
Method Article
Una técnica-Label libre para el Imaging espacio-temporal de las secreciones sola célula
En este artículo
Resumen
La comunicación entre celulares es fundamental para el control de diversas actividades fisiológicas dentro y fuera de la célula. En este trabajo se describe un protocolo para la medición de la naturaleza espacio-temporal de las secreciones de células individuales. Para ello, se utiliza un enfoque multidisciplinario que integra nanoplasmonic etiqueta libre de detección con imágenes de células vivas.
Resumen
Inter-cellular communication is an integral part of a complex system that helps in maintaining basic cellular activities. As a result, the malfunctioning of such signaling can lead to many disorders. To understand cell-to-cell signaling, it is essential to study the spatial and temporal nature of the secreted molecules from the cell without disturbing the local environment. Various assays have been developed to study protein secretion, however, these methods are typically based on fluorescent probes which disrupt the relevant signaling pathways. To overcome this limitation, a label-free technique is required.
In this paper, we describe the fabrication and application of a label-free localized surface plasmon resonance imaging (LSPRi) technology capable of detecting protein secretions from a single cell. The plasmonic nanostructures are lithographically patterned onto a standard glass coverslip and can be excited using visible light on commercially available light microscopes. Only a small fraction of the coverslip is covered by the nanostructures and hence this technique is well suited for combining common techniques such as fluorescence and bright-field imaging.
A multidisciplinary approach is used in this protocol which incorporates sensor nanofabrication and subsequent biofunctionalization, binding kinetics characterization of ligand and analyte, the integration of the chip and live cells, and the analysis of the measured signal. As a whole, this technology enables a general label-free approach towards mapping cellular secretions and correlating them with the responses of nearby cells.
Introducción
Comunicación entre celular es crucial para la regulación de muchas actividades fisiológicas dentro y fuera de la célula. Una variedad de proteínas y vesículas puede ser secretada que, posteriormente, activar procesos celulares complejos, tales como la diferenciación, la curación de heridas, la respuesta inmune, la migración, y la proliferación. 1-5 de fallos de funcionamiento de las vías de señalización inter-celulares han sido implicados en numerosos trastornos, incluyendo el cáncer, la aterosclerosis , y la diabetes, para nombrar unos pocos.
El ensayo óptima secreción celular debe ser capaz de mapear espacialmente y temporalmente la proteína secretada de interés sin interrumpir las vías de señalización pertinentes. De esta manera las relaciones causales entre los perfiles de concentración y la respuesta de las células receptoras se pueden deducir. Por desgracia, muchas de las técnicas a base de fluorescentes más comúnmente utilizados no cumplen con estos criterios. Proteínas de fusión fluorescentes se pueden utilizar para etiquetar el analito wentro de la célula, pero puede interrumpir la vía secretora, o si secretada, resulta en una luz difusa fuera de la célula, que es difícil de cuantificar. Ensayos basados en immunosandwich fluorescentes son las técnicas más comúnmente utilizadas para la detección de secreciones celulares pero típicamente requieren el aislamiento de células individuales 6-11. Además, la introducción del anticuerpo de detección típicamente se detiene o termina el experimento y el tamaño de las etiquetas de anticuerpos, 150 kDa para la IgG, es un impedimento para la señalización corriente abajo.
Debido a estos obstáculos es preferible que una técnica de etiqueta libre de ser utilizado para las secreciones de proteínas imagen y entre las tecnologías existentes de etiqueta libre, resonancia de plasmón superficial (SPR) y localizada resonancia de plasmón superficial (LSPR) sensores son excelentes candidatos 12-17. Estos sensores han sido ampliamente utilizados para estudios de unión de analito de proteínas, exosomas y otros biomarcadores. 18-24 de En el caso de LSPR, la nanostr plasmónicauctures pueden ser modelados litográficamente en cubreobjetos de vidrio y emocionado usando la luz visible a través de las configuraciones estándar de microscopía de campo amplio. Debido a su huella de nanoescala, la mayoría del sustrato de vidrio está disponible para las técnicas de imagen comunes como de campo brillante y microscopía de fluorescencia haciendo así estas sondas adecuado para la integración con la microscopía de células vivas 25-28. Hemos demostrado la medición en tiempo real de las secreciones de anticuerpos a partir de células de hibridoma utilizando nanoestructuras plasmónicas oro funcionalizado con resoluciones espaciales y temporales de 225 ms y 10 m, respectivamente. La configuración básica de chip se ilustra en la Figura 1. 28 La trayectoria de la luz de salida del microscopio se divide entre una cámara CCD utilizada para las imágenes y un espectrómetro junto fibra óptica para la determinación cuantitativa de ocupación fraccional de una matriz dada de nanoestructuras (Figura 2 ).
El protocol se presenta en este artículo se describe el diseño experimental para la medición en tiempo real de las secreciones de células individuales y al mismo tiempo el control de la respuesta de las células por medio de la microscopía de campo claro estándar. El enfoque multidisciplinario incluye la fabricación de nanoestructuras, la funcionalización de las nanoestructuras para la alta afinidad de unión de analitos, la optimización de la superficie tanto para minimizar la unión no específica y la caracterización de las constantes cinéticas uso de una superficie comercial Plasmon Resonance (SPR) instrumento, la integración de las líneas celulares sobre el sustrato, y el análisis de imágenes y datos espectrales. Anticipamos que esta técnica es una tecnología que permite la asignación de espacio-temporal de las secreciones celulares y sus relaciones causales con las células receptoras.
Protocolo
Fabricación 1. Nanoestructura
- Elija 25 mm cubreobjetos de vidrio de diámetro con un espesor aproximado de 170 mm (No. 1.5) como sustratos para la nanofabricación.
- Sumergir los cubreobjetos en solución Piranha (3: 1 relación de ácido sulfúrico y peróxido de hidrógeno) durante al menos 6 h. Lave la piraña cubreobjetos empapado con abundante ultrapura 18,2 mO agua destilada desionizada (DDW).
PRECAUCIÓN: ácido Piranha reacciona violentamente con materiales orgánicos y debe manejarse con sumo cuidado. - Depósito de 10 nm de cromo película delgada sobre los cubreobjetos por evaporación por haz de electrones para evitar efectos de carga durante el modelado y las imágenes de nanoestructuras.
- Haga girar la primera capa de la bicapa resistir consiste en copolímero de metacrilato de etilo lactato de metilo (MMA_EL6) a 2.000 rpm durante 45 segundos y luego hornear a 150 ° C. Gira la segunda capa de poli-metacrilato de metilo (950PMMA_A2) a 3.000 rpm durante 45 segundos y luego hornear a 180 ° C.
- Tamborileon resistir la bicapa utilizando la litografía por haz de electrones (EBL) a 25 kV con una dosis de 300 mu c área / cm 2. Desarrollar en alcohol isopropílico (IPA) / metilisobutilcetona (MIBK): 01.02 y enjuague en IPA.
- Depósito Ti (5 nm) / Au (80 nm) película sobre el sustrato usando un evaporador de haz de electrones.
- Después de la deposición de oro, levante la bicapa copolímero de resistir por inmersión del sustrato en acetona durante 4 hr.
- Inspeccionar el sustrato usando el microscopio electrónico de barrido (SEM) para confirmar la forma y tamaño nanoestructura, remover el cromo restante del sustrato a través de grabado en húmedo usando CR-7 reactivo de ataque durante 60 segundos a TA y luego enjuague en DDW.
- Diseñar un espaciamiento matriz de centro a centro de 33 micras de dejar espacio para imágenes de células entre matrices. Patrón de las nanoestructuras en 20 x 20 patrón para cada matriz con un paso de 300 nm utilizando un escritor de haz de electrones. Cada chip contiene 300 matrices de dimensión nanoestructura típica de la altura de 80 ± 2,5 nm y 70 ± 2,5 nm Diameter.
- Inspeccionar un subconjunto de matrices usando el microscopio de fuerza atómica (AFM) para la verificación de tamaño y uniformidad.
- Adjuntar un anillo de soporte, típicamente de silicio, a la parte posterior del cubreobjetos usando una epoxi de curado UV.
2. Chip de Limpieza y Aplicación de la auto-ensambladas monocapa
- Para la limpieza y la regeneración de los chips, la ceniza plasma a una potencia de 40 W en una mezcla de 300 mTorr 5% de hidrógeno, 95% de argón durante 45 segundos después de la limpieza de la cámara durante 5 min en las mismas condiciones.
- Funcionalizar las nanoestructuras de oro inmediatamente tras la calcinación plasma sumergiendo el chip en una solución etanólica de tiol de dos componentes que consiste en una proporción de 3: 1 de SH- (CH 2) 8 3 -EG -OH (SPO) y un componente, ya sea con una amina o grupo funcional carboxilo, a saber, SH- (CH 2) 11 -EG 3 -NH 2 (SPN) o SH- (CH 2) 11 -EG 3 -COOH (SPC).
- Deje la i de chipsn la solución de tiol O / N para formar una monocapa autoensamblada (SAM).
- Enjuague el chip con etanol y se seca con gas nitrógeno.
- Si es necesario, guarde el chip funcionalizado para un máximo de 2 semanas a 4 ° C.
- Cuando esté listo para su uso reaccionar el SPN o componente SPC con el ligando usando una química en función del ligando de elección (véase más adelante).
Nota: Los chips pueden ser regenerados y re-funcionalizado docenas de veces. Un chip dado puede ser usado por períodos que van desde 6 meses a más de un año. El espectro medido en una matriz dada se reproducen de forma fiable después de regeneraciones reiteradas incineración de plasma, seguido de biofuncionalización. 29
3. La funcionalización de superficies y Kinetic Caracterización
Nota: Utilice el chip funcionalizado en el instrumento SPR comercial para caracterizar las constantes de velocidad cinética entre el ligando y el analito, así como para estudiar la resistencia de SAM a no específica bncontrar. Hay una amplia gama de caudales y diseños de microfluidos que permiten funcionalización de la superficie eficiente. Puesto que tenemos una SPR disponible en el mercado nos estandarizado en torno a sus caudales recomendados. Observamos que estos caudales son típicos de todos los instrumentos SPR y así no son restrictivas. El instrumento SPR no es una necesidad ya que todos funcionalización se puede hacer directamente en el chip LSPR, pero redujo nuestra carga de trabajo, ya que es un instrumento de multiplexado mientras que nuestra configuración de microfluidos LSPR no lo es.
- Funcionalizar un chip de oro desnuda comercial con el SAM como se describe en la Sección 2.
- Si se utiliza un SAM basado SPC, activar el grupo carboxilo con una mezcla 1: 1 de 133 mM de 1-etil-3- (3-dimetilaminopropil) carbodiimida (EDC) y 33 mM de N hidroxisuccinimida (NHS) en DDW para 10 min.
- Conjugar el grupo carboxilo activado con el anticuerpo / ligando de interés durante 300 segundos usando una velocidad de flujo de 30 l / min. Preparar los ligandos entampón de fosfato pH 6, típicamente, pero esto puede variar dependiendo de la molécula.
- Después de la conjugación ligando, caudal 0,1 M de etanolamina en solución salina tamponada con fosfato (PBS) como un paso para el desactivador 300 segundos a una velocidad de 30 l / min. Etanolamina ayuda a minimizar la unión no específica.
- Introducir el analito de interés a un caudal de 100 l / min usando un intervalo de concentraciones y calcular las constantes de velocidad cinética utilizando un software de análisis de cinética.
- Si la unión no específica es problemático, aumentar la proporción de SPO para SPC o SPN.
Ajustes 4. LSPR Generales
- Ajustes del microscopio:
- Utilice una lámpara de 100 W halógena de Koehler iluminar la muestra. Use un filtro de paso largo (normalmente 593 nm de corte) en la trayectoria de la luz para eliminar las longitudes de onda que no contribuyen al cambio de resonancia (Figura 2).
- Para la recolección de datos LSPRi, utilizar un microscopio invertido con un Immersio aceite de 40Xn objetiva (1,4 NA) y una termoeléctricamente enfriaron 16 bits cámara CCD.
- Coloque un divisor de haz en el puerto de salida del microscopio para obtener Imágenes y espectros simultáneamente.
- Ajuste la platina del microscopio de temperatura controlada a 37 ° C y equilibrar durante 4 hr.
- Incorporar un conjunto de incubación adicional en el microscopio para regular la concentración de CO 2 y humedad al 5% y 95%, respectivamente.
- Preparación Chip y montaje:
- Funcionalizar el chip LSPR como se describe en la Sección 2 con las relaciones de SAM de dos componentes óptimos determinados a partir de los experimentos de SPR.
- Cargue el chip dentro de un soporte de microfluidos por encargo de la siguiente manera. Coloque el chip en una pieza inferior de aluminio. Sandwich del chip entre esta pieza inferior y una junta de silicona y una clara pieza superior de plástico. Utilice 4 tornillos para fijar el conjunto.
- Para una aplicación tiol típica a base de SPC, la caída de la capa 300 l una mezcla 1: 1tura de 133 mM de EDC y 33 mM de NHS en DDW para activar los grupos carboxilo del componente tiol SPC.
- Esperar 10 min y enjuagar manualmente la superficie con PBS 10 mM.
- Conjugar el grupo carboxilo activado con el ligando (típicamente un anticuerpo o fragmento de anticuerpo) de interés a gota recubrimiento de 300 l de solución de ligando.
- Esperar 30 min y enjuagar manualmente con PBS 10 mM.
- Caída de la capa 300 l de 0,1 M de etanolamina en PBS en el chip para minimizar la unión no específica. Espere 10 min.
- Lavar la etanolamina con PBS que contenía 0,005% de Tween 20 (PBS-T20).
- Coloque un pedazo de cuarzo por encima del chip para reducir las fluctuaciones en los datos relacionados con un menisco cambiante.
- Mantenga el chip mojado con tampón PBS-T20, mientras que el montaje en microscopio.
- Coloque el conjunto de chips LSPR firmemente en el soporte de la muestra etapa climatizada y conecte la tubería de microfluidos.
- Conecte el tubo de microfluidos para los bu montaje y flujoffer (o medio libre de suero para estudios de células) hasta un estado de equilibrio se alcanza.
- Permitir a la asamblea y el microscopio se equilibren durante al menos 2 horas.
- Alinear el chip usando la palanca de mando para que la matriz central está alineada con la fibra óptica para la espectroscopia. Los datos espectroscópicos se toma utilizando un espectrómetro y software de análisis espectral.
- Mantenga matrices en el foco durante todo el experimento utilizando el enfoque automático de software, Zeiss de enfoque definido o dispositivo de enfoque automático equivalente.
5. LSPR Imaging de secreciones anti-c-myc 9E10 de células de hibridoma
Nota: La línea celular de hibridoma utilizado para este estudio expresan anticuerpo anti-c-myc constitutivamente y por lo tanto no requieren un disparador químico
- Funcionalizar las nanoestructuras con el péptido c-myc. Esto tiene un valor K D de 1,77 nM para los anticuerpos anti-c-myc secretadas por el clon 9E10 células de hibridoma.
- Cultura de las células de hibridoma en completmedio de crecimiento e con 10% de suero bovino fetal (FBS) y 1% de antibióticos en un matraz T75 a 37 ° C bajo 5% de CO 2. Mantener una densidad celular de 4 × 10 5 células / ml.
- Para los estudios de secreción celular, las células de pellets desde el matraz T75 por centrifugación, se lavan dos veces con RPMI-1640 medio libre de suero (SFM) para eliminar los anticuerpos secretados y ajustar la densidad celular a 4 x 10 6 células / ml.
- Recoger las células y la prueba de viabilidad antes de introducirlos a los chips LSPR.
- Introducir 50 l de la solución de células manualmente en los chips LSPR con una micropipeta. Después de unos pocos minutos a 25 50 células se adhieren a la superficie de los chips LSPR.
- Lavar las células restantes en solución con frescos SFM mediante el sistema de perfusión de microfluidos.
- Seleccione los arrays LSPR para imágenes que están cerca, a menos de 10 m, pero no se superponen con las células.
- Para asegurarse de que la señal es específica para el antib secretada anti-c-mycodies introducen el medio de cultivo celular con y sin los anticuerpos presentes, así como con los anticuerpos, pero con sus sitios de unión bloqueados por la presencia de una concentración saturante de péptido c-myc en la solución.
- Calibrar los sensores al final de cada ejecución con una solución de saturación de anticuerpos anti-c-Myc (250 nm). Esto ayuda en la normalización de la respuesta de los sensores y determinar la ocupación fraccional basado en el perfil biofuncionalización de cada ejecución.
- Corregir la deriva en la dirección X e Y utilizando el software de alineación de la imagen.
Resultados
En un estudio típico de secreción de células vivas existen múltiples modos de recogida de datos que tiene lugar. La figura 3 muestra una superposición de una imagen LSPRi, lo que pone de relieve las matrices cuadradas, y una imagen iluminada de luz transmitida que pone de relieve la célula abajo a la izquierda. Los datos se recogen típicamente durante un período de 3 horas, seguido por la introducción de una solución saturante de analito para el cálculo de normalización se describe a continuación. Las imágenes de fluorescencia también se puede integrar en la rutina de recopilación de datos por la conmutación automática de un cubo de filtro. En la figura 4 una célula teñida con el colorante fluorescente membrana rodamina DHPE exhibe extensiones lamellipodia como (flechas). Si estas extensiones fueron a solaparse con los arreglos que les dan un falso positivo para la secreción de proteínas. Tener múltiples modos de imágenes puede ayudar a identificar estos hechos.
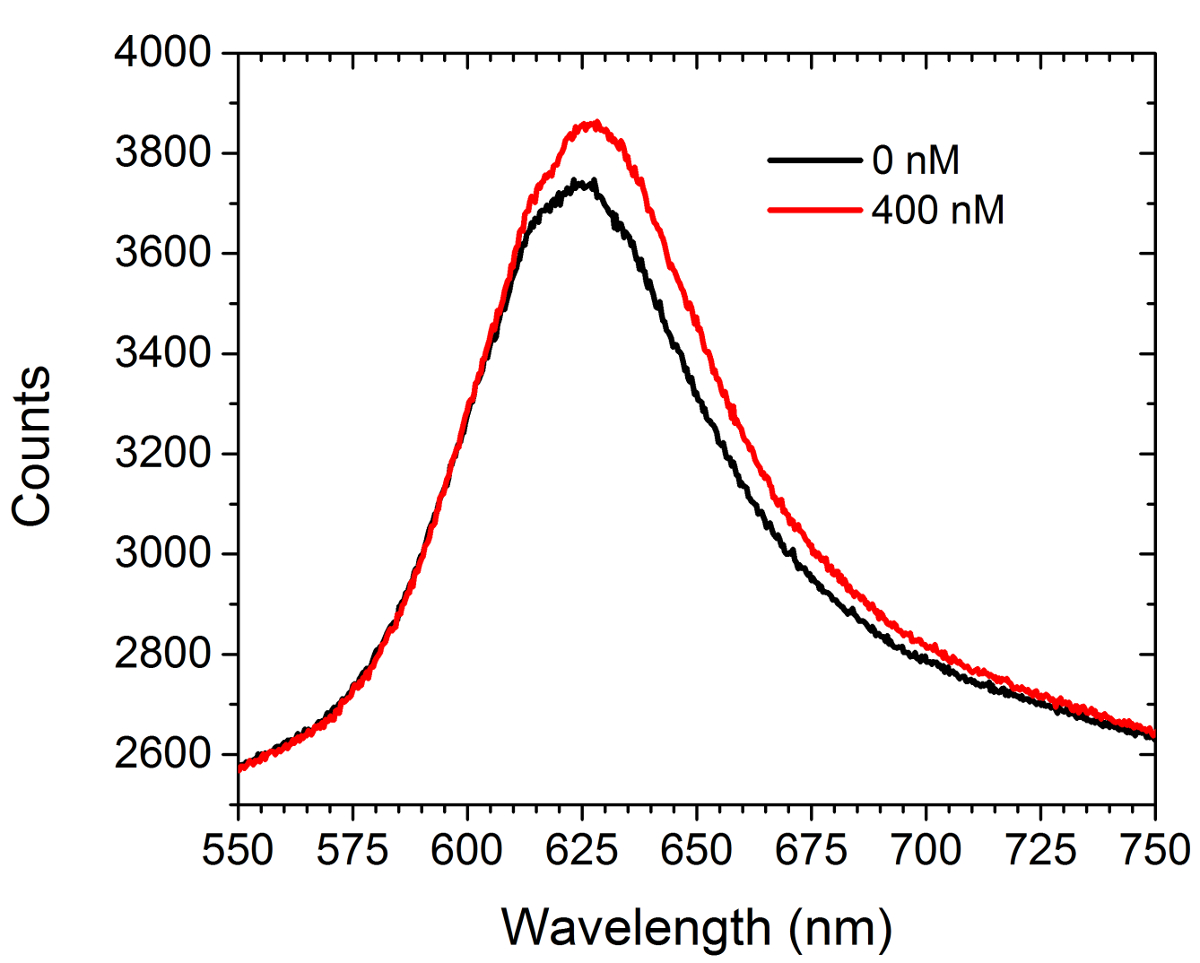
La Figura 5 muestra datos de espectrometría de antes y de popaer la introducción de una solución de saturación (400 nM) de adquirirse comercialmente anticuerpos anti-c-myc a los arrays funcionalizados c-myc. No hay células estaban presentes en este experimento. El espectro muestra tanto un desplazamiento hacia el rojo y un aumento en la intensidad. La diferencia entre las áreas bajo las dos curvas se traduce en un aumento en la intensidad de la imagen en el modo de matriz LSPRi en la cámara CCD. Un enfoque menos el análisis de datos cuadrados no lineales ha sido desarrollado para inferir la ocupación fraccional de ligandos unidos a la superficie de los espectros. 30,31
Al final del experimento, los valores de intensidad saturado (es decir, fraccionada ≈ ocupación 1) se utilizan para calcular una respuesta normalizada para cada matriz mediante la siguiente fórmula:
¿Dónde están la intensidad normalizada en el tiempo t punto, la intensidad inicial al comienzo del experimento, intensidad final saturada, y se mide la intensidad de la matriz en el momento t punto respectivamente.
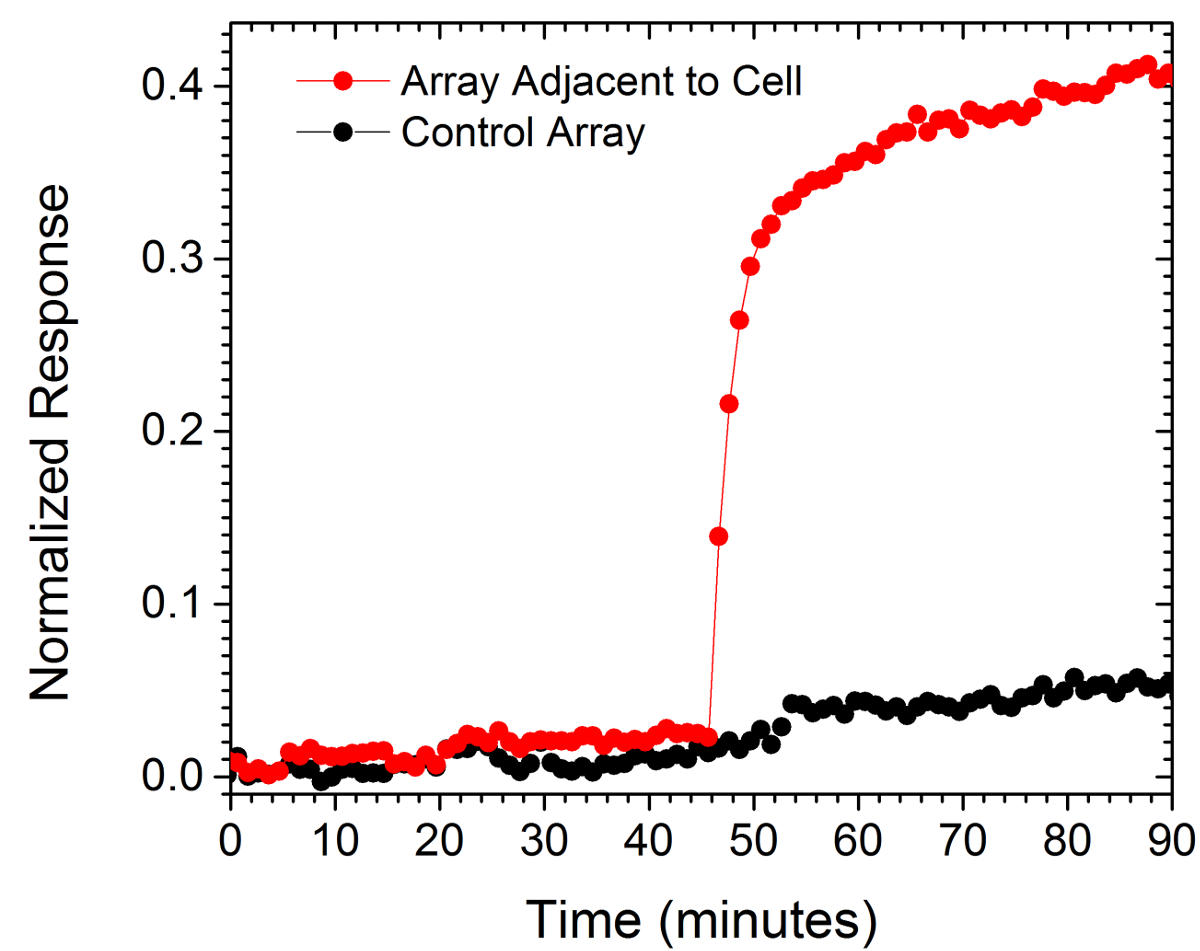
Los valores normalizados de dos matrices se muestran en la Figura 6. Una matriz estaba dentro de 10 micras de la célula bajo investigación mientras que el otro, utilizado como control, era una distancia de 130 m de la célula. El aumento repentino en la respuesta normalizada de la matriz más cerca de la celda respecto a la respuesta plana de la matriz de control es indicativo de una ráfaga localizada de anticuerpos secretados.

Figura 1. Diseño del sensor. Un dibujo que representa la geometría de un experimento típico de la secreción de células vivas. La célula (esferoide azul) se deposita sobre al chip LSPR que contiene matrices de nanoestructuras de oro biofuncionalizadas. En la vista ampliada-in, la secreción celular de interés, en este caso los anticuerpos mostrados como moléculas en forma de Y, se mide como se unen a la superficiede las nanoestructuras funcionalizados. Haga clic aquí para ver una versión más grande de esta figura.
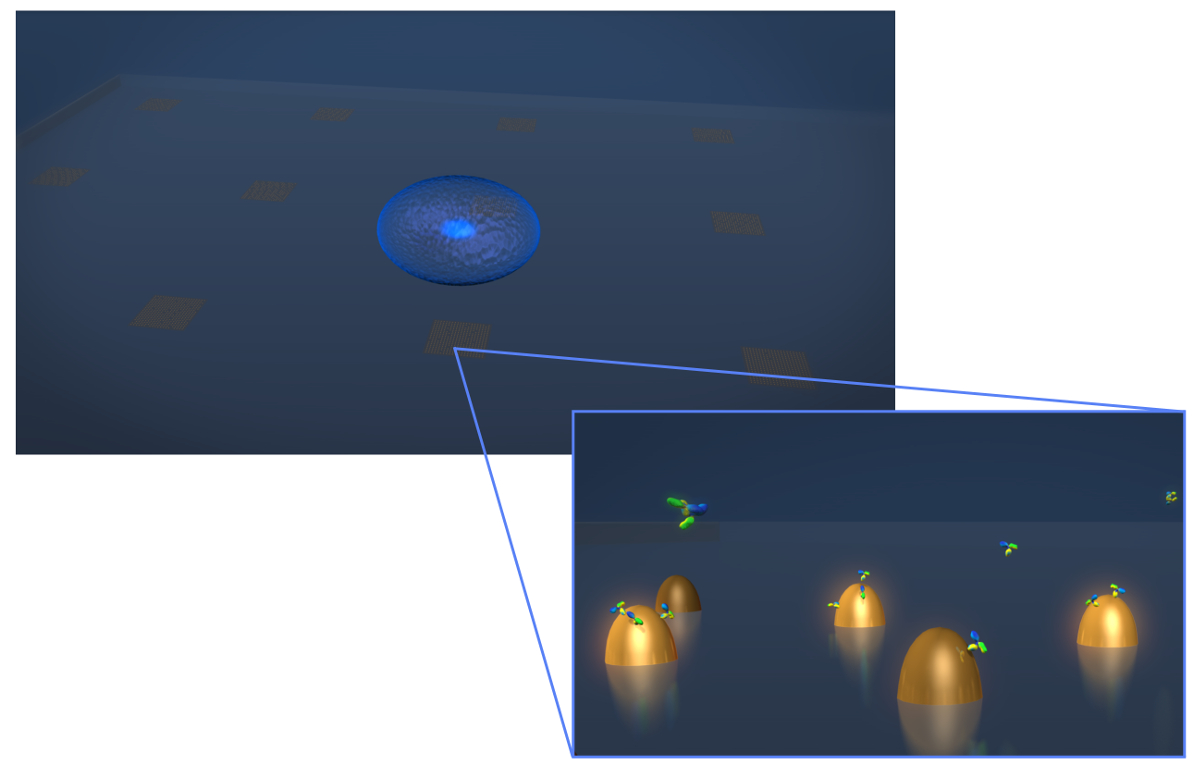{kind=link}

Figura 2. Configuración óptica. La luz iluminada por una lámpara halógena es primero filtrada por un filtro de paso largo (LP). La luz está polarizada linealmente (P1) y se ilumina la muestra a través de un objetivo de 40X / 1.4 NA. La luz dispersada es recogida por el objetivo y se pasa a través de un polarizador cruzado (P2). Un divisor del haz 50/50 (BS) se inserta en la trayectoria de la luz recogida para espectroscópico simultánea y análisis de imágenes. Arriba a la derecha:. Una imagen microscopía de fuerza atómica de 9 nanoestructuras individuales separadas por un lanzamiento de 300 nm Haga clic aquípara ver una versión más grande de esta figura.
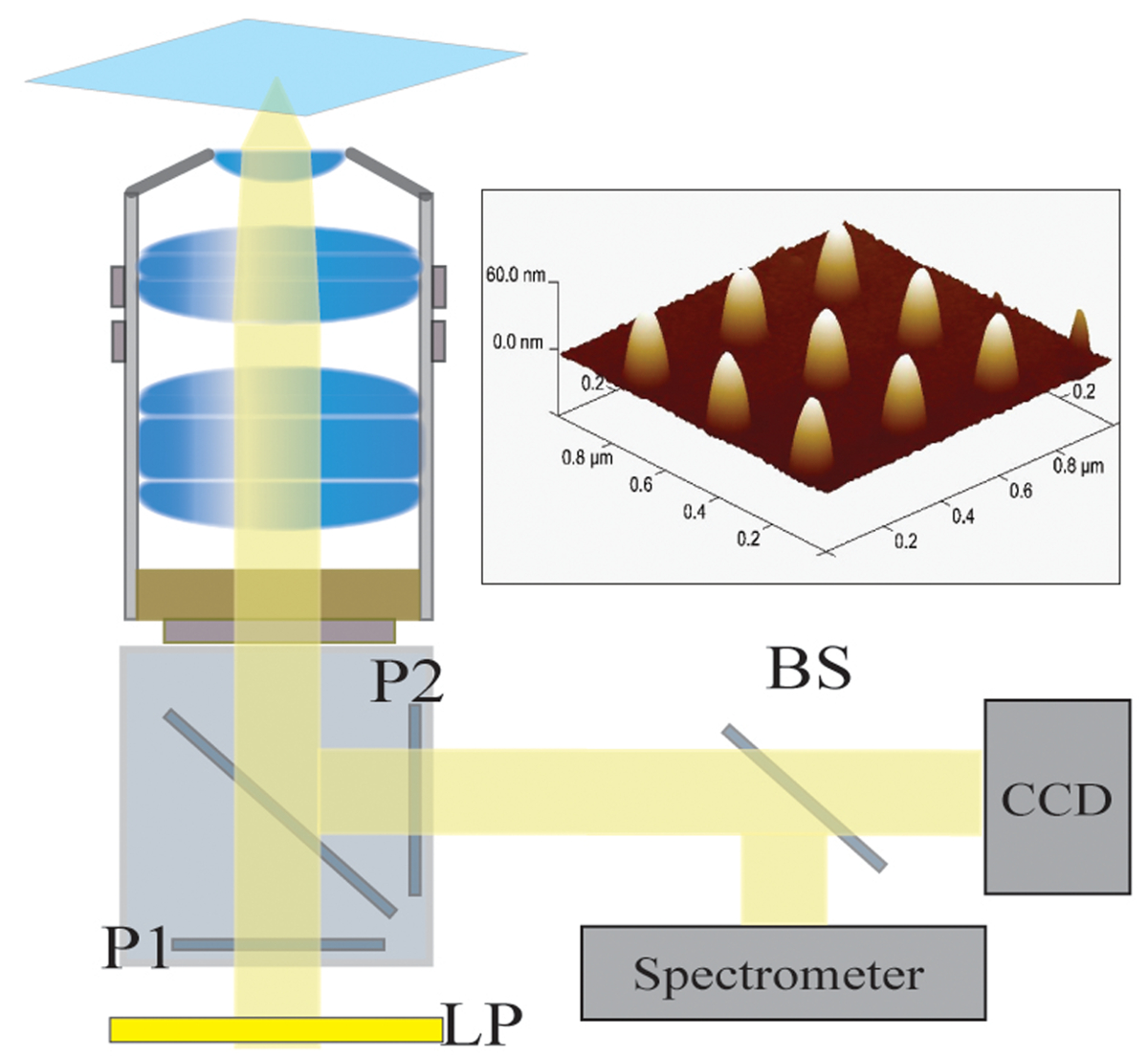{kind=link}

Figura 3. Vivo Cell LSPRi Estudio. Un fusionó imagen LSPRi luz transmitida y que muestra una sola célula de hibridoma (abajo a la izquierda), rodeada de 12 matrices. Esta es una imagen de contraste mejorado. La barra de escala es de 10 micras. Por favor haga clic aquí para ver una versión más grande de esta figura.
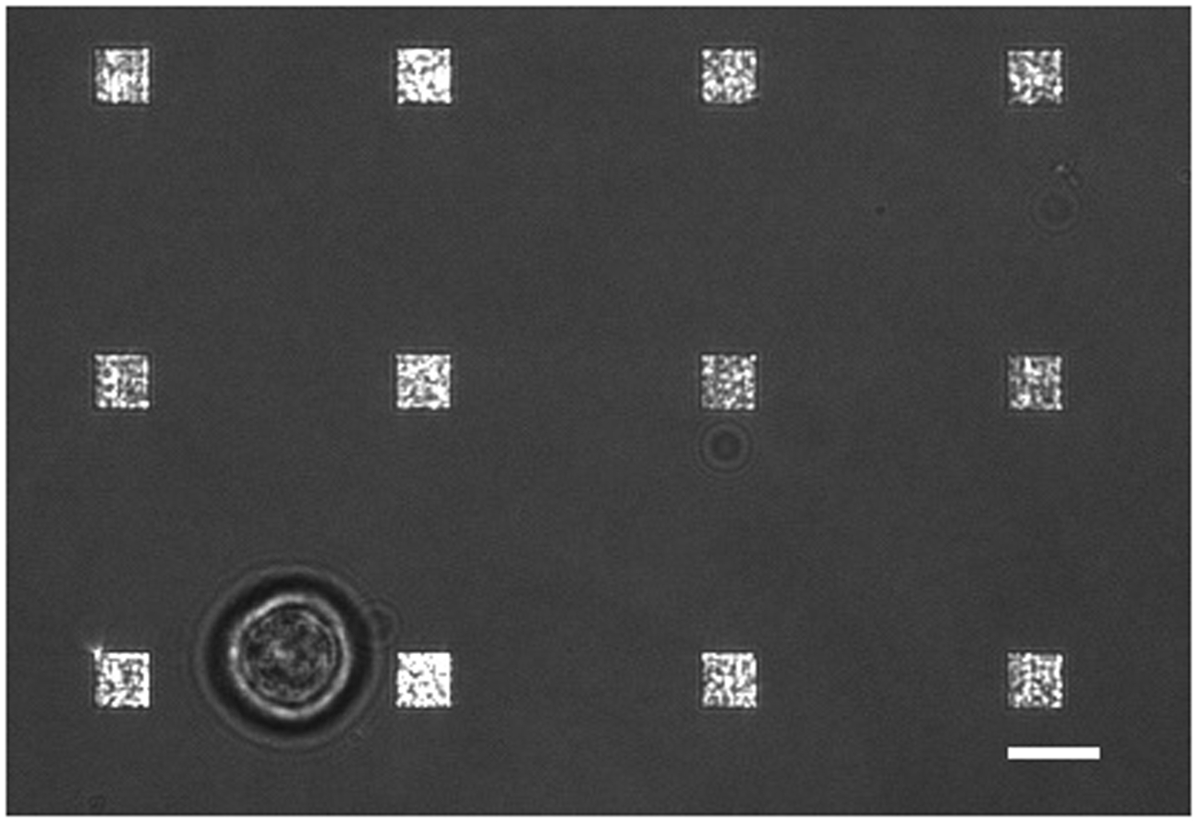{kind=link}

Figura 4. Estudio de fluorescencia de células vivas. Una imagen en color falso fluorescente de una sola célula de hibridoma se tiñeron con rodamina DHPE, que es un colorante de la membrana. En el modo de imágenes fluorescentes las matrices generalmente no son visibles, sin embargo, una matriz cercano es observable aquí como abcarecer de plaza en la esquina inferior derecha. La célula puede ser visto para ser separados de la matriz aunque extensiones como tentáculos (posiblemente filopodios o lamellipodia) se extiende hacia el exterior de las células (flechas). La barra de escala es de 10 micras. Por favor haga clic aquí para ver una versión más grande de esta figura.
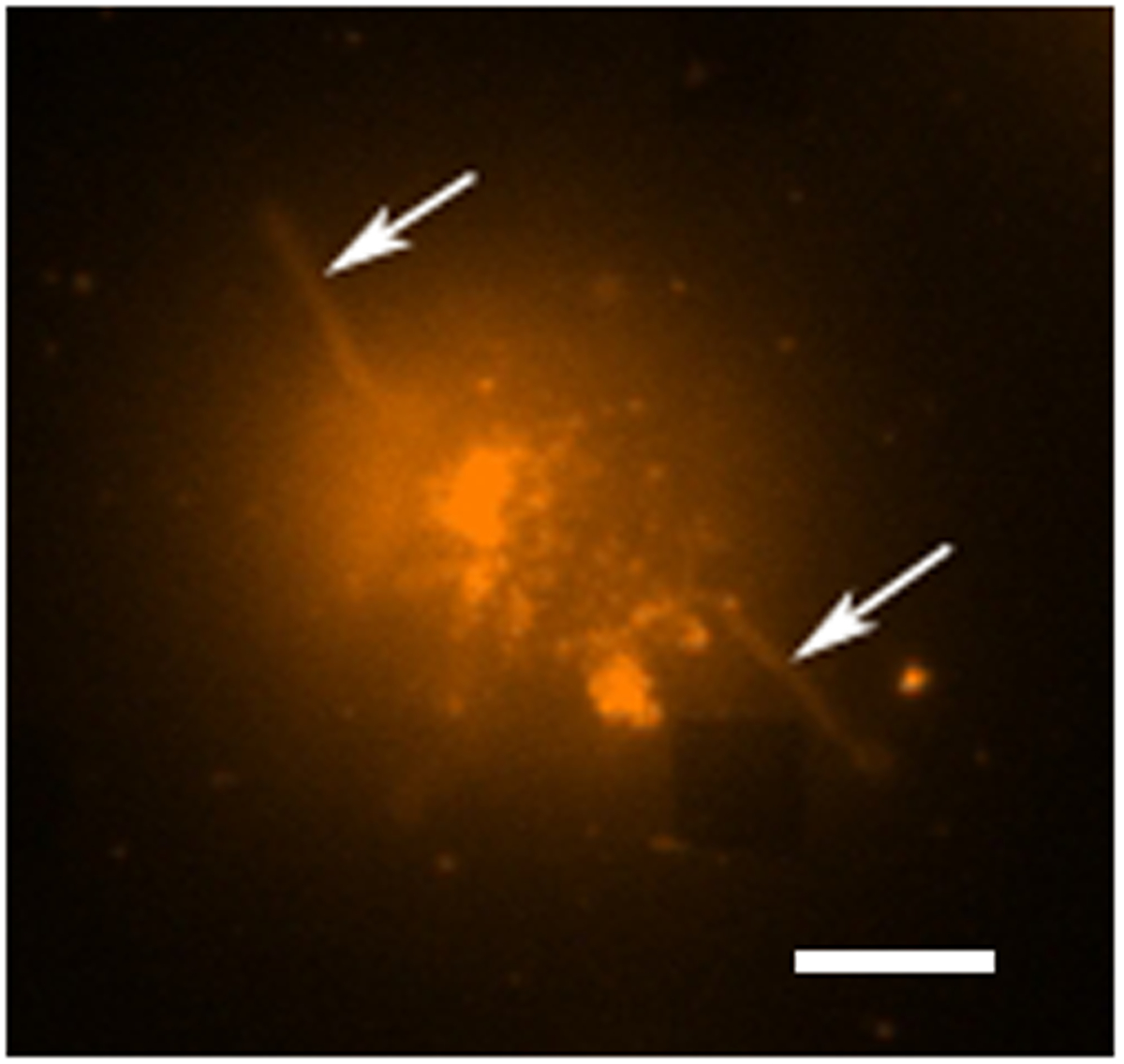{kind=link}

Figura 5. espectral modalidad. Los espectros obtenidos a partir de una matriz funcionalizado c-myc antes y después de la introducción de 400 nM solución de anticuerpos anti-c-myc. No hay células estaban presentes en este estudio. Por favor haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 6. Secreción sola célula. La respuesta de una matriz situada dentro de 15 micras de una sola célula y una situada 130 m de distancia (Control). La barra de escala es de 10 micras. Por favor haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
The LSPR imaging technique described in this work has numerous advantages over more traditional methodologies for detecting cell secretions. First, the time resolution of our technique is on the order of seconds whereas the commercial alternative, an immunosandwhich assay known as EliSpot, has a typical time resolution of 2 to 3 days.7,32 As a result we were able to resolve sudden changes in the rate of protein secretion, such as that shown in Figure 6. Second, having arrays distributed over the chip allows for the secreted signal to be tracked in space and time which enables more rigorous comparisons to diffusion-based models of cell secretion. In addition, arrays like the control array shown in Figure 6 can be used to subtract out global changes in the image that typically arise from instrumental factors such as focus drift. Third, our technique requires no modification of the cells. If desired, the experiment can incorporate commonly used tags such as fluorescent proteins, but if there is concern that such tags may negatively affect cell viability or homeostasis the label-free nature of our approach does not require them. Fourth, using the spectroscopic data we have demonstrated that quantitative information regarding the fractional occupancy of surface bound ligands can be calculated.
There are numerous alternative methods to EBL for fabricating metallic nanoparticles. However, we have found that the EBL provides considerable flexibility for optimizing nanostructure and array dimensions to best suit the optics and the cells under investigation. Also critical is the fact that the chips can be readily regenerated by plasma ashing. In this way, a typical chip can be used dozens of times. Biofunctionalization details must be modified for the specific application. The protocol presented here conjugated the surface with relatively small c-myc peptide ligands. Larger ligands such as whole antibodies typically require more spacing and thus a higher SPO to SPN/SPC ratio. Regardless, a well formed SAM layer is essential for preventing non-specific binding in live-cell experiments. In general, larger molecular weight analytes are more readily detected by LSPR. Thus, in its single-cell manifestation, this technique may not be appropriate for detecting the secretion of small proteins, such as cytokines.
The current setup has been used for studying individual non-adherent cells. There are significant number of secreted signaling proteins and vesicles to which the results reported in this work are directly applicable. For example carcinoembryonic antigen (CEA) which for decades now has been a diagnostic marker for cancer. Colon cancer cells are known to secrete CEA at the rates of thousands of molecules/cell/hr and the molecular weight is 180 kDa which exceeds that of IgG antibodies. CEA is believed to be involved in autocrine and paracrine signaling pathways but the spatio-temporal nature of these secretions have never been measured. Our technique can directly address these signaling questions. An extension of this work will be to measure the spatio-temporal nature of CEA secretion from single cells.33 Future work will also focus on integrating LSPRi with two and three dimensional cell cultures of adherent cells. By incorporating multiplexed arrays capable of detecting a number of secreted proteins in parallel, this technique has the potential to open a new window into cell secretions and how they influence neighboring cells.
Divulgaciones
We thank George Anderson for helpful comments and discussions. This work was supported by the Naval Research Laboratory’s Institute for Nanoscience and the National Research Council Research Associateship Award.
Agradecimientos
The authors have nothing to disclose.
Materiales
| Name | Company | Catalog Number | Comments |
| 25 mm diameter glass coverslips | Bioscience Tools | CSHP-No1.5-25 | 170±5 µm is optimal |
| Poly-methyl methacrylate | Microchem | PMMA 950 A4 | |
| Ethyl lactate methyl metacrylate | Microchem | MMA EL6 | |
| Electron beam evaporator | Temescal | FC-2000 | |
| Electron beam lithography | Raith | Series 150 | |
| Ethanol | Sigma-Aldrich | 459836 | |
| Acetone | Sigma-Aldrich | 320110 | |
| CR-7 chromium etchant | Cyantek | CR-7 | |
| Scanning electron microscope | Zeiss | Ultra 55 | |
| Atomic force microscope | Veeco | Nanoscope III | |
| Plasma ashing system | Technics | Series 85 RIE | |
| SH-(CH2)8-EG3-OH (SPO) | Prochimia | TH 001-m8.n3-0.2 | |
| SH-(CH2)11-EG3-COOH (SPC) | Prochimia | TH 003m11n3-0.1 | |
| SH-(CH2)11-EG3-NH2 (SPN) | Prochimia | TH 002-m11.n3-0.2 | |
| Surface plasmon resonance system | Biorad | XPR36 | |
| Bare gold chip | Biorad | GLC chip | Plasma ashed to remove the monolayer |
| 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide | Thermo | 22980 | |
| N-hydroxysuccinimide (NHS) | Thermo | 24510 | |
| Pentylamine-Biotin | Thermo | 21345 | |
| Ethanolamine | Sigma-Aldrich | E9508 | |
| Neutraavidin | Thermo | 31000 | |
| Phosphate buffered saline | Thermo | 28374 | |
| Tween 20 | Sigma-Aldrich | P2287 | |
| Inverted microscope | Zeiss | Axio Observer | Microscope is equipped with 40X oil immersion objective; CO2 and humidity incubation from Pecon GmbH |
| CCD camera | Hamamatsu | Orca R2 | Thermoelectrically cooled (16 bit) |
| Spectrometer | Ocean Optics | QE65Pro | |
| Spectrasuite | Ocean Optics | version1.4 | |
| c-myc peptide HyNic Tag | Solulink | SP-E003 | |
| monoclonal anti-c-myc antibody | Sigma-Aldrich | M4439 | |
| Hybridoma cell line | ATCC | CRL-1729 | |
| Antibiotic Antimycotic Solution (100×) | Sigma-Aldrich | A5955 | |
| Serum free media RPMI 1640 | Invitrogen | 11835-030 | |
| Fetal bovine serum | ATCC | 30-2020 | |
| Rhodamine DHPE | Life Technologies | L-1392 |
Referencias
- Ludwig, A. -. K., Giebel, B. Exosomes: Small vesicles participating in intercellular communication. The International Journal of Biochemistry & Cell Biology. 44, 11-15 (2012).
- Friedl, P., Gilmour, D. Collective cell migration in morphogenesis, regeneration and cancer. Nature Reviews Molecular Cell Biology. 10, 445-457 (2009).
- Letterio, J. J., Roberts, A. B. Regulation of immune responses by TGF-beta. Annual Review of Immunology. 16, 137-161 (1998).
- Werner, S., Grose, R. Regulation of wound healing by growth factors and cytokines. Physiological Reviews. 83, 835-870 (2003).
- Werner, S., Krieg, T., Smola, H. Keratinocyte-fibroblast interactions in wound healing. Journal of Investigative Dermatology. 127, 998-1008 (2007).
- Bailey, R. C., Kwong, G. A., Radu, C. G., Witte, O. N., Heath, J. R. DNA-encoded antibody libraries: A unified platform for multiplexed cell sorting and detection of genes and proteins. Journal of the American Chemical Society. 129, 1959-1967 (2007).
- Gazagne, A., et al. A Fluorospot assay to detect single T lymphocytes simultaneously producing multiple cytokines. Journal of Immunological Methods. 283, 91-98 (2003).
- Han, Q., et al. Polyfunctional responses by human T cells result from sequential release of cytokines. Proceedings of the National Academy of Sciences of the United States of America. 109, 1607-1612 (2012).
- Han, Q., Bradshaw, E. M., Nilsson, B., Hafler, D. A., Love, J. C. Multidimensional analysis of the frequencies and rates of cytokine secretion from single cells by quantitative microengraving. Lab on a Chip. 10, 1391-1400 (2010).
- Ma, C., et al. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nature Medicine. 17, 738-743 (2011).
- Shirasaki, Y., et al. Real-time single-cell imaging of protein secretion. Scientific Reports. 4, (2014).
- Milgram, S., et al. On chip real time monitoring of B-cells hybridoma secretion of immunoglobulin. Biosensors and Bioelectronics. 26, 2728-2732 (2011).
- Abbas, A., Linman, M. J., Cheng, Q. A. New trends in instrumental design for surface plasmon resonance-based biosensors. Biosensors & Bioelectronics. 26, 1815-1824 (2011).
- Ermakova, A., et al. Detection of a Few Metallo-Protein Molecules Using Color Centers in Nanodiamonds. Nano Letters. 13, 3305-3309 (2013).
- Haes, A. J., Van Duyne, R. P. A nanoscale optical blosensor: Sensitivity and selectivity of an approach based on the localized surface plasmon resonance spectroscopy of triangular silver nanoparticles. Journal of the American Chemical Society. 124, 10596-10604 (2002).
- Horowitz, V. R., Aleman, B. J., Christle, D. J., Cleland, A. N., Awschalom, D. D. Electron spin resonance of nitrogen-vacancy centers in optically trapped nanodiamonds. Proceedings of the National Academy of Sciences of the United States of America. 109, 13493-13497 (2012).
- Sepulveda, B., Angelome, P. C., Lechuga, L. M., Liz-Marzan, L. M. LSPR-based nanobiosensors. Nano Today. 4, 244-251 (2009).
- Barbillon, G., et al. Biological and chemical gold nanosensors based on localized surface plasmon resonance. Gold Bulletin. 40, 240-244 (2007).
- Endo, T., et al. Multiple label-free detection of antigen-antibody reaction using localized surface plasmon resonance-based core-shell structured nanoparticle layer nanochip. Analytical Chemistry. 78, 6465-6475 (2006).
- Endo, T., Kerman, K., Nagatani, N., Takamura, Y., Tamiya, E. Label-free detection of peptide nucleic acid-DNA hybridization using localized surface plasmon resonance based optical biosensor. Analytical Chemistry. 77, 6976-6984 (2005).
- Haes, A. J., Hall, W. P., Chang, L., Klein, W. L., Van Duyne, R. P. A localized surface plasmon resonance biosensor: First steps toward an assay for Alzheimer's disease. Nano Letters. 4, 1029-1034 (2004).
- Jonsson, M. P., Jonsson, P., Dahlin, A. B., Hook, F. Supported lipid bilayer formation and lipid-membrane-mediated biorecognition reactions studied with a new nanoplasmonic sensor template. Nano Letters. 7, 3462-3468 (2007).
- Park, J. H., et al. A regeneratable, label-free, localized surface plasmon resonance (LSPR) aptasensor for the detection of ochratoxin A.. Biosensors & Bioelectronics. 59, 321-327 (2014).
- Mayer, K. M., Hao, F., Lee, S., Nordlander, P., Hafner, J. H. A single molecule immunoassay by localized surface plasmon resonance. Nanotechnology. 21, (2010).
- Endo, T., Yamamura, S., Kerman, K., Tamiya, E. Label-free cell-based assay using localized surface plasmon resonance biosensor. Analytica Chimica Acta. 614, 182-189 (2008).
- Huang, Y. X., Cai, D., Chen, P. Micro- and Nanotechnologies for Study of Cell Secretion. Analytical Chemistry. 83, 4393-4406 (2011).
- Oh, B. R., et al. Integrated Nanoplasmonic Sensing for Cellular Functional Immunoanalysis Using Human Blood. ACS Nano. 8, 2667-2676 (2014).
- Raphael, M. P., Christodoulides, J. A., Delehanty, J. B., Long, J. P., Byers, J. M. Quantitative Imaging of Protein Secretions from Single Cells in Real Time. Biophysical Journal. 105, 602-608 (2013).
- Raphael, M. P., et al. A New Methodology for Quantitative LSPR Biosensing and Imaging. Analytical Chemistry. 84, 1367-1373 (2011).
- Raphael, M. P., et al. Quantitative LSPR imaging for biosensing with single nanostructure resolution. Biophysical Journal. 104, 30-36 (2013).
- Raphael, M. P., et al. A new methodology for quantitative LSPR biosensing and imaging. Analytical Chemistry. 84, 1367-1373 (2012).
- Henn, A. D., et al. Modulation of single-cell IgG secretion frequency and rates in human memory B cells by CpG DNA, CD40L, IL-21, and cell division. Journal of Immunology. 183, 3177-3187 (2009).
- Bramswig, K. H., et al. Soluble Carcinoembryonic Antigen Activates Endothelial Cells and Tumor Angiogenesis. Cancer Research. 73, 6584-6596 (2013).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados