtung der Kammeroberfläche
HINWEIS: Dieser Schritt sollte innerhalb von 1 h nach der Plasmabehandlung der Objektträger (Schritt 1.2.2) erfolgen, um eine gute Beschichtungseffizienz zu gewährleisten.- 1 ml 1%ige Fluorsilanlösung (1H,1H,2H,2H-Perfluordecyltrichlorsilan) in fluoriertem Öl (HFE-7500) frisch zubereiten und in eine Spritze füllen. Schieben Sie die Beschichtungslösung durch einen PTFE-Spritzenvorsatzfilter und eine 27G x 0,75 Zoll große Nadel, die mit einem 0,3 mm x 0,76 mm PTFE-Mikroschlauch verbunden ist, in die Beobachtungskammer.
- Nach 1 Minute Inkubation wird die Beschichtungslösung mit Stickstoffdruck unter einem Abzug aus der Kammer gespült. Spülen Sie die Kammer mit fluoriertem Öl (nur HFE-7500) mit einer anderen Spritzenbaugruppe.
- Lagern Sie die mit fluoriertem Öl gefüllte Kammer mit geschlossenen Einlässen bei Raumtemperatur (RT). Nach jedem Experiment werden Zellen und Tröpfchen direkt ausgewaschen, um eine gute Konservierung der Beschichtung zu gewährleisten.
HINWEIS: Das Protokoll kann hier pausiert werden, und die Kammern können mehrere Monate gelagert und wiederverwendet werden.
- Kammerhalter mit Magneten
- Um die magnetischen Nanopartikel auszurichten, legen Sie während der Tröpfchenverkapselung und Bildgebung ein statisches Magnetfeld an die Beobachtungskammer an. Platzieren Sie dazu die Kammer in einem speziell angefertigten 3D-gedruckten Mikroskopiehalter (siehe Abbildung 2D und die Datei in Bounab et al.17 Supplementary Data 4), der zwei Neodym-Magnete an den Längsseiten der Kammer hält.
2. Funktionalisierung von Nanopartikeln
HINWEIS: Der Prozess für die Funktionalisierung der Nanopartikel ist für jedes Zytokin ähnlich, der einzige Unterschied besteht in der Zugabe von zytokinspezifischen Capture-Antikörpern. Die Funktionalisierung für jedes Zytokin wird in verschiedenen, individuellen Reaktionsröhrchen parallel durchgeführt. Vor diesem Protokoll wurden der TNFα-Capture-Antikörper und der IL-1β-Detektionsantikörper intern mit Biotin bzw. Alexa Fluor 647 markiert. Die Konjugation erfolgte gemäß dem Protokoll des Herstellers, das auf der Website des Herstellers zu finden ist (siehe Links in der Materialtabelle), und die Antikörper wurden aliquotiert und bei -20 °C gelagert.
- Geben Sie 50 μl Streptavidin-funktionalisierte Nanopartikel (Durchmesser (Ø) 300 nm) in das Röhrchen, das für die TNFα-Detektion bestimmt ist, 50 μl für IL-1β und 100 μl für IL-6. Verdünnen Sie die Nanopartikellösung 1:1 (v/v) in phosphatgepufferter Kochsalzlösung (PBS).
- In jedes Röhrchen wird 1/20 (v/v) des jeweiligen Volumens der biotinylierten Capture-Antikörper (Stammkonzentrationen bei 0,5 mg/ml) gegeben und 30 min bei RT inkubiert.
HINWEIS: Wenn Sie der Nanopartikellösung kleine Volumina hinzufügen, legen Sie das Volumen oben auf das Röhrchen und waschen Sie es mehrmals mit dem Großteil der Lösung ab. Dies sorgt für eine gute Durchmischung und verhindert die Bildung von Zuschlagstoffen. - 1/100 (v/v) 1 mM D-Biotin-Lösung in das Röhrchen geben und 5 min bei RT inkubieren. Daraus ergibt sich eine finale Biotinkonzentration von 10 μM.
HINWEIS: Überschüssiges Biotin blockiert freie Bindungsseiten auf den Nanopartikeln und reduziert die unerwünschte Aggregatbildung. - Sammeln Sie die Partikel, indem Sie einen Neodym-Magneten nahe an die Röhre halten. Warten Sie, bis der Überstand klar ist, und entsorgen Sie den Überstand.
HINWEIS: Die während des gesamten Assays verwendeten Magnete weisen sehr starke Anziehungskräfte auf, die bei versehentlichem Zusammenbrechen zweier Magnete zu körperlichen Schäden führen können. - Um die unspezifische Adsorption an der Nanopartikeloberfläche zu reduzieren, resuspendieren Sie die Nanopartikel sofort im 0,5-fachen des endgültigen Volumens von Schritt 2.1 von Pluronic F-127 (10 %) und im 0,5-fachen des Volumens PBS. Inkubieren Sie die Lösung 30 Minuten lang bei RT.
- Sammeln Sie die Partikel mit dem Magneten, entsorgen Sie den Überstand und resuspendieren Sie das 1-fache Volumen des Speicherpuffers (RPMI 1640, 5 % Knockout-Serumersatz, 1 % Pen/Strepto, 1 % rekombinantes humanes Serumalbumin (HSA), 25 mM HEPES, 0,1 % Pluronic F-127). Inkubieren Sie die Lösung 30 Minuten lang bei RT.
HINWEIS: Das Protokoll kann hier pausiert werden, und die Partikel können nun bis zu 1 Woche bei 4 °C gelagert werden. - Unmittelbar vor der Verkapselung resuspendieren Sie die Partikel durch Pipettieren und mischen Sie die konjugierten Nanopartikel in einem Verhältnis von 2:1:1 (v/v) für IL-6:TNFα:IL-1β.
HINWEIS: Die unterschiedlichen Verhältnisse funktionalisierter Nanopartikel hängen vom verwendeten Antikörperpaar für jedes Zytokin ab und wurden experimentell durch Kalibrierproben bestimmt, um einen optimalen Dynamikbereich zu erhalten. - Mit vollständigem Medium (RPMI 1640, 10 % FBS, 1 % Pen/Strepto, 25 mM HEPES) waschen, indem die Partikel mit dem Magneten aufgefangen, der Überstand verworfen und wieder suspendiert werden. Wiederholen Sie diesen Schritt, aber halten Sie nur das 0,5-fache des Volumens des vollständigen Mediums aus Schritt 2.7 erneut an.
- Geben Sie die unterschiedlich markierten IL-6-, TNFα- und IL-1β-Nachweisantikörper in die Lösung, um eine Endkonzentration von jeweils 10 nM zu erreichen. Die Lösung ist nun bereit, für die Tröpfchenexperimente verwendet zu werden.
3. Vorbereitung der Zellen
HINWEIS: PBMC wurden aus einem Buffy-Mantel isoliert, der von der Blutbank Zürich erhalten wurde. Die Zellen wurden eingefroren und in Kryofläschchen (1 x 107 Zellen/Fläschchen) in flüssigem Stickstoff über mehrere Monate gelagert.
- Auftauen der Zellen
- 1 h vor Beginn des Experiments lassen Sie das gesamte Medium und den MACS-Puffer (2 mM EDTA, 0,5 % BSA in DPBS, steril gefiltert) bei RT erwärmen. Bereiten Sie das Röhrchen mit den Zellen vor, indem Sie 9 mL des vollständigen Mediums in ein 15-ml-Röhrchen geben und es im Wasserbad bei 37 °C aufbewahren.
- Entnehmen Sie ein PBMC-Kryofläschchen (mit ~1 x 107 Zellen) aus seiner Lagerung in flüssigem Stickstoff. Schwenken Sie das Kryoröhrchen im Wasserbad bei 37 °C, bis nur noch eine geringe Menge Eis übrig ist.
- Wischen Sie das Rohr mit 70% EtOH ab und übertragen Sie es in den Laminar-Flow-Schrank. Geben Sie 1 ml vorgewärmtes vollständiges Medium in das Kryofläschchen, mischen Sie es vorsichtig und überführen Sie alle Zellen in das Röhrchen mit dem warmen vollständigen Medium. Das Kryofläschchen kann mit 1 mL warmem Komplettmedium gewaschen werden, um die maximale Anzahl von Zellen zurückzugewinnen.
- Die Zellen werden bei 500 x g für 5 min bei RT geschleudert, der Überstand verworfen und das Zellpellet mit 1 mL des vollständigen Mediums vorsichtig mit einer Pipette resuspendiert. Fügen Sie 9 ml des vollständigen Mediums hinzu.
- Die Zellen werden 5 Minuten lang bei RT bei 500 x g gedreht. Der Überstand wird verworfen und wie zuvor in 1 ml vollständiges Medium resuspendiert.
- Zählen Sie die Zellen mit dem verfügbaren Zellzähler. In diesem Fall kam ein automatisierter Zellzähler zum Einsatz. Die Zellen wurden gezählt, indem 10 μl Zellsuspension mit 10 μl Trypanblau gemischt und 10 μl der Mischung in den Zellzählobjektträger überführt wurden.
- Fleckenbildung und FcR-Blockierung
- Berechnen Sie die Gesamtzahl der Zellen und das Volumen, das zur Resuspendierung der Zellen benötigt wird, bei 2 x 106 lebenden Zellen/ml. Bereiten Sie die Zellfärbelösung (CellTrace Violet) vor, indem Sie das Stammmaterial (5 mM) 1000x in PBS (Arbeitskonzentration von 5 μM) verdünnen.
- Die Zellen werden bei 500 x g 5 min lang bei RT gedreht. Der Überstand wird verworfen und die Zellen werden in dem in Schritt 3.2.1 berechneten Volumen der Zellfärbelösung resuspendiert. Inkubieren Sie die Zellen bei 37 °C für 5 min.
- Am Ende der Inkubation wird der verbleibende Farbstoff in der Lösung durch Zugabe eines vollständigen Mediums (mindestens das 2-fache Volumen der Farbstofflösung) abgeschreckt. Die Zellen bei 500 x g für 5 min bei RT drehen.
- Der Überstand wird verworfen, das Zellpellet in 60 μl MACS-Puffer resuspendiert und 20 μl humaner FcR-Block pro 1 x 107 Zellen hinzugefügt. Inkubieren Sie die Zellen 10 Minuten lang bei RT.
- Füllen Sie das Röhrchen auf 10 mL mit MACS Buffer und drehen Sie die Zellen bei 500 x g für 5 min bei RT.
- Den Überstand verwerfen und die Zellen in 1 ml vollständigem Medium resuspendieren. Zählen Sie die Zellen wie in Schritt 3.1.6 beschrieben.
- Zellstimulation mit LPS
- Verdünnen Sie die Zellen anhand der Zellzahl mit 1 x 106 Zellen/ml und überführen Sie 2 ml Zellen in jeder Vertiefung in einer 6-Well-Platte mit extrem geringer Bindung.
- Verdünnen Sie LPS in einem vollständigen Medium und geben Sie es in die Vertiefung, die die Zellen enthält, für eine Endkonzentration von LPS von 1 μg/ml. Inkubieren Sie die Zellen 6 Stunden lang bei 37 °C.
- Vorbereitung für die Verkapselung
- Am Ende der Stimulationszeit wird die Zellsuspension in ein neues 15-ml-Röhrchen überführt.
- Geben Sie 1 ml des vollständigen Mediums in die leere Vertiefung. Trennen Sie mit einem Zellschaber die restlichen Zellen. Übertragen Sie die Zellen in ein neues 15-ml-Röhrchen. Waschen Sie die Vertiefung mit 1 mL des vollständigen Mediums und geben Sie sie in ein weiteres 15-ml-Röhrchen.
- Schleudern Sie die beiden Röhrchen bei 500 x g für 5 min bei RT und überführen Sie 1 mL der unverdünnten Überstandslösung (aus dem ersten Röhrchen mit den nicht gewaschenen Zellen) in ein neues Röhrchen zur weiteren Analyse (z. B. ELISA).
- Den Rest der Überstände entsorgen.
- Resuspendieren Sie die Pellets in 0,5 ml vollständigem Medium, kombinieren Sie die Zellen aus derselben Vertiefung und überführen Sie sie in ein Zentrifugenröhrchen. Zählen Sie die Zellen wie in Schritt 3.1.6 beschrieben.
- Drehen Sie die Zellen bei 500 x g für 5 min bei RT und verwerfen Sie den größten Teil des Überstands (es verbleiben ca. 100 μl). Ohne das Pellet erneut zu resuspendieren, sehr vorsichtig 200 μl des vollständigen Mediums hinzufügen.
- Entsorgen Sie den Überstand. Resuspendieren Sie die Zellen in einem vollständigen Medium bei einer Konzentration von 6,6 bis 13,3 x 106 Zellen/ml, um eine durchschnittliche Zellzahl pro Tröpfchen von λ = 0,2-0,4 für die Verkapselung zu erreichen, wie in Schritt 8.6 definiert.
HINWEIS: Die Schritte 3.4.6 und 3.4.7 sollten unmittelbar vor der Verkapselung durchgeführt werden, um eine Zytokinsekretion in den Überstand zu vermeiden. Die Anzahl der Zellen pro Tröpfchen folgt einer Poisson-Verteilung: , wobei P den Anteil der Tröpfchen mit X Zellen und λ die mittlere Anzahl von Zellen pro Tröpfchen darstellt.
, wobei P den Anteil der Tröpfchen mit X Zellen und λ die mittlere Anzahl von Zellen pro Tröpfchen darstellt.
4. Verkapselung und Tröpfchenproduktion
ANMERKUNG: Die Verkapselung von Zellen in Tröpfchen wird durch einen mikrofluidischen Tröpfchengenerator-Chip ermöglicht, dessen Herstellung an anderer Stelle ausführlich beschrieben wird17. Alternativen sind im Handel erhältlich (siehe Beispiel in der Materialtabelle). Ein geeignetes Tröpfchengenerator-Chipdesign weist zwei Einlässe für wässrige Phasen, einen Einlass für die Ölphase und einen Auslass für die erzeugten Tröpfchen auf. Darüber hinaus sollte ein geeigneter kommerzieller Tröpfchengenerator-Chip die Produktion von Wasser in fluorierten Öltröpfchen mit einem Volumen von 40-60 pL ermöglichen. Das hier beschriebene Protokoll führt zu Wasser/Öl-Emulsionen (Tröpfchen) mit einem Durchmesser von 50 μm. Die Verwendung verschiedener Optionen zur Änderung des Protokolls kann zu größeren oder kleineren Tröpfchen führen.
- Vorbereitung der Spritzenpumpe (Abbildung 2A)
- Füllen Sie eine 1-ml-Spritze mit 500 μl kontinuierlicher Phase, bestehend aus 2 % 008-Fluortensid in fluoriertem Öl HFE-7500. Verbinden Sie eine 27G x 0,75 Zoll Nadel mit einem 0,30 mm x 0,76 mm PTFE-Mikroschlauch und montieren Sie die Baugruppe auf der Spritze und anschließend auf der Spritzenpumpe.
HINWEIS: Stellen Sie sicher, dass keine Luft in der Spritze oder Kanüle verbleibt, da dies konsistente Durchflussraten verhindert. - Bereiten Sie zwei maßgefertigte Pipettenspitzenkonnektoren für die wässrigen Phasen vor (Abbildung 2B): Stanzen Sie mit einem Ø0,75 mm Biopsienstanzer ein Loch in die Mitte eines ~5 mm hohen PDMS-Ausschnitts mit Ø6 mm. Ziehen Sie ~3 cm PTFE-Schlauch (0,56 mm Innendurchmesser, 1,07 mm Außendurchmesser) durch das Loch im PDMS-Ausschnitt und schieben Sie die Baugruppe in die Oberseite einer 200-μl-Pipettenspitze. Verbinden Sie die andere Seite des Rohrs mit einer 23Gx 1,25-Zoll-Nadel. Versiegeln Sie den Stecker, indem Sie UV-härtenden Kleber auf die Pipette auftragen und mit UV-Licht aushärten.
HINWEIS: Da UV-Licht schädlich für das Auge ist, tragen Sie zum Schutz eine UV-Schutzbrille. - Füllen Sie zwei 1-mL-Spritzen mit 500 μl leichtem Mineralöl, befestigen Sie zwei 23G-Nadeln mit den individuell angefertigten Aufsätzen und montieren Sie beide auf die Spritzenpumpe.
- Aspirieren Sie 30 μl Nanopartikel und 30 μl Zelllösung mit der Spritzenpumpensteuerungssoftware in die Pipettenspitzen der wässrigen Phasen.
- Bereiten Sie eine Beobachtungskammer vor, indem Sie die Oberfläche mit Wasser reinigen, um Schmutz und Staub zu entfernen, und trocknen Sie sie mit Präzisionstüchern. Klemmen Sie die Kammer in den bedruckten Kammerhalter, der mit zwei Neodym-Magneten ausgestattet ist.
HINWEIS: Stellen Sie sicher, dass die Magnete in die richtige Richtung zeigen (sich gegenseitig anziehen), um ein längliches Aggregat zu bilden. - Winkeln Sie die Kammer leicht an (30°). Öffnen Sie beide Anschlüsse und stecken Sie ein Papiertuch in die obere Öffnung, um die überschüssige äußere Phase während des Befüllens aufzunehmen.
- Füllen Sie eine 1-ml-Spritze mit 500 μl kontinuierlicher Phase, bestehend aus 2 % 008-Fluortensid in fluoriertem Öl HFE-7500. Verbinden Sie eine 27G x 0,75 Zoll Nadel mit einem 0,30 mm x 0,76 mm PTFE-Mikroschlauch und montieren Sie die Baugruppe auf der Spritze und anschließend auf der Spritzenpumpe.
- Tröpfchenproduktion und Kammerbefüllung
- Verbinden Sie die kontinuierliche Phase über einen Schlauch mit dem oberen Einlass des Mikrofluidik-Chips (Abbildung 2A,C,F). Spülen Sie den Chip für ca. 30 s mit kontinuierlicher Phase mit einer Durchflussrate von 1800 μL/h.
- Verbinden Sie die Pipettenspitzen der wässrigen Lösungen mit den beiden mittleren Einlässen (Abbildung 2A,C,F).
- Starten Sie den Fluss der wässrigen Lösung mit je 200 μL/h und lassen Sie die Kanäle und den Auslass mit Flüssigkeit füllen. Bei der Verwendung von magnetischen Nanopartikeln sollte eine homogene, braun-rot gefärbte Lösung aus dem Chipauslass fließen.
- Sobald die Flüssigkeit am Auslass erscheint, starten Sie den Phasenfluss des fluorierten Öls bei 800 μL/h und warten Sie, bis eine stabile Tröpfchenproduktion hergestellt ist, die durch den Austritt einer homogenen, grauen, glänzenden Lösung am Auslass bestätigt wird.
- Sobald eine stabile Tröpfchenproduktion hergestellt ist, sammeln Sie die produzierten Tröpfchen, indem Sie einen PTFE-Mikroschlauch (0,3 mm Innendurchmesser x 0,76 mm Außendurchmesser) an die Auslassöffnung anschließen und sie in eine Beobachtungskammer leiten, indem Sie den Mikroschlauch durch das Ferrule-Modul einer fingerfesten einteiligen Armatur führen (Abbildung 2A).
- Wenn eine ordnungsgemäße Tröpfchenproduktion stattfindet, sollte eine homogene, glänzende Flüssigkeit die Kammer mit einer geraden Vorderseite von unten nach oben füllen.
- Sobald die Kammer gefüllt ist, stoppen Sie den Durchfluss und verschließen Sie die Anschlüsse mit Anschlussstopfen mit fingerfestem Druck.
HINWEIS: Achten Sie darauf, die Kammer nicht zu fest zu schließen. Das Einfangen oder Einströmen von Luft kann zu Bewegungen der Tröpfchen führen und somit die Nachführung während der Messung beeinträchtigen. - Spülen Sie den Chip nach der Tröpfchenproduktion mit fluoriertem Öl und blasen Sie alle Erinnerungen an Flüssigkeit mit Stickstoff aus, um seine Funktion zu erhalten. Chips können mehrfach wiederverwendet und monatelang gelagert werden, solange sie nicht verstopft sind.

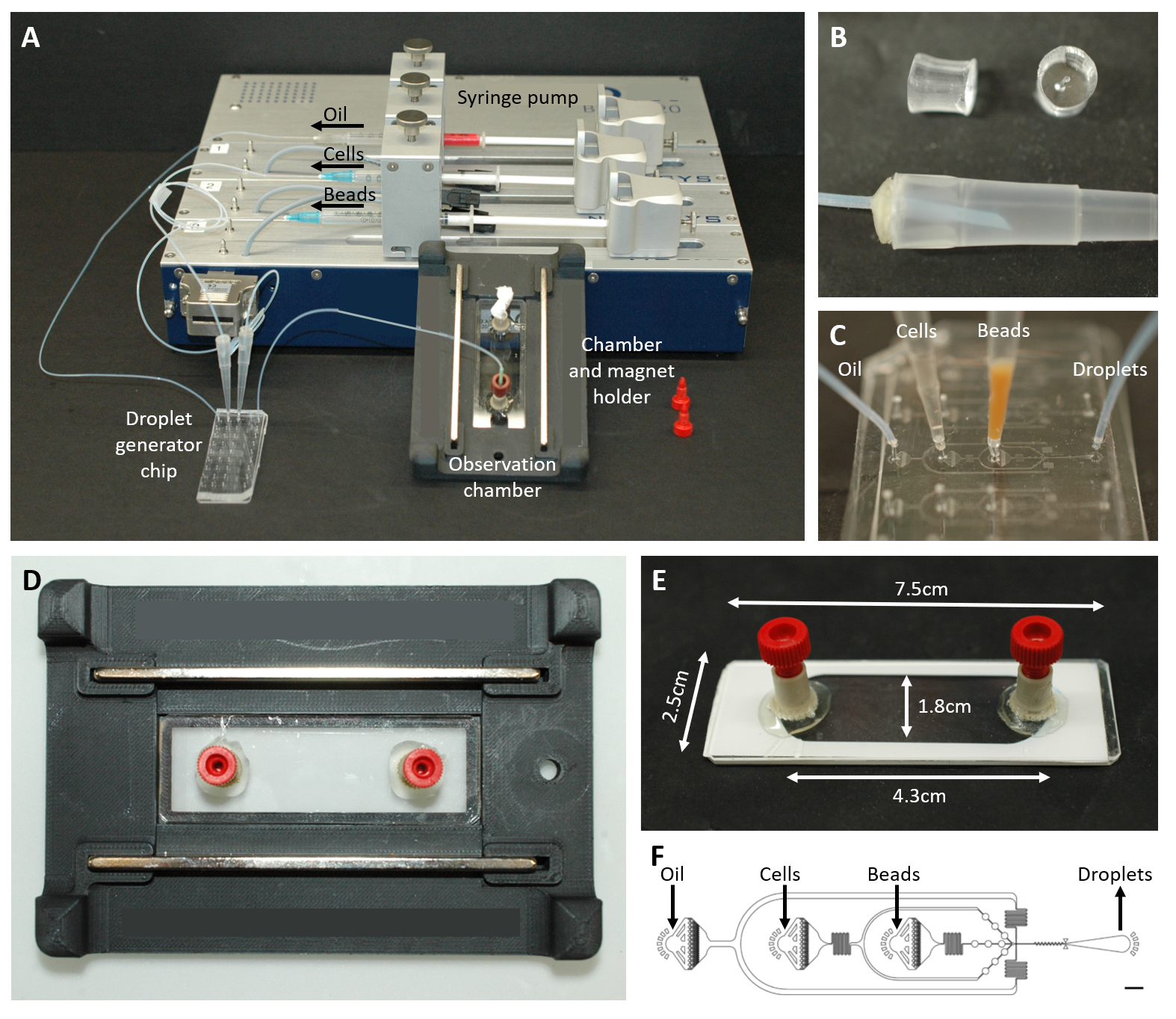
Abbildung 2: Überblick über den mikrofluidischen Aufbau. (A) Aufbau für die Tröpfchenverkapselung mit der Spritzenpumpe, dem Tröpfchenerzeugungschip sowie der Beobachtungskammer und dem Mikroskophalter. (B) Bild des gestanzten PDMS-Steckers (oben), der einen Anschluss an eine 200-μl-Pipettenspitze (unten) bildet, wie in Protokollschritt 4.1.2 beschrieben. (C) Bilder der Verbindung von Schläuchen und Pipettenspitzen mit dem Tröpfchenerzeugungschip. (D) Bild der Kammer, die sich in dem kundenspezifischen 3D-gedruckten Mikroskophalter mit zwei Magneten oben und unten befindet. (E) Foto der Beobachtungskammer (mit weißem Klebeband zur Veranschaulichung). (F) Layout des mikrofluidischen Chips für die Tröpfchenerzeugung (Maßstab: 750 μm). Diese Zahl wurde von17 geändert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
5. Bilderfassung und -messung
HINWEIS: Die Bildaufnahme erfolgt an einem inversen Standard-Epifluoreszenzmikroskop, das in einem Inkubator untergebracht ist und Messungen bei 37 °C ermöglicht. Die hier beschriebenen Einstellungen sind spezifisch für ein Nikon Eclipse Ti2 Mikroskop, das mit der NIS Elements Software (V. 5.30.04) läuft und mit einer Orca Fusion Kamera ausgestattet ist, kann aber im Allgemeinen an alle anderen Fluoreszenzmikroskope und Kameras angepasst werden.
- Einstellen von Messparametern
- Um die Größe des Bildes festzulegen, wählen Sie eine Array-Größe von 10 x 10 Bildern aus. Dieses Array wird etwa 50.000 bis 70.000 Tröpfchen enthalten. Verwenden Sie 1 % Überlappung und aktivieren Sie die Überblendung für das Zusammenfügen von Bildern.
- Um die Anzahl der gemessenen Kanäle einzustellen, wählen Sie den DAPI-Kanal für die Zelldetektion, FITC, TRITC, Cy5-Kanäle für den Zytokinnachweis (Beadlines) und den BF-Kanal für die Tröpfchendetektion. Verwenden Sie Pixel-Binning 2 x 2 und 16 Bit für die Bittiefe. Passen Sie die Kameraeinstellungen an, um Pixelwerte für die Tröpfchenintensität zu erzielen, die nicht für jeden Fluoreszenzkanal das Maximum der Kamera erreichen.
HINWEIS: Die genauen Belichtungszeiten und Lampenintensitäten für jeden Kanal hängen vom verwendeten Modell und den Reagenzien ab und werden vor der Erstellung von Kalibrierkurven (Schritt 7) festgelegt. Die Verwendung der gleichen Erfassungseinstellungen bei Kalibrierung und Zellmessungen ist wichtig für eine genaue Quantifizierung. - Um die zeitaufgelöste Messung einzurichten, wählen Sie alle 30 Minuten eine Messung aus, um insgesamt 9 Messungen zu erhalten.
HINWEIS: Die Messparameter können für verwendete Zellen, Stimulanzien, Reagenzien, gemessene Zytokine, Inkubationstemperatur und Mikroskopmodelle unterschiedlich sein.
- Starten der Messung
- Montieren Sie den Kammerhalter mit einem Tisch im Well-Plate-Format auf das Mikroskop (Abbildung 2D) und schalten Sie mit dem 10x-Objektiv auf den Hellfeldkanal (BF) um.
- Konzentrieren Sie sich auf die immobilisierten Tröpfchen in BF und stellen Sie sicher, dass die Baugruppe in einer perfekten Ebene montiert ist, indem Sie herumschwenken und bei Bedarf anpassen. Begeben Sie sich für die folgenden Schritte in die Mitte der Kammer.
- Aktivieren Sie das automatische Fokussiersystem (PFS) und stellen Sie es auf die optimale Messebene auf dem BF-Kanal ein, so dass Tröpfchenkanten als schwarze, scharfe Kreise erscheinen, die leicht von der Ölphase und dem Hintergrund unterschieden werden können.
HINWEIS: Messungen sind auch ohne automatisches Fokussiersystem möglich, aber wenn das Mikroskop damit ausgestattet ist, empfehlen wir dringend, es zu verwenden. Dies verbessert die Messqualität bei großen und stark zusammengefügten Bildern. - Durchlaufen Sie alle Fluoreszenzkanäle und stellen Sie für jeden die optimale Messebene ein. Stellen Sie bei Relocation-Messungen auf den Kanälen FITC, TRITC und Cy5 sicher, dass das Nanopartikelaggregat perfekt fokussiert ist, bei den DAPI-Kanälen stellen Sie sicher, dass die Zellen fokussiert sind.
HINWEIS: Optimale Fokusebenen und Z-Werte können für alle gemessenen Kanäle unterschiedlich sein. Stellen Sie sicher, dass Sie für jeden Kanal individuelle PFS-Offsets speichern. - Bevor Sie mit der Messung beginnen, gehen Sie durch alle Kanäle, um die einzelnen Brennpunkte zu überprüfen, und warten Sie 5 Minuten, bis sie sich äquilibriert haben, da während des Aufwärmens der Lösungen zunächst Bewegungen auftreten können.
- Starten Sie die Messung. Überprüfen Sie nach der Erstellung des ersten Bildes auf Unregelmäßigkeiten (Schärfe, sich bewegende Tröpfchen, falsche Kanäle usw.). Starten Sie die Erfassung bei Bedarf erneut oder füllen Sie im Falle von Luft die Kammer wieder auf (beginnen Sie mit Schritt 4.1.4). Lassen Sie die Baugruppe die Tröpfchen über einen Zeitraum von 4 Stunden abbilden.
6. Bildanalyse
- Installieren Sie die Bildanalysesoftware (DropMap Analyzer App v 4.023) in MatLab (https://github.com/ESPCI-LCMD/MiMB) und übertragen Sie die generierte .nd2-Datei aus dem Experiment auf einen Analysecomputer.
- Öffnen Sie die Anwendung. Wählen Sie die angegebenen Einstellungen aus, andernfalls belassen Sie den Standardwert: CH1: DAPI, WD (ganzes Droplet) ausgewählt; CH2: FITC, BL (Wulstlinie) ausgewählt; CH3: TRITC, BL ausgewählt; CH4: Cy5, BL ausgewählt; Maximaler Tropfendurchmesser (μm): 70; Tropfenerkennung: Voll; Sendungsverfolgung: Ja. Drücken Sie auf die Schaltfläche Start (Fruchtsymbol), um den Speicherort der .nd2-Datei auszuwählen und die Analyse zu starten.
- Nach einigen Minuten zeigt das Programm einen Beispielausschnitt des Bildes an (Abbildung 3A). Drücken Sie die Leertaste , bis Sie eine für die Tröpfchenerkennung geeignete Tröpfchenerkennung gefunden haben, und drücken Sie dann die Eingabetaste. Zeichnen Sie im selben Bildausschnitt ein Rechteck in einem repräsentativen Bereich, um Schwellenwertparameter zur Erkennung von Tröpfchen zu finden.
- Nach einigen Minuten öffnet sich ein weiteres Fenster, in dem die Intensitätsverteilung des DAPI-Kanals angezeigt wird. Ziehen Sie den Schieberegler per Drag & Drop, um nur das Signal der gefärbten Zellen zu erkennen, und klicken Sie in der oberen rechten Ecke auf Fertig .
- Nach der Segmentierung des Bildes in einzelne Tröpfchen mit Durchmessern kleiner als Max. Tropfendurchmesser (μm) führt das Programm nun die folgenden Schritte für jedes Tröpfchen, jeden Zeitpunkt und jeden Fluoreszenzkanal ohne weitere Benutzereingaben durch (siehe Abbildung 3).
- Die Software berechnet die bewegten Pixel des Tröpfchens zwischen den Zeitpunkten (Tröpfchen, die sich um mehr als 40 Pixel bewegen, werden automatisch ausgeschlossen).
- Die Software misst den durchschnittlichen Fluoreszenzwert des gesamten Tröpfchens und erkennt und misst die mittlere Intensität der Perlenlinie, indem sie das hellste Pixel auf einer horizontalen Linie findet und alle Pixelintensitäten auf einer vertikalen Linie von oben nach unten des Tröpfchens mittelt. Dies geschieht automatisch und wird verwendet, um die durchschnittlichen Werte für die Verlagerung der Perlenlinie (Abbildung 3B) gemäß der Gleichung zu berechnen:

- Die Software berechnet den Prozentsatz der Gesamtpixel im Tröpfchenbereich über dem auf dem DAPI-Kanal festgelegten Schwellenwert.
- Die resultierende .xslx-Datei enthält die folgenden Spalten, die für die weitere Analyse von Interesse sind: DropIdX (ID des Tröpfchens, das über die Zeit verfolgt wird), TrueCentroid_ t*2-1 und t+2 (x- bzw. y-Koordinaten des Tröpfchenmittelpunkts für den Zeitpunkt t), DiameterMicrons (Tröpfchendurchmesser in μm), TrackingMove (Anzahl der über die gesamte Messzeit bewegten Pixel), FluoChannel_BL_Ratio_t (Verlagerungswert für FluoChannel zum Zeitpunkt t), DAPI_WD_PosPxlCount_t (Anzahl der Pixel über dem Schwellenwert im gesamten Tröpfchen im DAPI-Kanal zum Zeitpunkt t).

Abbildung 3: Bildanalyse durch die Bildanalysesoftware. (A) Tröpfchen werden im Hellfeldkanal (BF) mit Hilfe einer Hough-Transformation erkannt, wobei jedes Tröpfchen mit einem roten Kreis markiert wird. Maßstabsbalken: 200 μm. (B) Innerhalb jedes Tröpfchens wird die Nanopartikel-Beadline durch die hellsten Pixel in der horizontalen Ebene identifiziert und die Fluoreszenzintensitäten für alle Pixel, die sich von oben nach unten erstrecken, gemittelt. Zusätzlich wird die Zelle durch einen Pixelprozentsatz >0 über dem Schwellenwert für den gesamten Tröpfchenbereich identifiziert. Maßstab: 20 μm. (C) Die Analysatorsoftware vergleicht die Fluoreszenzintensität der Nanopartikel mit dem Tröpfchenhintergrund für die Kanäle FITC, TRITC und Cy5 über alle gemessenen Zeitpunkte für jedes einzelne Tröpfchen. Dargestellt sind die Zeitpunkte 0, 4 (120 min) und 9 (240 min). Um die korrekte Tröpfchen- und Zellerkennung manuell zu überprüfen, werden auch die DAPI- und BF-Kanäle angezeigt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
7. Kalibrierung
HINWEIS: Für eine quantitative Auslesung muss die Kalibrierung der Zytokinkonzentrationen auf die Fluoreszenzverlagerungswerte einmal durchgeführt werden, da Unterschiede zwischen verschiedenen Versuchsaufbauten auftreten können. Alle erforderlichen Schritte sind in den vorherigen Protokollabschnitten beschrieben, auf die verwiesen wird.
- Bereiten Sie Nanopartikel vor, wie in Schritt 2 beschrieben.
- Rekonstituieren Sie die humanen rekombinanten Proteine IL-6, TNFα und IL-1β gemäß den Anweisungen des Herstellers.
HINWEIS: Stellen Sie sicher, dass gefrorene Aliquots nur einmal aufgetaut und sofort verwendet werden. - Bereiten Sie eine 2-fache Verdünnungsreihe für alle drei Proteine zusammen unter Verwendung eines vollständigen Mediums (10 % FBS, 1 % Pen/Strep, 25 mM HEPES) mit einer Ausgangskonzentration von 80 nM bis hinunter zu 0,625 nM vor.
- Führen Sie die Verkapselung wie in Schritt 4 beschrieben mit funktionalisierten Nanopartikeln in der ersten und RPMI erst in der zweiten wässrigen Phase durch. Diese Messung dient als Blank und die gemessene Standardabweichung wird für die spätere Datenanalyse verwendet.
- Warten Sie 5 Minuten und stellen Sie die Tröpfchen wie in Schritt 5 beschrieben dar. Nehmen Sie 3 Bilder mit einer Array-Größe von 2 x 2 in den jeweiligen Fluoreszenzkanälen auf.
- Wiederholen Sie die Schritte 7.4 und 7.5 mit allen vorbereiteten Kalibrierlösungen, beginnend mit der niedrigsten und endend mit der höchsten Konzentration.
- Analysieren Sie die Bilder, wie in Schritt 6 beschrieben. Verwenden Sie nicht die WD-Option für den DAPI-Kanal, und legen Sie Tracking auf Nein fest.
- Die Analyse gibt Fluoreszenzverlagerungswerte für jedes gemessene Tröpfchen in einem Bild aus. Extrahieren Sie den Median und die Standardabweichung für jeden Fluoreszenzkanal. Mittelwerten Sie den Median und die Standardabweichung für jedes gemessene Bild pro Konzentration.
- Erstellen Sie eine Kalibrierungskurve, indem Sie die gemittelte mediane Verlagerung gegen die gemessenen Konzentrationen jedes rekombinanten Proteins darstellen.
- Passen Sie die Kurven mit einer einphasigen Assoziation an:
 ,
,
mit Y = Verschiebung bei x, Y0 = Verschiebung der Blindmessung und x der verwendeten Konzentration. Die erhaltene Kalibrierkurve wird verwendet, um die Verlagerungswerte zu quantifizieren, wie in Schritt 8 beschrieben.
HINWEIS: Passen Sie nur die Werte bis zur höchsten gemessenen Verlagerung an und schließen Sie Werte aus höheren Konzentrationen mit niedrigerer gemessener Verlagerung aus. Eine Abnahme der gemessenen Relokationswerte bei höheren Konzentrationen ist zu erwarten und tritt aufgrund des Hook-Effekts und der begrenzten Bindungskapazität der Nanopartikel auf.
8. Datenanalyse
- Schließen Sie Tröpfchen mit einem TrackingMove-Wert größer als 10 aus, d.h. die sich im zeitlichen Verlauf der Messung um mehr als 10 Pixel bewegt haben.
- Identifizieren Sie Tröpfchen, die gefärbte Zellen enthalten (DAPI-Kanal), im ersten Zeitpunkt, indem Sie in der Spalte DAPI_WD_PosPxlPercent_1 nach Tröpfchen mit Werten über 0 sortieren.
- Identifizieren Sie Tröpfchen, die sekretierende Zellen enthalten, indem Sie die folgenden 3 Kriterien auf die Fluoreszenzverlagerung jedes Fluoreszenzkanals (FluoChannel_BL_Ratio_t Säulen) anwenden.
- Identifizierung von Tröpfchen mit steigenden Verlagerungswerten, indem nach einer positiven Steigung über die Messzeit sortiert wird.
- Identifizierung von Tröpfchen mit Verlagerungswerten, die die Nachweisgrenze (LOD) erreichen. Ein Tröpfchen wird ausgewählt, wenn die maximale Fluoreszenzverschiebung über die Messzeit größer ist als die LOD, die wie an anderer Stellebeschrieben berechnet wird 25
 : , wobei μRelocation t0 der Median aller Relocation-Werte zum Zeitpunkt 0 und σBLK die Standardabweichung des während der Kalibrierung gemessenen Blindwerts ist, jeweils zytokinspezifisch sind.
: , wobei μRelocation t0 der Median aller Relocation-Werte zum Zeitpunkt 0 und σBLK die Standardabweichung des während der Kalibrierung gemessenen Blindwerts ist, jeweils zytokinspezifisch sind. - Überprüfen, ob der Anstieg des Verlagerungswerts signifikant ist, indem überprüft wird, ob die Änderung zwischen der maximalen und der minimalen gemessenen Fluoreszenzverlagerung über die Messzeit besser ist als:
 .
.
- Identifizieren Sie mitsekretierende Zellen, indem Sie die in Schritt 8.3 beschriebenen Kriterien erfüllen. für mehr als einen Fluoreszenzkanal gleichzeitig.
- Wiederholen Sie Schritt 8.3. für alle Tröpfchen, die keine Zelle enthalten (DAPI_WD_PosPxlPercent_1 = 0). Verwenden Sie diese Droplets, um den falsch-positiven Prozentsatz zu berechnen.
- Bestimmen Sie den genauen λ-Wert der Messung, indem Sie 200 - 500 Tröpfchen nach dem Zufallsprinzip auswählen und mit der Funktion Überprüfen und Sortieren der Bildanalysesoftware untersuchen. Zählen Sie die Anzahl der Zellen in diesen Tröpfchen und berechnen Sie:
λ = Anzahl der gezählten Zellen / Anzahl der analysierten Tröpfchen - Berechnen Sie die Gesamtzahl der verkapselten Zellen für die Messung durch:
Gesamtzellzahl = λ × Anzahl der analysierten Tröpfchen - Berechnen Sie den Prozentsatz der sezernierenden Zellen anhand der ermittelten Zellzahl. Berechnen Sie zusätzlich den falsch-positiven Prozentsatz für jedes Zytokin (in der Regel weniger als 3%-5% bezogen auf die Anzahl der real positiven Ergebnisse pro Kanal) und verwenden Sie sie als interne Kontrolle für experimentelle Konsistenz und Reproduzierbarkeit.
- Um die sezernierten Zytokinkonzentrationen zu berechnen, rechnen Sie die Relokationswerte mit den etablierten Kalibriergleichungen aus Schritt 7.10 in die Konzentration um.
- Berechnen Sie die Sekretionsrate (SR) zwischen den Zeitpunkten mithilfe der folgenden Gleichung.

- Berechnen Sie die durchschnittliche Sekretionsrate über die Messung, indem Sie die einzelnen Sekretionsraten zwischen den Zeitpunkten mitteln. Wenn die maximal messbare Verlagerung vor Ende der Messung erreicht wurde, stellen Sie die Konzentration auf die maximal messbare Konzentration ein (dieser Wert ist zytokinspezifisch und entspricht der maximalen Konzentration, die in der Kalibrierkurve in Schritt 6.10 gemessen und verwendet wurde) und berechnen Sie keine weitere Konzentration. Berechnen Sie die Sekretionsrate und den Durchschnitt nur bis zu diesem Zeitpunkt.
HINWEIS: Wenn weniger als 50 sezernierende Zellen in einem Fluoreszenzkanal nachgewiesen werden, sollten die Tröpfchen mit der Verifizierungs- und Sortierfunktion visuell untersucht werden, und Tröpfchen mit Fluoreszenz- oder Nanopartikelaggregaten können von der Analyse ausgeschlossen werden. - Um weitere Parameter aus der Sekretionskurve jeder einzelnen Zelle zu extrahieren, führen Sie für jede Zelle und jedes Zytokin mit einem benutzerdefinierten Python-Skript (auf Anfrage erhältlich) eine Anpassung des kleinsten Quadrats an die Zeit-Konzentrationskurve durch. Die angepasste Funktion ist eine sigmoidale Kurve nach der unten beschriebenen Formel (Anpassungen mit R2<0,95 sind von den folgenden Schritten ausgeschlossen):

wobei C dem Konzentrationsplateau [nM], t50 der Verschiebung des halben Maximums [s] und m der Hill-Steigung [min-1] entspricht. Aus diesen Parametern werden die folgenden Kurvendeskriptoren extrahiert, wie unten beschrieben.- Cmax [nM]: Maximal gemessene Konzentration.
 : Zeitpunkt des Beginns der Sekretion, wenn die Passform 10% von C erreicht.
: Zeitpunkt des Beginns der Sekretion, wenn die Passform 10% von C erreicht. : Sekretionsrate als angenäherte lineare Steigung der Kurve zwischen 10 % und 90 % der Zeit-Konzentrations-Kurve.
: Sekretionsrate als angenäherte lineare Steigung der Kurve zwischen 10 % und 90 % der Zeit-Konzentrations-Kurve.
Ergebnisse
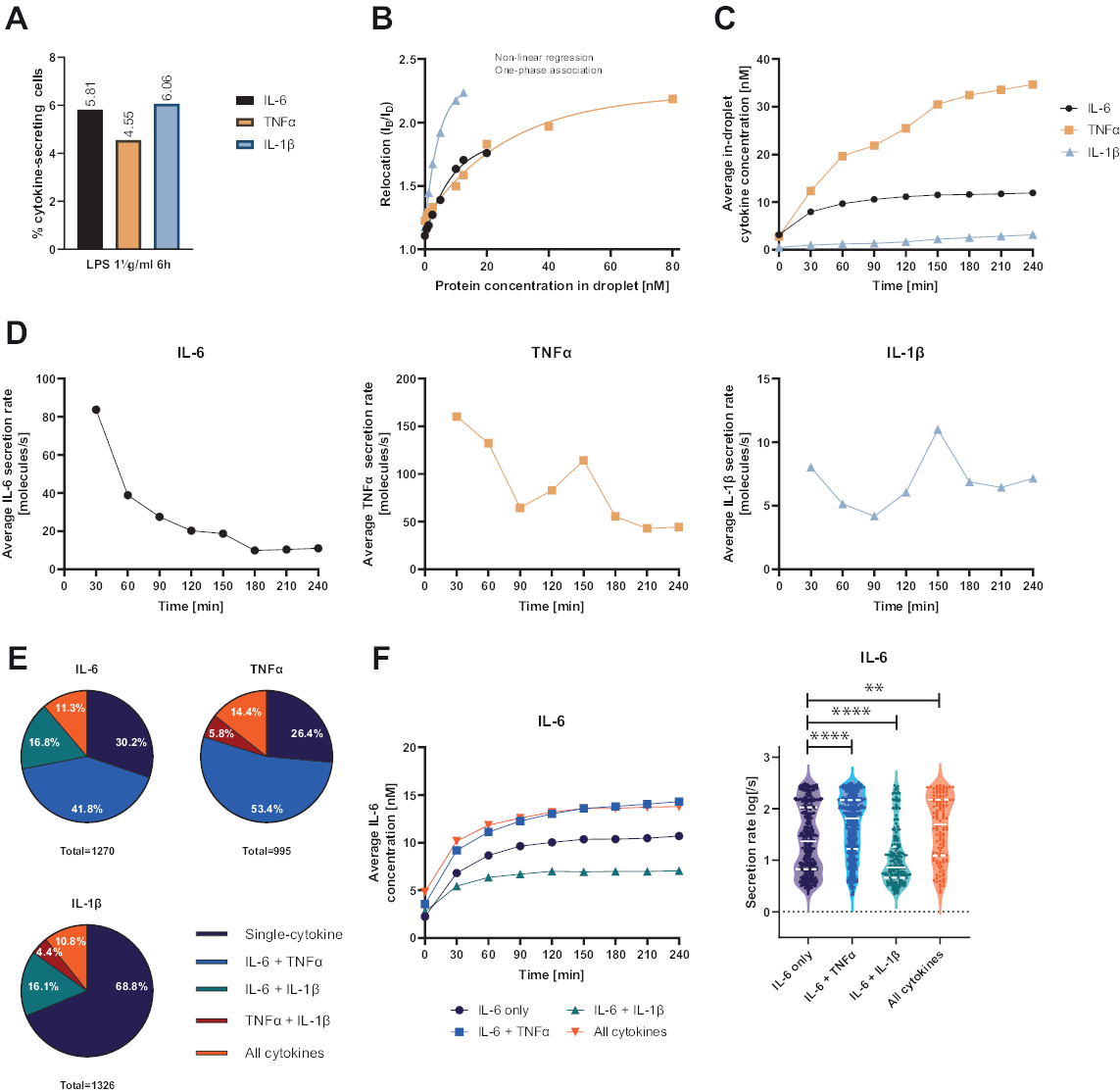
Die vorgestellte funktionale Einzelzellplattform ermöglichte die Messung mehrerer Parameter. Zunächst wird ähnlich wie bei Standardtechniken am Ende der Messung die Häufigkeit der sezernierenden Zellen dargestellt (Abbildung 4A). Nach der Stimulation mit 1 μg/ml Lipopolysaccharid (LPS) für 6 Stunden von mononukleären Zellen des peripheren Blutes (PBMC) sezernierten 5,81% der Zellen IL-6 (n= 1270), 4,55% TNFα (n= 995) und 6,06% IL-1β (n= 1326).
Um die Zytokinsekretion zu quantifizieren, wurden Kalibrierkurven mit bekannten Konzentrationen rekombinanter Zytokine erstellt (Abbildung 4B). Diese Kalibrierkurven ermöglichen die Quantifizierung der Zytokinkonzentrationen in Tröpfchen über die Zeit. Beispielhaft erreichte die durchschnittliche IL-6-Konzentration in Tröpfchen nach 90 min für LPS-stimulierte PBMC ein Plateau, während die durchschnittliche IL-1β-Konzentration in Tröpfchen ab 90 min schneller anstieg, was die dynamische Auflösung der Plattform und die Möglichkeit zeigt, Zellsubpopulationen zu extrahieren, die spezifische Zytokine sezernieren (Abbildung 4C). Da sich die Konzentration zwischen den Messpunkten ändert, ist es möglich, dynamische Sekretionsraten pro Zytokin zu berechnen. Unter Berücksichtigung der durchschnittlichen Sekretionsrate für jedes Zytokin (Abbildung 4D) zeigten IL-6-sezernierende Zellen eine konstante Abnahme der durchschnittlichen Sekretionsrate, während TNFα- und IL-1β-sezernierende Zellen beide einen Anstieg der Sekretionsrate nach 90 min Messzeit und einen zweiten Rückgang nach 150 min zeigten.
Darüber hinaus ist es möglich, Zellen in Abhängigkeit von den sezernierten und co-sezernierten Zytokinen in Subpopulationen zu gruppieren (Abbildung 4E). Hier werden IL-6 und TNFα von 30,2 % bzw. 26,4 % der Zellen, die IL-6 bzw. TNFα sezernieren, einfach sezerniert, während einfach sezernierende IL-1β-Zellen 68,8 % aller IL-1β-sezernierenden Zellen ausmachten. Darüber hinaus können die Auswirkungen der Co-Sekretion auf die sezernierten Konzentrationen und Sekretionsraten geklärt werden (Abbildung 4F). Bei der Betrachtung von IL-6-sezernierenden Zellen wurden unterschiedliche Mengen an IL-6 sezerniert, wenn die Zellen zusätzlich TNFα oder IL-1β produzierten. In ähnlicher Weise unterschied sich die Verteilung der gemittelten Sekretionsraten über die Messung statistisch zwischen den Zellen, die neben TNFα (höhere Sekretionsraten) und IL-1β (niedrigere IL-6-Sekretionsraten) nur IL-6 oder IL-6 sezernierten.

Abbildung 4: Repräsentative Ergebnisse der IL-6-, TNFα- und IL-1β-sezernierenden PBMC nach 6-stündiger Stimulation mit 1 μg/ml LPS. (A) Prozentualer Anteil der PBMC, die IL-6, TNFα und IL-1β am Ende der 4-stündigen Messung sezerniert. (B) Multiplex-Zytokin-Kalibrierungskurven werden mit bekannten Konzentrationen rekombinanter Zytokine erstellt. Dies ermöglicht die Quantifizierung von Zellexperimenten, indem aus dem Verlagerungswert die Zytokinkonzentration im Tröpfchen berechnet wird. Die Punkte wurden mit einer nichtlinearen einphasigen Assoziationskurvenanpassung angepasst, r2=0,9926 (IL-6), 0,9901 (TNFα), 0,9990 (IL-1β). (C) Durchschnittliche sekretierte Konzentrationen von IL-6, TNFα und IL-1β, die durch Seernierung von PBMC über die Messzeit von 4 h freigesetzt wurden. (D) Durchschnittliche Sekretionsraten von IL-6, TNFα und IL-1β über die Messzeit von 4 Stunden. (E) Relativer Prozentsatz der mitsezernierenden Zellen, die IL-6, TNFα oder IL-1β und Kombinationen davon sezernieren. Normalisiert auf alle sekretierenden Zellen, die für jedes Zytokin nachgewiesen wurden. (F) Gemittelte IL-6-Konzentrationen über die Messzeit und Verteilungen der durchschnittlichen Sekretionsrate (log) für IL-6-sezernierende Zellen mit Co-Sekretionsauflösung (n=383 für nur IL-6, n=531 für IL-6 + TNFα, n= 213 für IL-6 + IL-1β und n=143 für IL-6+TNFα+IL-1β). Statistische Unterschiede in den Verteilungen der Sekretionsrate wurden mit zweiseitigen, ungepaarten, nichtparametrischen Kolmogorov-Smirnov-Tests mit einer Konfidenz von 95% bewertet, wobei der p-Wert dargestellt ist. ** (p <0,002) und **** (p <0,0001). Die volle Linie stellt den Median und die gestrichelte Linie die Quartile dar. nZellen insgesamt = 21 866. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Um zusätzliche Informationen auf Einzelzellebene zu extrahieren, kann eine Sigmoidfunktion an die Konzentrations-Zeitpunkte jeder Zelle und jedes Zytokins angepasst werden (Abbildung 5). Ein beispielhafter Datensatz der Konzentration über die Zeit für eine Zelle und die entsprechende sigmoidale Anpassung ist in Abbildung 5A dargestellt. Hier liefert das Anpassungsverfahren der kleinsten Quadrate die folgenden Parameter: C, entsprechend dem oberen Plateauwert der Kurve, t50 , der die zeitliche Verschiebung der Kurve von Null quantifiziert, und die Hügelsteigung m, die die Steilheit des ansteigenden Teils der Sigmakurve mit 10 % und 90 % Konzentrationswerten beschreibt, die während der gesamten Messung erreicht wurden. Aus diesen Anpassungsparametern können einige Kurvendeskriptoren extrahiert werden, wie in Schritt 7.12 beschrieben. Dies ergibt Cmax, den höchsten Konzentrationswert der Daten, tstart, die Startzeit der Sekretion, definiert als das Erreichen von 10 % des Konzentrationswerts des oberen Plateaus, und SRlin, die Sekretionsrate während des ansteigenden Teils der Kurve.
Um Zellsubpopulationen zu klassifizieren, wurden die Kurvendeskriptoren, die aus allen Einzelzellanpassungen gewonnen wurden, in jeweils drei Kategorien eingeteilt: DieCmax-Werte wurden in niedrig, mittel und hoch gruppiert,wobei die t-Werte in frühe, mittlere und späteSR-Lin in langsame, mittlere und schnelle Sekretoren eingeteilt wurden. Um diese Klassifizierung zu veranschaulichen, werden vier exemplarische Einzelzellsekretionskurven und ihre entsprechenden Kurvendeskriptoren gezeigt (Abbildung 5A-D), wobei Kurve A die Eigenschaften eines frühen niedrigen Sekretors mittlerer Rate aufweist, Kurve B ein früher, langsamer und hoher Sekretor, Kurve C ein früher schneller Sekretor mit hohem Sekretor und Kurve D eine späte niedrige Sekretion. Es ist wichtig zu beachten, dass die Cutoffs für diese Kriterien zell-, zytokin- und assayparameterspezifisch sind und für jede Forschungsfrage angepasst werden müssen. Darüber hinaus wurde hier nur die IL-6-Sekretion von PBMC nach 1 μg/mL LPS-Stimulation für 6 h berücksichtigt, was bedeutet, dass die meisten Zellen mit 80% bzw. 79% frühe und hohe Sekretoren waren (Abbildung 5E-F). In Bezug auf die Sekretionsrate wurde eine bipolare Reaktion beobachtet, wobei 55% der IL-6-sezernierenden Zellen langsame Sekretoren und 39% schnelle Sekretoren sind (Abbildung 5G).
Um das Sekretionsverhalten weiter zu charakterisieren, wurden die Kurvendeskriptoren für jede Zelle gegeneinander aufgetragen und verschiedene Cluster extrahiert (Abbildung 5H-J). Es gibt keine klare Korrelation zwischent-Start undC-max (Abbildung 5H): Die beiden größten Populationen waren frühe niedrige Sekretoren und hohe Sekretoren unabhängig vom Sekretionsstart. Unter Berücksichtigung der Beziehung zwischent-Start und SRlin (Abbildung 5I) waren die meisten Zellen frühe langsame Sekretoren mit einer klaren Population von frühen hohen Sekretoren und wenigen langsamen/mittleren bis späten Sekretoren. Hinsichtlich der Korrelationen von SRlin und Cmax (Abbildung 5J) waren fast keine schnellen niedrigen bis mittleren Sekretoren vorhanden, mit nur einer größeren Population von schnellen niedrigen Sekretoren. Darüber hinaus gab es eine große Population von schnellen Sekretoren, die nicht von der maximal gemessenen Konzentration abhingen, und zwei Populationen von hohen Sekretoren, die entweder langsam oder schnell sezernierten. Zusammenfassend lässt sich sagen, dass die Untersuchung des Zusammenhangs zwischen den Kurvendeskriptoren für einzelne Zellen zu einer wesentlich detaillierteren Analyse führt und möglicherweise neue biologische Erkenntnisse aus Einzelzellsekretionsmessungen extrahieren kann.
Mit der oben vorgestellten Analyse haben wir die Sekretionsdynamik von co-sekretierenden Zellen extrahiert (Abbildung 6). Zwei Beispielkurven zeigen eine unterschiedliche Dynamik der Co-Sekretion von IL-6 und TNFα aus zwei Einzelzellen mit einem gleichzeitigen Start beider Zytokine (Abbildung 6A) oder einem sequentiellen Sekretionsstart, wobei IL-6 zuerst sezerniert wird (Abbildung 6B). Zur Klassifizierung aller co-sekretierenden Zellen wurde eine Sekretionsverzögerung von 60 min definiert, wobei alle Zellen, die innerhalb dieses Bereichs mit der Sekretion beginnen, als simultane Sekretoren und alle Zellen mit längerer Verzögerung als sequentielle Sekretoren betrachtet werden. Diese Analyse ermöglichte auch die Beobachtung, welches Zytokin zuerst sezerniert wurde. Für IL-6 und TNFα wurde in 76 % der Zellen hauptsächlich eine gleichzeitige Co-Sekretion beobachtet (Abbildung 6C), während für IL-6 und IL-1β in 86 % der Zellen eine sequentielle Co-Sekretion beobachtet wurde, wobei IL-6 in den meisten Fällen das erste Zytokin war, das sezerniert wurde (Abbildung 6D).
Betrachtet man den Startzeitpunkt der Sekretion für die verschiedenen Zytokine für alle einzelnen co-sekretierenden Zellen, so konnte in den durchgeführten Experimenten keine eindeutige Korrelation zwischen den Startzeiten der Sekretion beobachtet werden. Für die Co-Sekretion von IL-6 und TNFα (Abbildung 6E) war ein größerer vertikaler Cluster um 0 min vorhanden, was den co-sezernierenden Zellen entspricht, die häufiger mit IL-6 beginnen. Für die Co-Sekretion von IL-6 und IL-1β (Abbildung 6F) begannen die meisten Zellen zu Beginn der Messung mit der Sekretion von IL-6, während IL-1β hauptsächlich später sezerniert wurde. Zusammenfassend lässt sich sagen, dass die hier vorgestellte Analyse die Identifizierung verschiedener Sekretor-Subpopulationen und komplexer Zytokin-Co-Sekretionsdynamiken ermöglichte.

Abbildung 5: Detaillierte Analyse verschiedener dynamischer Sekretionsmuster für einzelne IL-6-sezernierende Zellkurven. (A) Repräsentative Einzelzell-Zytokin-Konzentrationsdaten über die Messzeit mit der angepassten Sigmoidkurve und den extrahierten Parametern. (B-D) Drei beispielhafte Einzelzell-Zytokin-Konzentrationskurven für die verschiedenen Zytokinsekretortypen, die für die IL-6-Sekretion nach LPS-Stimulation gefunden wurden. (E-G) Prozentuale Anteile von IL-6-sezernierenden Zellen, die mit folgenden Kriterien (n=633) in die verschiedenen Sekretortypen eingeteilt werden: E. Cmax: niedrig <5 nM, hoch >19,5 nM, F. tStart: früh <30 min, spät >120 min, G. SRlin: langsam <250 Moleküle/s, schnell >750 Moleküle/s. (H-J) Beziehung zwischen den drei Sekretionskurvendeskriptoren Cmax, tstart und SRlin für jede einzelne Zelle (n=633). Die große Population bei Cmax=20nM resultiert aus dem Erreichen der oberen Nachweisgrenze des Assays. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 6: Extraktion von Co-Sekretionsmustern aus Einzelzell-Konzentrationskurven. (A-B) Repräsentative Konzentrationskurven für Einzelzellen, die IL-6 und TNFα (A) gleichzeitig bzw. (B) sequentiell co-sezernieren. (C-D) Prozentsatz der Zellen, die eine gleichzeitige und sequentielle Co-Sekretion von IL-6 und TNFα (n=249) bzw. IL-6 und IL-1β (n=72) aufweisen. Die sequentielle Sekretion wird durch die Verzögerung zwischen den Zytokinsekretionsstarts von mehr als 60 Minuten definiert. Farben zeigen an, welches der Zytokine zuerst mit der Sekretion begonnen hat. (E-F) Beziehung zwischen den Startzeiten der Sekretion für die verschiedenen Zytokine für jede sekretierende Zelle (nIL6-TNFα=249, nIL6-IL1β=72). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Diskussion
Die Freisetzung und Sekretion von Zytokinen wird in der Immunologie und klinischen Medizin häufig untersucht3. Eine unausgewogene Zytokinsekretion kann zu nachteiligen Auswirkungen für Patienten führen, die an Infektionen, aber auch an neurologischen Erkrankungen, Entzündungen oder Krebs leiden 26,27,28. Obwohl ihre Bedeutung für Gesundheit und Krankheit gut belegt ist, bleibt die Untersuchung von Zytokinen und ihren sezernierenden Zellen eine Herausforderung, da die derzeitigen Methoden nicht in der Lage sind, Zytokine, die aus einer einzelnen Zelle stammen, zeitaufgelöst genau nachzuweisen und zu quantifizieren. Für den hier vorgestellten Workflow wurde ein etabliertes Stimulationsprotokoll mit PBMC verwendet und deren Sekretion von IL-6, TNF-α und IL-1β gemessen. Die Entscheidung, PBMCs anstelle von einzelnen, gereinigten Subpopulationen zu verwenden, ergab sich aus der vorherigen Anwendung zur Untersuchung von Zytokinfreisetzungssyndromen (CRS)23, einer Erkrankung, die durch stark erhöhte Plasmakonzentrationen von proinflammatorischen Zytokinen, einschließlich IL-6, TNF-α und IL-1β gekennzeichnet ist29. Da CRS in der Regel nicht nur an eine Population gebunden ist, verwendeten wir PBMCs, wie sie in vivo vorhanden wären. Zelluläre Subpopulationen können jedoch einzeln gereinigt und beurteilt werden, wenn die wissenschaftliche Fragestellung diesen Schritt erfordert. Die Inkubationszeit, die Stimulationsbedingungen und die dynamischen Assay-Bereiche wurden optimiert, um die Sekretion für die drei interessierenden Zytokine zu messen. Der Arbeitsablauf und die hier vorgestellten Daten zeigen, wie die zeitaufgelöste Einzelzellsekretion mehrerer Zytokine eingerichtet, kalibriert, quantifiziert, gemessen und analysiert werden kann. Dieses Protokoll liefert eine Blaupause dafür, wie eine multifunktionale Analyse der Zytokinsekretion die große funktionelle und dynamische Vielfalt des bei Patienten sezernierten Zytokins ermöglichen könnte.
Mehrere entscheidende Aspekte des beschriebenen Assay-Protokolls ermöglichen eine einzigartige biologische Auslesung. Zunächst ermöglichte die Einzelzellverkapselung in mikrofluidischen Tröpfchen die Extraktion von Daten für jede einzelne Zelle. Ereignisse von Mehrfachzellverkapselungen können je nach Fragestellung durch Bildanalyse detektiert und ein- oder aussortiert werden. Zweitens ermöglichte die Einbeziehung mehrerer unabhängiger fluoreszierender In-Droplet-Immunoassays und die Ausrichtung der funktionalisierten Nanopartikel die quantitative Messung von bis zu drei Zytokinkonzentrationen parallel. Dieses Multiplexing ermöglichte die Analyse von Zytokin-Co-Sekretionsmustern auf Einzelzellebene. Drittens ermöglichte die Immobilisierung der Tröpfchen die zeitliche Messung und Korrelation der Zytokinsekretion für jede sekretierende Zelle und ermöglichte die Unterscheidung zwischen ko-okkurrenter und sequentieller Sekretion. Die Zeitauflösung lieferte auf einzigartige Weise Daten zu Sekretionsmustern und Subpopulationen verschiedener Sekretortypen. Schließlich ermöglichte die parallelisierte Bildanalyse die effiziente Extraktion und Verfolgung großer Datenmengen aus Messungen mit über 20.000 einzelnen Zellen. Die Extraktion aus einzelnen Sekretionskurven ermöglichte darüber hinaus die Entdeckung phänotypischer Subpopulationen und Funktionalitäten.
Neben seiner einzigartigen Auslesung hat der Assay auch technische Vorteile gegenüber der Standard-Zytokinanalyse. Dank der geringen Größe der Verkapselungskompartimente von etwa 60 pL können absolute Mengen an sezernierten Zytokinen direkt aus der biologischen Quelle nachgewiesen werden, wobei die Nachweisgrenzen der Zellsekretion entsprechen. Bei der Miniaturisierung des Assays werden auch kleinere Mengen teurer Bioreagenzien verwendet. Darüber hinaus erfordert der Aufbau nur sehr wenig Spezialgeräte, die in Biologie- und Bioengineering-Laboren oft bereits vorhanden sind. Fluoreszenzmikroskope sind weit verbreitet, und Spritzenpumpen werden häufig in biotechnischen Laboratorien eingesetzt oder können zu relativ geringen Kosten erworben werden. Ist eine Zellkultur vorhanden, belaufen sich die Kosten für die komplette Ausrüstung für die Durchführung der Experimente auf rund 148.000 Euro, wobei der Großteil auf das automatisierte Epifluoreszenzmikroskop entfällt (130.000 Euro). Ein solches Instrument ist jedoch häufig in biologischen Laboratorien zu finden, und der Rest der Kosten wird auf die Spritzenpumpe (13.000 Euro, aber es gibt günstigere Alternativen) und kleinere Geräte verteilt. Die Herstellung des Tröpfchenchips und der Beobachtungskammer ist sehr gut beschrieben17 und kann außerhalb einer Reinraumumgebung mit der erforderlichen Infrastruktur, wie z. B. Öfen und Plasmareinigern, durchgeführt werden, die in den meisten biotechnologischen Laboratorien vorhanden sind. Alternativ stehen verschiedene Anbieter zur Verfügung, um interessierte Labore mit Tröpfchengenerator-Chips zu versorgen. Aufgrund der geringen benötigten Volumina ist der Assay kostengünstig und einfach einzurichten.
Um ein Höchstmaß an Reproduzierbarkeit zu gewährleisten, haben wir einige kritische Schritte für den Erfolg des Protokolls identifiziert. Ein häufiges Problem für Erstanwender ist die Tröpfchenbewegung während der Messung. Während die Analysesoftware einzelne Tröpfchen bis zu einem gewissen Grad verfolgen kann, führt übermäßige Bewegung zu einem Verlust der Einzelzellauflösung und ungenauen Ergebnissen. Bewegungen können vermieden werden, indem ordnungsgemäß luftdichte Messkammern, die richtige Tröpfchengröße und die richtigen Kammergrößen, eine kurze Gleichgewichtsphase vor Beginn der Messung und die richtige Tensidkonzentration verwendet werden. Ein weiterer kritischer Schritt ist die genaue Fokussierung vor Beginn der Messung. Eine unsachgemäße Fokussierung führt zu deutlich verringerten Fluoreszenz-Relocation-Werten und einer Unterschätzung der Menge des sezernierten Zytokins. Abhängig von der Fragestellung und dem vorliegenden Protokoll ist schließlich das richtige Timing zwischen den verschiedenen Schritten von größter Bedeutung für die Reproduzierbarkeit. Insbesondere die Wartezeit zwischen dem Befüllen der Kammer und dem Start der Messung sollte konstant sein, da sonst das Messfenster der sezernierten Zytokine verpasst werden könnte.
Zu den Einschränkungen der vorgestellten Technologie gehört die eingeschränkte Fähigkeit, die Zellen nach der Verkapselung weiter zu manipulieren. Daher ist es derzeit nicht möglich, Stimulanzien, Antikörper oder zusätzliche Reagenzien hinzuzufügen oder zu entfernen. Da die Zellen in ihrem isolierten Bioreaktor verkapselt sind, können während der Messung keine Wechselwirkungen zwischen den Zellen (kontaktbasierte oder parakrine Signalwege) stattfinden. Diese Einschränkung kann teilweise durch vorherige Masseninkubationen überwunden werden. Darüber hinaus sind auch verstärkte autokrine Effekte durch sezernierte Zytokine möglich, die nicht quantifiziert oder mit Sicherheit ausgeschlossen werden können, da nur Antikörper-detektierte sezernierte Zytokine gemessen werden. Die isolierte Sicht auf die Zytokinsekretion muss also immer im Kontext der entsprechenden Fragestellung und Anwendung beschrieben werden. Diese Einschränkung könnte jedoch auch für die detaillierte Untersuchung von verkapselten Multiples, Dubletten und Tripletts verwendet werden, wenn dies von Interesse ist. Dies würde ein interessantes Setup bieten, das nützlich ist, um Zell-Zell-Kontakt oder parakrine Fragen zu untersuchen. Schließlich ist auch der Dynamikbereich des Assays begrenzt und muss an die spezifische Anwendung angepasst werden. Hier haben wir den dynamischen Bereich des Assays an die erwartete sezernierte Menge der gemessenen Zytokine angepasst.
Um die Fähigkeiten und die Anwendbarkeit des Assays weiter zu verbessern, könnten in Zukunft mehrere Entwicklungen in Bezug auf biologische, technische und Datenanalyseaspekte in Angriff genommen werden. Auf der biologischen Seite könnte die Messung von zusätzlichen Zytokinen, anderen sezernierten Proteinen, Stoffwechsel- oder Zelloberflächenmarkern durch Anpassung des Assays integriert werden. Darüber hinaus kann dieser Assay in einen Arbeitsablauf neben anderen zellbasierten Assays integriert werden, um die Messwerte zu erweitern (z. B. Durchflusszytometrie, Färbung oder Sequenzierung). Darüber hinaus könnte die Benutzerfreundlichkeit des Assays vereinfacht werden, z. B. durch die Schaffung eines integrierten mikrofluidischen Chips für die Tröpfchenerzeugung und -beobachtung, wodurch möglicherweise eine breitere Anwendung außerhalb von biotechnologischen Laboratorien in einem klinischen Umfeld ermöglicht wird. In Bezug auf die Datenanalyse könnte die Extraktion und Verfolgung von Informationen aus Bildern durch eine verbesserte Automatisierung und die Verwendung von Ansätzen des maschinellen Lernens erweitert werden, z. B. um das Vorhandensein und die Position der Zelle(n) und der Beadline in jedem Tröpfchen ohne Fluoreszenzmarkierung zu erkennen. Dadurch würden zusätzliche Fluoreszenzkanäle eröffnet, die für Immunoassays verwendet werden könnten, was dazu führen würde, dass noch mehr Zytokine parallel gemessen werden könnten.
Der vorgestellte Assay und die zugehörigen Protokolle und Analysen können für verschiedene potenzielle Anwendungsfälle im Zusammenhang mit der Dynamik der Zytokinsekretion eingesetzt werden. Genauer gesagt könnte der Assay möglicherweise grundlegende immunologische Fragen beantworten, wie z. B. die Identifizierung von zelltyp- und aktivierungsspezifischen Zytokinsekretionsprofilen, die Polyfunktionalität von Zytokin-sezernierenden Zellen oder die Zeitlichkeit und Aufrechterhaltungsmechanismen von Zytokin-Gleichgewichten. Darüber hinaus könnte die Plattform in klinischen Anwendungen die Entschlüsselung der Rolle von Zytokinen während aktiver oder chronischer Entzündungsreaktionen ermöglichen, wie sie bei COVID-19 beobachtet wurden30, oder ein Werkzeug für die Stratifizierung von Patienten und die Personalisierung von Behandlungen auf der Grundlage einzigartiger Signaturen bieten, wie z. B. bei der Autoinflammation31. Zusammenfassend lässt sich sagen, dass die quantitative zeitaufgelöste Bestimmung der Zytokinsekretion aus einzelnen Zellen eine dringend benötigte Methode ist, da sie aufklärt, wie ein bestimmtes Medikament, eine Infektion, eine genetische Veränderung oder eine Ex-vivo-Stimulation eine bestimmte Reaktion hervorruft.
Offenlegungen
Spezifische Aspekte, wie z.B. die Beadline-Messungen von Zellen, wurden patentiert.
Danksagungen
Dieses Projekt wurde unterstützt durch das Stipendium #2021-349 des Strategischen Schwerpunktbereichs Personalisierte Gesundheit und verwandte Technologien (PHRT) des ETH-Bereichs (Eidgenössische Technische Hochschulen), das Starting Grant des Europäischen Forschungsrats (Grant #803'336) und den Schweizerischen Nationalfonds (Grant #310030_197619). Darüber hinaus danken wir Guilhem Chenon und Jean Baudry für ihre Arbeit und Entwicklung des ersten DropMap-Analysators.
Materialien
| Name | Company | Catalog Number | Comments |
| 008-FluoroSurfactant | RAN Biotechnologies | 008-FluoroSurfactant-10G | |
| 2-Stream flow-focusing droplet maker, 30 µm nozzle, PFOS hydrophobic surface treatment | Wunderli chips | ||
| Alexa Fluor 647 NHS Ester | ThermoFisher | A20006 | https://www.thermofisher.com/ch/en/home/references/protocols/cell-and-tissue-analysis/labeling-chemistry-protocols/fluorescent-amine-reactive-alexa-fluor-dye-labeling-of-igm-antibodies.html |
| Anti-Human IL-1β (Monoclonal Mouse), AF647 labelled in-house | PeproTech | 500-M01B | |
| ARcare92524 double-sided adhesive tape | Adhesvies Reasearch | ARcare92524 | |
| Bio-Adembeads Streptavidin plus 300nm | Ademtech | Cat#03233 | |
| Biotinylated Goat Anti-Human IL-1β | PeproTech | 500-P21BGBT | |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | A3059 | |
| Cell Scraper | TPP | 99002 | |
| CellTrace Violet Cell Proliferation Kit | Invitrogen | C34557 | Cell staining solution |
| Chromafil Xtra PTFE-45/25 syringe filters | Macherey-Nagel | 729205 | |
| Costar 6-well Clear Flat Bottom Ultra-Low Attachment | Corning | 3471 | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10283 | |
| D-Biotin | Fluorochem | M02926 | |
| DPBS, no calcium, no magnesium | Gibco | 14196-094 | |
| epT.I.P.S. Standard 2-200 µl | Eppendorf | 30000889 | |
| Ethylenediaminetetraacetic acid disodium salt solution | Sigma-Aldrich | 3690 | |
| EZ-LINK-NHS-PEG4-Biotin | ThermoFisher | A39259 | https://www.thermofisher.com/order/catalog/product/20217 |
| FcR Blocking Reagent, human | Miltenyi Biotec | 130-059-901 | |
| Fetal Bovine Serum | Gibco | 10270-106 | |
| Handy dish soap | Migros | 5.01002E+11 | |
| HEPES (1 M) | Gibco | 15630-080 | |
| HFE-7500 Oil 3M TM Novec | Fluorochem | B40045191 | |
| Idex F-120 Fingertight One-Piece Fitting, Standard Knurl, Natural PEEK, 1/16" OD Tubing, 10-32 Coned | Cole-Parmer | GZ-02014-15 | |
| IL-6 Monoclonal Antibody (MQ2-13A5 - Rat), FITC | ThermoFisher | 11-7069-81 | |
| IL-6 Monoclonal Antibody (MQ2-39C3), Biotin | ThermoFisher | 13-7068-85 | |
| KnockOut Serum Replacement | ThermoFisher | 10828-010 | |
| Loctite AA 3491 curable UV glue | Henkel AG & Co | 3491 | |
| Microscope slides (76x26x1mm, clear white) | Menzel Gläser | ||
| Mineral oil light | Sigma-Aldrich | 330779 | |
| NanoPort Assembly Headless, 10-32 Coned, for 1/16" OD | Idex | N-333 | |
| Neodymium block magnet | K&J Magnetics | BZX082 | |
| Omnifix-F Spritze, 1 ml, LS | Braun | 9161406V | |
| Penicillin-Streptomycin (10,000 U/mL) | Gibco | 15140-122 | |
| Phosphate buffered saline | Sigma-Aldrich | P4417 | |
| Pluronic F-127, 0.2 µm filtered (10% Solution in Water) | ThermoFisher | P6866 | |
| Precision wipes | Kimtech Science | 5511 | |
| PTFE microtubing 0.30 × 0.76 mm | FisherScientific | 1191-9445 | |
| PTFE microtubing 0.56 × 1.07 mm | FisherScientific | 1192-9445 | |
| Recombinant Human IL-1β | Peprotech | Cat#200-01B | |
| Recombinant Human IL-6 | Peprotech | Cat#200-06 | |
| Recombinant human serum albumine (HSA) | Sigma-Aldrich | A9731 | |
| Recombinant Human TNF-α | Peprotech | Cat#300-01A | |
| Reusable biopsy punch diameter 0.75 mm and 6 mm | Stiefel | 504529 and 504532 | |
| RPMI 1640 Medium, no phenol red | Gibco | 11835-030 | |
| Standard LPS, E. coli K12 | InvivoGen | tlrl-eklps | |
| Sterican needles 23 G for 0.56 mm diameter microtubing | FisherScientific | 15351547 | |
| Sterican needles 27 G for 0.30mm diameter microtubing | FisherScientific | 15341557 | |
| TNF alpha Monoclonal Antibody (MAb11), PE | ThermoFisher | 12-7349-81 | |
| TNF-alpha Monoclonal Antibody (MAb1), biotinylated in-house | ThermoFisher | 14-7348-85 | |
| Trypan Blue Stain (0.4%) for use with the Countess Automated Cell Counter | Invitrogen | T10282 | |
| Vacuum Filtration "rapid"-Filtermax | TPP | 99500 | |
| Devices | |||
| Cameo 4 automatic cutting machine | Silhouette | ||
| Cetoni Base 120 + 3x NEMESYS Low Pressure Syringe Pumps | Cetoni | NEM-B101-03 A | |
| Countess II Automated Cell Counter | ThermoFisher | ||
| Inverted Epi-fluorescence microscope Ti2 | Nikon | ECLIPSE Ti2-E, Ti2-E/B*1 | |
| OKO Lab Cage Incubator, dark panels | OKO Lab | ||
| ORCA-Fusion Digital CMOS camera | Hammatsu | C14440 | |
| SOLA Light Engine | Lumencor | sola 80-10247 |
Referenzen
- Chen, L., et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 9 (6), 7204-7218 (2017).
- Cicchese, J. M., et al. Dynamic balance of pro- and anti-inflammatory signals controls disease and limits pathology. Immunol Rev. 285 (1), 147-167 (2018).
- Liu, C., et al. Cytokines: From clinical significance to quantification. Adv Sci. 8 (15), e2004433 (2021).
- Rojas, J. M., Avia, M., Martín, V., Sevilla, N. IL-10: A multifunctional cytokine in viral infections. J Immunol Res. 2017, 6104054 (2017).
- Kohanawa, Y. M. A regulatory effect of the balance between TNF-α and IL-6 in the granulomatous and inflammatory response to Rhodococcus aurantiacus infection in mice. J Immunol. 177 (1), 642-650 (2006).
- Geginat, J., et al. Plasticity of human CD4 T cell subsets. Front Immunol. 5, 630 (2014).
- Sallusto, F. Heterogeneity of human CD4+ T cells against microbes. Ann Rev Immunol. 34 (1), 317-334 (2016).
- Chetaille Nézondet, A. L., Poubelle, P. E., Pelletier, M. The evaluation of cytokines to help establish diagnosis and guide treatment of autoinflammatory and autoimmune diseases. J Leukocyte Biol. 108 (2), 647-657 (2020).
- Sims, J. T., et al. Characterization of the cytokine storm reflects hyperinflammatory endothelial dysfunction in COVID-19. J Allergy Clin Immunol. 147 (1), 107-111 (2021).
- Yasen, A., et al. Single-cell RNA sequencing reveals the heterogeneity of infiltrating immune cell profiles in the hepatic cystic echinococcosis microenvironment. Infection and Immunity. 89 (12), (2021).
- Jiang, Y., et al. Single-cell RNA sequencing highlights intratumor heterogeneity and intercellular network featured in adamantinomatous craniopharyngioma. Sci Adv. 9 (15), (2023).
- Tanguay, S., Killion, J. J. Direct comparison of ELISPOT and ELISA-based assays for detection of individual cytokine-secreting cells. Lymphokine Cytokine Res. 13 (4), 259-263 (1994).
- Bucheli, O. T. M., Sigvaldadóttir, I., Eyer, K. Measuring single-cell protein secretion in immunology: Technologies, advances, and applications. Eur J Immunol. 51 (6), 1334-1347 (2021).
- Brower, K. K., et al. Double emulsion flow cytometry with high-throughput single droplet isolation and nucleic acid recovery. Lab Chip. 20 (12), 2062-2074 (2020).
- Brower, K. K., et al. Double emulsion picoreactors for high-throughput single-cell encapsulation and phenotyping via FACS. Anal Chem. 92 (19), 13262-13270 (2020).
- Luo, X., Chen, J. Y., Ataei, M., Lee, A. Microfluidic compartmentalization platforms for single cell analysis. Biosensors. 12 (2), 58 (2022).
- Bounab, Y., et al. Dynamic single-cell phenotyping of immune cells using the microfluidic platform DropMap. Nat Protoc. 15 (9), 2920-2955 (2020).
- Eyer, K., et al. Single-cell deep phenotyping of IgG-secreting cells for high-resolution immune monitoring. Nat Biotechnol. 35 (10), 977-982 (2017).
- Gaa, R., et al. Versatile and rapid microfluidics-assisted antibody discovery. mAbs. 13 (1), 198130 (2021).
- Gerard, A., et al. High-throughput single-cell activity-based screening and sequencing of antibodies using droplet microfluidics. Nat Biotechnol. 38 (6), 715-721 (2020).
- De Jonghe, J., et al. spinDrop: a droplet microfluidic platform to maximise single-cell sequencing information content. Nat Comm. 14 (1), 4788 (2023).
- Wheeler, M. A., et al. Droplet-based forward genetic screening of astrocyte-microglia cross-talk. Science. 379 (6636), 1023-1030 (2023).
- Portmann, K., Linder, A., Oelgarth, N., Eyer, K. Single-cell deep phenotyping of cytokine release unmasks stimulation-specific biological signatures and distinct secretion dynamics. Cell Rep Meth. 3 (7), 100502 (2023).
- Portmann, K., Linder, A., Eyer, K. Stimulation-induced cytokine polyfunctionality as a dynamic concept. eLife. 12, 89781 (2023).
- Armbruster, D. A., Pry, T. Limit of blank, limit of detection and limit of quantitation. Clin Biochem Rev. 29, S49-S52 (2008).
- Yang, J., et al. New insight into neurological degeneration: Inflammatory cytokines and blood-brain barrier. Front Mol Neurosci. 15, 1013933 (2022).
- Kim, P. S., Ahmed, R. Features of responding T cells in cancer and chronic infection. Curr Opin Immunol. 22 (2), 223-230 (2010).
- Becher, B., Spath, S., Goverman, J. Cytokine networks in neuroinflammation. Nat Rev Immunol. 17 (1), 49-59 (2017).
- Cosenza, M., Sacchi, S., Pozzi, S. Cytokine release syndrome associated with T-cell-based therapies for hematological malignancies: Pathophysiology, clinical presentation, and treatment. Int J Mol Sci. 22 (14), 7652 (2021).
- Hu, B., Huang, S., Yin, L. The cytokine storm and COVID-19. J Med Virol. 93 (1), 250-256 (2021).
- Marcuzzi, A., et al. Autoinflammatory diseases and cytokine storms-Imbalances of innate and adaptative immunity. Int J Mol Sci. 22 (20), 11241 (2021).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten