
Method Article
Una base de sangre de prueba para la detección de ROS1 y RET transcripciones de la fusión de circulación de ácido ribonucleico con reacción en cadena de polimerasa Digital
En este artículo
Resumen
Detección de ácido ribonucleico (cRNA) de la sangre que circula es una necesidad en el diagnóstico clínico. Aquí describimos los métodos que caracterizan a cRNA de pacientes con cáncer pulmonar de células no pequeñas mediante reacción en cadena de polimerasa digital sensible y específico. Las pruebas de cumplen requisitos de diseño para detectar variantes de fusión dentro de las 72 horas.
Resumen
Hemos desarrollado nuevos métodos para el aislamiento y la caracterización del tumor derivado circulante el ácido ribonucleico (cRNA) para biopsia líquida base de sangre. Detección robusta de cRNA de sangre representa una solución a una necesidad fundamental en diagnóstico clínico. La prueba comienza con la colección de sangre en tubos de colección de sangre que contienen conservantes que estabilizan cRNA. Libres de células exosomal y RNA asociada a plaquetas es aislada de plasma en este sistema de prueba. El cRNA es revés transcrito a ADN complementario (cDNA) y amplificación mediante reacción en cadena de polimerasa digital (dPCR). Las muestras se evalúan para el biomarcador de destino, así como un gen de control. Validación de la prueba incluyó límite de detección, exactitud y robustez estudios con muestras analíticas. El método desarrollado como resultado de estos estudios reproducible detectar múltiples variantes de fusión para ROS1 (proto-oncogene del C-Ros variantes 1; 8) y RET (reorganizado en proto-oncogene de la transfección; 8 variantes). Se ha optimizado el flujo de trabajo de procesamiento de muestra para que constantemente se pueden generar resultados dentro de 72 horas de la recepción de la muestra.
Introducción
Hasta 25% de cáncer de pulmón de células no pequeñas (CPCNP) pacientes pueden no tener suficiente tejido disponible para probar en el momento del diagnóstico. Incluso en casos donde el tejido está disponible, puede no ser de suficiente cantidad y calidad para llevar a cabo aconsejable pruebas moleculares1,2. En casos donde hay suficiente tejido de una biopsia para perfiles moleculares, los pacientes pueden tener que esperar varias semanas o más para obtener los resultados, o iniciar el tratamiento sin resultados moleculares3,4. Sin embargo, es crítico que diagnóstico molecular informativo esté disponible dado el advenimiento de múltiples opciones de tratamiento para pacientes con CPCNP diagnosticados. La prueba de ADN libre circulante (cfDNA) de biopsia líquida es una solución a los retos de tejido tradicional prueba4,5,6. Opciones actuales de prueba para las mutaciones útiles en NSCLC uso cfDNA y un flujo de trabajo basado en la dPCR similar para la generación de resultado rápido, incluir el receptor del factor de crecimiento epidérmico (EGFR) sensibilización de mutaciones ΔE746-A750 y L858R, mutación de EGFR resistencia T790M , Variantes del proto-oncogén KRAS (KRAS) y la variante de B-Raf proto-oncogene (BRAF) V600E. Aunque no ampliamente adoptado por el campo, derivado de tumor ARN mensajero (ARNm) de circulación aislados de biopsia líquida también puede proporcionar información clínica importante7,8,9. Anteriormente hemos desarrollado y divulgado sobre los métodos de detección multiplexada de las equinodermos microtúbulos asociados proteína como 4-anaplástico linfoma Receptor tirosina quinasa (EML4-ALK) fusión variantes del plasma de sangre10. En este estudio, hemos ampliado estos métodos para incluir objetivos de RNA multiplexados de orden superior para ROS1 y RET, cubriendo ocho variantes de fusión dentro de cada ensayo. El objetivo fue desarrollar una técnica rápida, sensible, específica y reproducible para la detección de estas variantes de la fusión del plasma de pacientes con NSCLC.
Se inicia el proceso de prueba en un consultorio médico usando RNA estabiliza sangre colección tubos11. Estos tubos contienen una preservativa así como inhibidores de la Rnasa de la célula. Las muestras son prioridad enviado durante la noche a las centralizada acreditado por la Universidad de CAP de patólogos americanos/clínica laboratorio enmiendas para la mejora (CLIA)-laboratorio acreditado (laboratorio clínico) para el procesamiento por personal competente. Una vez recibida por el laboratorio clínico, cada paso del proceso se lleva a cabo bajo aprobado procedimientos operativos estándar (SOP). Sangre se centrifuga para recuperar el plasma, que se utiliza para aislar el RNA que es de circulación libre en la sangre o dentro de encapsular moléculas, como los exosomas y plaquetas7,8,9. Para aislar el ARN de estos compartimientos, seleccionamos el sistema de recuperación de RNA basada en las comparaciones de varios métodos de extracción. El ARN aislado es concentrado y atrás transcrito a cDNA. Varias enzimas transcriptasa reversa y cartillas gene-específicas fueron evaluadas durante la optimización del método de síntesis de cDNA para maximizar ROS1 y RET destino transcripción conversión10. Esto es crítico para la baja abundancia circulando transcripciones, como variantes del tumor derivado de la fusión. Por último, hemos optimizado dPCR concentraciones cartilla y sonda para permitir la detección de multiplexado de RET o ROS1 variantes de fusión y el control del gen, glucuronidasa-β (GUSB). Entonces combinamos las mejores condiciones de cada uno de los estudios de optimización en un protocolo cerrado final antes de realizar los estudios de validación analítica descritos en este informe. Este protocolo y estos resultados proporcionan la base para un flujo de trabajo rápido y sensible para la detección rutinaria de variantes raras de la fusión en circulación.
Protocolo
Se siguen las instrucciones del fabricante para los reactivos enumerados a continuación, a menos que lo contrario se describe. Los ensayos PCR están disponibles en el mercado productos diseñados para detectar fusiones ROS1 y RET.
1. trabajo con RNA en preparación para transcripción reversa (RT)-dPCR: buenas prácticas de laboratorio
- Crear un ambiente libre de Rnasa al trabajar con el ARN.
- Utilizar aerosoles disponibles en el mercado diseñados para inactivar contaminantes RNasas.
- Uso certificado libre de Rnasa reactivos, puntas y tubos. Utilizar puntas de barrera para las pipetas para evitar la introducción de RNasas o contaminación cruzada de las muestras.
- Siempre use una capa del laboratorio para evitar que partículas de ropa en su muestra. Designar un laboratorio capa específico para usar con el procesamiento de RNA.
- Guantes para evitar la contaminación de la muestra de RNasas en piel. Cambiar guantes con frecuencia.
Nota: Asumir las superficies del laboratorio están contaminadas con Rnasa ya que están expuestos al medio ambiente. Guantes en contacto con la piel, pelo, picaportes, manijas de congelador, rotuladores/markers, etc. se supone que ya no son libres de Rnasa. - Descontaminar las pipetas, benchtops, centrifugadoras y otras superficies de trabajo con un ARNasa inactivación aerosol antes de usarlo.
- Si es posible, mantener un conjunto de equipos para uso con el ARN.
- Minimizar las interrupciones del flujo de aire en las áreas de laboratorio cuando se trabaja con muestras de RNA para evitar que las partículas caigan en muestras o contaminar el área de trabajo.
- Tienda purificado RNA a-80 ° c.
- Evitar congelar-deshiela múltiple de muestras de RNA, ya que esto puede causar degradación.
2. generación de Material analítico de ARN para los controles positivos
- Uso de ADN sintético diseño había publicado secuencias de mRNA para variantes de fusión de interés10.
- Para una variante de fusión determinado, seleccione una secuencia de fusión mRNA que incluye el sitio de fusión además de suficiente longitud flanqueando en cada lado para cubrir el amplicon PCR.
- Seleccionar secuencias nucleotídicas entre 50-250 nt para mímico el tamaño de RNA con plasma enriquecido de plaquetas en circulación.
- Agregar una secuencia de promotor de T7 (5'-CAGAGATGCATAATACGACTCACTATAGGGAGA-3') en el extremo 5' de la secuencia de destino.
- Ordenar secuencias sintéticas como fragmentos trenzado doble del ácido desoxirribonucléico (ADN).
- Reconstituir el ADN sintético en tampón Tris-EDTA (TE) a una concentración final de 10 ng/μl.
- Convertir 60 ng de ADN sintético ARN mediante transcripción en vitro .
- Purificar las transcripciones del RNA con reactivo de fenol/guanidina basado en12.
- Incluyen la ADNasa I, libre de ARNasa para extraer ADN plantilla residual.
- Medir concentración de purificada en vitro RNA usando un fluorómetro disponible comercialmente con tintes de RNA específicas y normas. Asegúrese de que el ARN está dentro del rango aceptable para las normas solicitadas. Se puede requerir dilución.
- Confirman éxito transcripción por electroforesis del gel utilizando un gel de agarosa 2% mezclado con la mancha de gel de RNA y una escalera de RNA, incluyendo el rango de tamaño de 50-250 nt de alta gama.
- Carga 500 ng de cada uno en vitro RNA en un gel.
- Correr el gel a 5 V/cm.
- Visualizar bandas solo usando iluminación y documentar los resultados.
- Confirmar el tamaño esperado de la transcripción para cada una de las variantes de fusión (basadas en diseño en el paso 2.1.2).
- Confirmar la detección de cada uno en vitro RNA por RT-dPCR mediante análisis PCR específica variante emparejado (vea los pasos 5 a 8 del presente Protocolo).
- Opcional: Prepare una mezcla equimolar de en vitro RNA que contiene cada una de las variantes de fusión y el control del gen GUSB.
- Si se realiza el paso 2.9: confirmar la detección de cada una de las variantes de fusión incluidas en la mezcla de control de dPCR mediante ensayos PCR específicos de la variante (véanse los pasos 5 a 8 del presente Protocolo).
- Determinar concentración deseada entrada para controles positivos analíticos analizando las concentraciones oscilan entre 0.25 y 2.5 de fg de10. Elegir la concentración base de salida número de copia deseado.
- Tras la confirmación, preparar alícuotas de solo uso de 10 μl de ARN analítica para el control positivo (paso 4.4) y tienda a-80 ° c.
3. donantes muestras
- Recoger las muestras de sangre entera humana 10 mL en tubos 10 mL sangre (BCT) que contiene un conservante de RNA libres de células.
Nota: Todos los donantes humanos se consentimiento para uso de investigación y no información de identificación de donantes específicos será recopilada o utilizada durante la prueba. - Proceso de muestras de sangre entera dentro del plazo especificado por el fabricante BCT.
- Plasma humano normal combinado puede adquirirse de una fuente comercial para uso en el control positivo analítico. Preparar un solo uso, alícuotas de 1 mL de plasma humano normal agrupado los tienda a-80 ° c para su uso con el control positivo (paso 4.4).
4. recuperación de la ARN del Plasma de la circulación
Nota: Es importante trabajar rápidamente durante este procedimiento.
- Centrifugar tubos de sangre total a 200 x g durante 20 min.
- Recoger hasta 4 mL del plasma del tubo de la colección de sangre centrifugada utilizando una pipeta serológica. Tenga cuidado de no molestar o aspirar la capa de la capa anteada.
- Aislar el RNA circulante mediante un kit disponible comercialmente que puede capturar exosomas, plaquetas y ARN sin células de plasma. Aislar el RNA de la muestra control positivo junto a cada lote.
-
Preparar el Control positivo por cada lote de muestras clínicas como sigue:
- Descongelar 1 mL combinaron normal plasma humano alícuota (paso 3.3).
- Descongele la parte alícuota 10 μl analítica RNA (paso 2.12).
- Preparar el control positivo agregando 10 μl RNA analítica en la muestra de plasma humano normal una vez que el etanol se ha agregado al plasma lisado.
- Eluir las muestras con 100 μl de agua libre de nucleasa. Proceder de inmediato con ARN limpieza y concentración.
- Las muestras pueden almacenadas en hielo húmedo y cubiertas, para hasta una hora.
- Concentrado de RNA usando método de la columna base y eluir en 9 μl de agua libre de ARNasa.
- Proceder de inmediato al paso 5, o mantener las muestras en hielo húmedo hasta una hora.
5. reversa de la transcripción del RNA a cDNA
- Convertir muestra circulación concentrada del RNA a cDNA utilizando un kit de reacción de transcripción reversa disponible en el mercado, incluyendo iniciadores al azar (véase tabla 1 de componentes).
Nota: Iniciadores específicos gene son opcionales y pueden diseñarse para variantes de la prueba. Imprimaciones están diseñados en base a la blanco de secuencia de ARN. Utilizar secuencias variante de fusión del punto 2.1.- Incluyen ninguna muestra de control de la transcriptasa inversa y ninguna muestra de control de RNA (véase tabla 1).
- Aislar el cDNA de la reacción de transcripción reversa usando una ADN disponible en el mercado concentrador spin columna.
Nota: Este paso facilita la extracción de las enzimas, cartillas y libre Deoxinucleótidos trifosfatos (dNTPs). - Utilice cDNA inmediatamente en reacciones de PCR o almacenar a-80 ° c.
6. digital PCR
Nota: Esta PCR es específico de gota PCR digital (véase Tabla de materiales).
-
Precauciones de la mezcla PCR.
- Guantes un laboratorio desechables capa y nitrilo.
- Uso PCR mezclar los reactivos en una área de preparación de reactivos dedicados. No manipule el cDNA en el área de preparación sólo reactivo.
- Cubrir las puntas de prueba trabajando para protegerlos de la luz. Luz excesiva puede foto bleach el tinte fluorescente conectado a la sonda.
- Transporte de mezclas, cubiertas y protegido de la luz, en un área separada de pre-amplificación antes de ADNc debe ser agregado.
- CDNA a probar de añadir a la mezcla PCR en una campana de limpieza PCR situado en zona de pre-amplificación.
- Preparar mezclas PCR para un volumen final de reacción de 20 μl según tabla 2.
- Distribuir la mezcla PCR + cDNA para placas PCR.
Nota: Como una guía se recomienda el uso de un diseño de la placa. - Cubrir la placa con un sellador de placas desmontables.
- Centrifugue las placas brevemente para recoger muestras en la parte inferior de los pozos.
- Mezclar en agitador de placa en una posición baja para 10 s.
- Centrifugue las placas brevemente para recoger la muestra en la parte inferior del pozo.
- Retire el sellador de placa. Realizar generación de gotas de mezcla de cDNA PCR ya sea con un sistema de generación de gotas manuales o automáticos.
- Para la generación de la gota manual, transferencia de mezcla PCR de 20 μl a pozos de muestra en el cartucho de la generación de gotas. Añadir 70 μl de aceite de la generación de gotas. Cubierta con junta y transferencia cartucho de goma al generador manual gotita para iniciar la generación de gotas. Tras generación gotita, transferencia de gotas a una placa fresca de PCR utilizando puntas recomendadas por el fabricante. Aspirar y dispensar las gotas lentamente, durante 5 a 6 s cada una, sin tocar la abertura de la punta del cartucho de gotita o placa.
- Para la generación automatizada de la gotita, selle la placa con papel de aluminio y transferencia para el generador de gotas. Asegurar todos los consejos, cartuchos, y las placas están en su lugar antes de comenzar la generación de gotas.
- Siguientes gota generación y transferencia de las gotas a una placa PCR fresca, sellar con un sellador de placas de aluminio y placas de ciclo térmico utilizando la configuración en el cuadro 3.
- Después el termociclador ejecutar es completa, leer la placa utilizando un lector de gota. Crear un diseño de placa para el software lector que identifica la ubicación de controles, muestras, etc.y la carga en el software para empezar a leer.
7. Análisis y revisión y generación de resultados
- Analizar placa leer resultados utilizando software disponible comercialmente.
- Desplácese hasta el menú Analyze para ver diagramas de dos dimensiones (2D) amplitud.
- Evaluar la calidad de los datos examinando los datos de la gota.
- Evaluar los datos para los números de evento aceptado total usando el menú de eventos. Si hay menos de 10.000 eventos por pozo, evaluar cuidadosamente los datos de problemas adicionales.
- Verificar datos para amplitudes de fluorescencia aberrante. Las diferencias de amplitud significativa y las diferencias de concentración entre muestras replicadas indican mala manipulación o mezcla de muestras.
- Hacer notas de racimos de gota con patrones de rociado sobre un eje de 45 grados, que es indicativo de gotitas de mala calidad o las muestras problema.
- Examinar los datos de Control positivo, No de la transcriptasa reversa (RT No) y No RNA Control (NRC) en primer lugar. Seleccionar todas las muestras de control y examinar la calidad del racimo por parcela 2D. Para umbral apropiado, una clara separación entre grupos de la gota debe ser evidente.
- Para cada variante de ensayo, establecer el umbral basado en pozos de control.
- Establecer umbrales en parcelas 2D utilizando la herramienta de punto de mira para separar la población de gotas de la negativa doble de la población de gen control (marcada con sonda de 5'-hexachloro-fluoresceína-CE phosphoramidite), eje y y población gene variant, si presentan) marcada con fluoresceína amidite sonda de OR 6-carboxyfluorescein), eje x.
- Suma de copias de cada pozo replicar para una sola muestra.
- Expresar los resultados como el número de copias variables detectada.
Nota: Para determinar el valor de corte analítico para llamar a una muestra positiva o negativa, ejecutar normal muestra de donante sano establecer (al menos 10 muestras individuales) a través del proceso finalizado y establecer el límite por encima de cualquier señal de fondo perceptible para la mutación de interés. Además, establecer el número de copias del gen de control necesaria para llamar a un resultado positivo o negativo. Este corte de gen de control funciona como un control interno de calidad (QC) para evaluar la cantidad y calidad de cada muestra de RNA que se procesa.
8. verificación de las condiciones de reacción RT-dPCR utilizando líneas celulares (opcionales)
- Para comprobar la detección de variantes de fusión, utilice comercialmente disponibles líneas celulares que expresan la fusión de ROS1 o RET ARNm de interés. Proceda del siguiente modo:
- Homogeneizar las células congeladas en flash en una solución de lisis Guanidinio basado directamente en el estado congelado. Incluso breve deshielo antes de homogeneización puede causar la pérdida y degradación del RNA.
- Aislar el RNA utilizando columnas de spin de membrana de silicona diseñados para RNA.
- Medir la concentración de las muestras de RNA utilizando un fluorómetro con estándares y reactivos específicos de RNA.
- Diluir el RNA aislado en un fondo de tipo ARN de plasma u otra fuente comercial.
- Llevar a cabo pasos de transcripción inversa de RNA a cDNA, PCR Digital y análisis de datos y revisión y generación de resultados mencionados en el presente Protocolo para confirmar la detección de la variante deseada.
Resultados
Este protocolo describe un sistema de prueba para la detección de variantes de fusión RNA para el uso en la medición de las mutaciones del conductor dentro del plasma de pacientes de CPCNP (figura 1A). La fusión mRNA fueron productos de la expresión de los cambios de RET y ROS1 más comunes en la población de CPCNP identificó13,14,15,16,17. Análisis PCR multiplexados entonces fueron diseñados para detectar las ocho variantes de transcripción más frecuentes para cada destino en NSCLC en una sola reacción. Los desplazamientos más comunes en el lugar geométrico de ROS1 generan asociaciones con las 5' porciones de CD74, SDC4, SLC34A2, EZR o TPM3 genes (figura 1B). Los desplazamientos más comunes en el lugar geométrico RET conducen a yuxtaposición con KIF5B, para que el ensayo cubre a seis uniones exón. Otros socios RET que incluyen los CCDC6 y TRIM33 (figura 1C). En total, los ensayos cubren aproximadamente el 88% de ROS1 y 99% de las alteraciones del RET sabidas para ocurrir en la población de pacientes de CPCNP17.
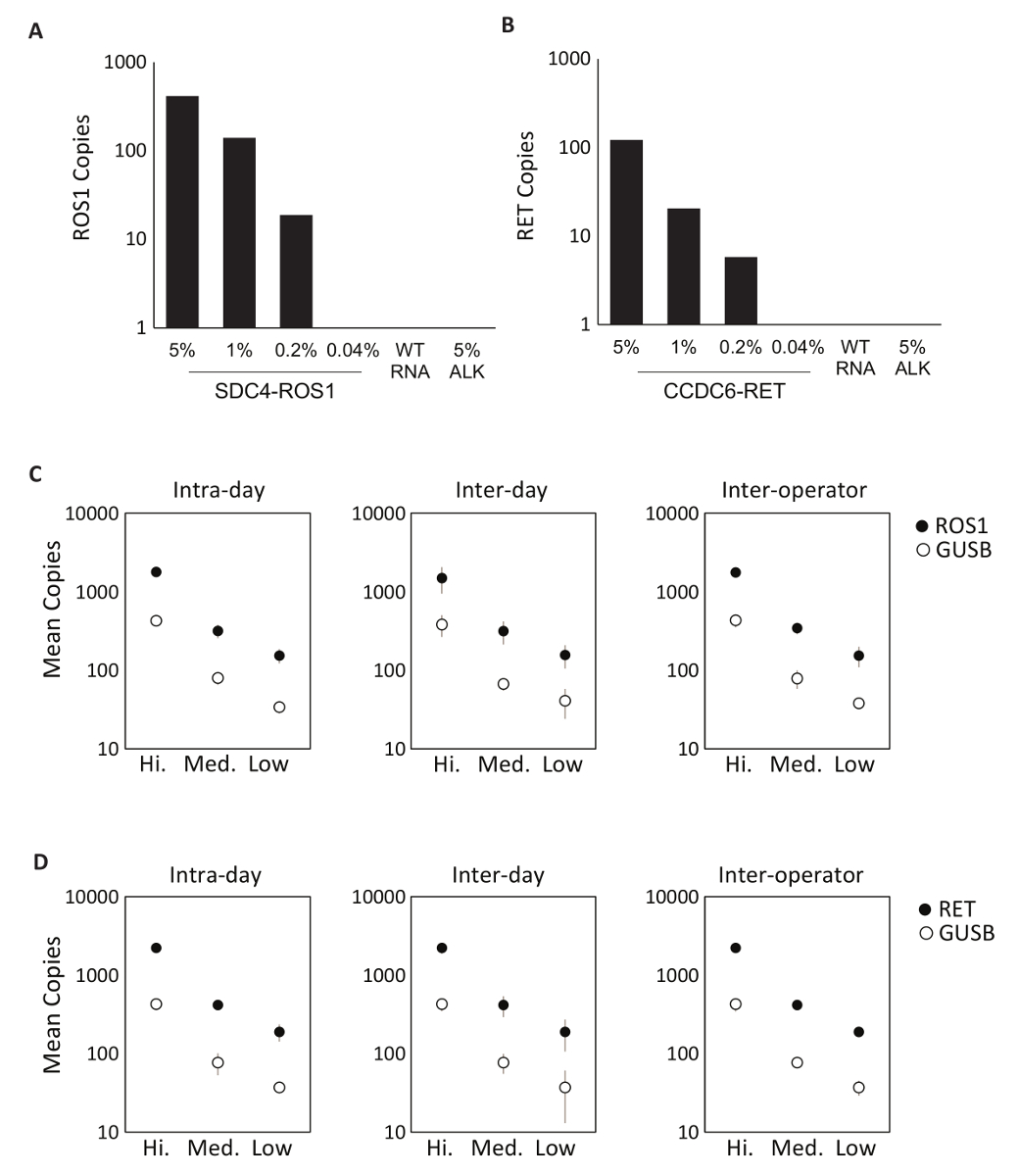
La especificidad de los componentes del ensayo se evaluó primero con ocho individuales en vitro RNAs que contienen la secuencia de ARNm para las transcripciones de la fusión por la ROS1 o RET multiplexado ensayos. Cada especie de RNA fue probado contra cada ensayo de variantes individual que comprende la versión multiplexada. No había ninguna reactividad cruzada de estos ensayos, demostrando así 100% especificidad analítica en el diseño multiplexado ensayos (datos no mostrados). Para determinar el límite inferior de detección del Protocolo de prueba, derivado de líneas celulares que expresan una variante de la fusión incluida en el ensayo de ARN total se mezclaron en un fondo de ARN normal en concentraciones de 5%, 1%, 0.2% y 0,04%. El multiplexado RET y ROS1 variante análisis PCR detectados tan poco como 0,2% variante de fusión (figura 2A-B). Además, una preparación de 5% objetivo línea celular derivada RNA (expresando una transcripción de la fusión de EML4-ALK) no se detectó con los ensayos multiplexados ROS1 y RET, lo que demuestra especificidad (figura 2A-B).
Prueba de precisión del proceso RT dPCR fue realizada para ambos ROS1 y analítica RET. material de control compuesto por equimolar en vitro RNAs fue procesado en tres concentraciones (alta, media y baja) mediante transcripción reversa y dPCR en tres diferentes ocasiones dentro del mismo día (intradía), en tres días consecutivos (entre días) y con dos operadores (inter-operador). Resultados de pruebas de precisión demostró una detección precisa de la transcripción de la fusión de intereses, así como un gen de control, GUSB, que se incluye como una métrica de control de calidad interna (figura 2-D).
Además del control interno GUSB, cada lote de muestras clínicas se ejecutó con un conjunto de controles de lote. Control positivo fue desarrollado de una mezcla de análisis in vitro RNA que representa cada una de las variantes de fusión en RT dPCR, así como analítica en vitro RNA para GUSB. Este ARN se enriquecieron en el plasma humano normal lisado durante la extracción de RNA y fue procesado junto con las muestras clínicas en todo el protocolo. El no control de la transcriptasa inversa (sin RT) era un control negativo para confirmar la ausencia de material contaminante en el flujo de trabajo de extracción de RNA y demostrar la especificidad de los cebadores de RNA. El control de la RT No fue generado usando el mismo material como control positivo, pero no incluye enzima dentro de la reacción de síntesis de cDNA. El no control de RNA (NRC) es un control negativo para confirmar la ausencia de contaminación de las transcripciones en los componentes de la reacción reversa de la transcripción. Este control fue introducido en el flujo de trabajo en la etapa de síntesis de cDNA, y se agregó agua en la reacción en lugar de una plantilla de RNA. Los controles RT sin y NRC deben ser negativos en ambos canales, si los resultados deben ser entregados. La tabla 1 enumera los componentes de la reacción reversa de la transcripción para cada control. Se muestran ejemplos de los diagramas 2D para cada uno de estos controles para la ROS1 (figura 3 A-C) y ensayos multiplexores RET ( C-e-Gde lafigura 3 ). Variantes de fusión fueron detectadas usando una sonda de amidite (FAM) de fluoresceína y se representan en el eje y, mientras que el gen control, GUSB, fue detectado utilizando una sonda de 5'-hexachloro-fluoresceína-CE phosphoramidite (hexadecimal) y es en el eje x. Estos controles por lotes se evaluaron en el transcurso de 21 días para determinar la robustez del ensayo. Gotas positivas fusión y GUSB control gen gotas observaron ROS1 y RET en todas las ejecuciones de 21 ejecutadas en el transcurso del estudio (figura 3D, H). Todos los controles negativos (RT sin y NRC) dieron resultados negativos a través de los 21 días (datos no mostrados).
La capacidad de solucionar problemas es un componente crítico de cualquier protocolo de pruebas a ejecutar en el ámbito de laboratorio clínico. Aquí, nos proporcionan ejemplos del mundo real de resultados óptimos utilizando el protocolo de RT-dPCR. El primero es un diagrama 2D ejemplo demostrando la importancia del no control de la transcriptasa inversa (figura 4A). En este ejemplo, mutantes gotas positivas estaban presentes aunque no hubo ninguna conversión de cDNA debido a la falta de la enzima. Este resultado fue probablemente debido a la dPCR cartillas amplificación de ADN genómico de objetivo. En este caso, diseño de un ensayo que abarca el intrón evitará que la amplificación de la DNA genomic. Alternativamente, una enzima DNasa Rnasa-libres se puede utilizar para eliminar el ADN contaminante, pero no es recomendable para la detección de objetivos raros, ya que podría producirse alguna degradación del RNA durante la incubación con la enzima. La siguiente parcela 2D ejemplo fue un NRC con gotas positivas en ambos canales (figura 4B). Esto indica contaminación en algún punto de la instalación de dPCR RT. En este caso, la recomendación es desechar cualquier potencialmente contaminados los reactivos utilizados en las pruebas de fondo descontaminar todo el equipo y vuelva a probar con componentes de la reacción fresca. La trama 2D ejemplo tercera presentada como un aerosol de gotas a lo largo de una línea de 45° (figura 4C). Esto es causado a menudo por corte y une de gotitas. Cuidado gotita manejo antes de ciclos térmicos es esencial, como las gotas son propensas a dañar. Se recomienda el uso de generación automatizada de la gotita, cuando esté disponible. Si transferir manualmente genera gotas, asegúrese de elegir las puntas ancha recomendadas y emplear la técnica de pipeteo cuidadoso. Transferencia de gota requiere aspiración lenta y dispensación, con cada lugar durante 5-6 segundos, y es esencial que el apertura de punta de la pipeta toque el cartucho de la gota o bien. Despachador, mantener la punta de la pipeta en el nivel del líquido y aumentar poco a poco como gotas dispensada (ver vídeo demostración). El ejemplo de la parcela 2D final demuestra una falta de separación entre las poblaciones de gotas positivas y negativas (figura 4). Esto puede tener varias causas. Inhibidores de la PCR de fuerte, como detergentes utilizados en almacenadores intermediarios de la lisis y un exceso de ADN altamente degradado, pueden causar pérdida de la separación. En este caso, considerar la adición de un paso de limpieza entre la síntesis de cDNA y dPCR (como se describe en el paso 5 del presente Protocolo). Finalmente, falta de separación también puede ser debido a las condiciones óptimas de amplificación, y también se debe considerar la optimización del paso de la polimerización en cadena.
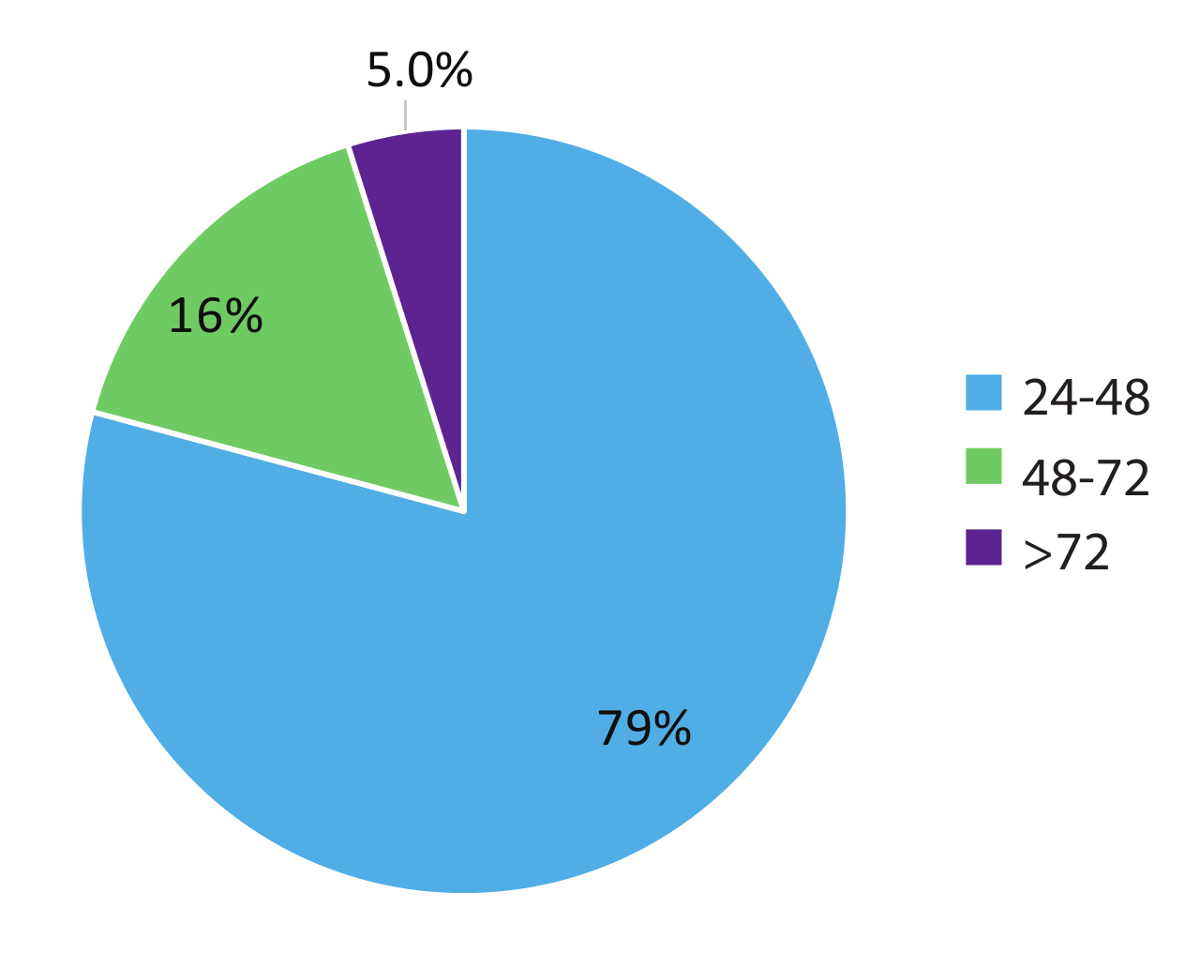
Datos de paciente de mundo real 984 figura 5 representan tiempos de vuelta de la muestra y demuestra el carácter rápido de este flujo de trabajo de prueba. Resultados se informó que el médico tan pronto como dentro de 48 horas (79% de los casos) de la recepción de la muestra y en el 95% de los casos, dentro de las 72 horas. En conclusión, el uso de estabilizado circulación RNA sangre colección tubos, procedimientos de extracción de RNA optimizados de la sangre y RT-dPCR ejecutar según un protocolo optimizado con la apropiada interno y controles por lotes, puede proporcionar un sistema de prueba rápida para la detección precisa de fusión RNA variantes relevantes en el CPCNP.

Figura 1 : Resumen de pasos de procesamiento muestra sangre para la detección de variantes de fusión mediante ensayos específicos de RET más prevalente y ROS1 variantes en el CPCNP. (A) prueba de la muestra se inicia cuando se extrae sangre entera y un BCT viene en el kit de colección de muestras para el laboratorio clínico. Circular de RNA se recupera de múltiples fuentes dentro del plasma enriquecido con plaquetas, atrás transcripción con imprimación específica gen y purificaron para el uso en dPCR. Las muestras se procesaron mediante un sistema comercialmente disponible que consiste en la generación de gotitas (emulsión), amplificación y conteo de gotas. Datos se analizaron utilizando el software disponible en el mercado. Resultados de la prueba son entonces documentados y comunicados al médico solicitando a prueba. El proceso está diseñado para trabajar dentro de un plazo de 72 horas desde la recepción de la muestra a liberación de resultado. Ocho variantes para ROS1 (B) y (C) RET están cubiertas dentro de los ensayos multiplexados. Adaptado de la página web Biodesix con autorización. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2 : Validación analítica. Líneas celulares expresan (A) SDC4 ROS1 fusión y fusión (B) CCDC6-RET se diluyeron en un fondo de ARN total humano de tipo salvaje (WT RNA). Con cada variante de la fusión, el límite de detección se estableció en 0,2% frecuencia variante con criterios previamente definidos para cada ensayo de variante. Todas las muestras por encima de este umbral también contienen por lo menos 21 copias del gen de control. Estándar de EML4-ALK (ALK) de 5% en un fondo de tipo RNA fue probada para demostrar la especificidad del ensayo, que fue confirmada por un resultado negativo. Estándares analíticos de RNA multiplexados se midieron en alta, media y bajas concentraciones de ROS1 (C) y (D) RET. precisión se evaluó en tres carreras en el mismo día (intradía), tres carreras en tres días consecutivos (entre días) y con dos operadores independientes (inter-operador). Se muestran los medios número de copias y desviaciones estándar. Adaptado de la página web Biodesix con autorización. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3 : Procesamiento de datos de robustez y ejemplos de Control de lote. Parcela 2D de la dPCR de ensayo ROS1 multiplexado no resultados para control positivo (A), (B) control de la transcriptasa reversa y (C) no hay control de plantilla del RNA. (D) los controles fueron funcionados en 21 días consecutivos (excepto fines de semana y festivos). Significa copia número +/-desviaciones estándar para ROS1 positivo control fueron 439 +-141. No de la transcriptasa reversa y sin controles de plantilla fueron también cada día, y eran todos negativos (datos no mostrados). Parcela 2D de la RET multiplexado ensayo dPCR no resultados para control positivo (E), (F) control de la transcriptasa reversa y (G) no hay control de plantilla del RNA. (H) controles fueron funcionados en 21 días consecutivos (excepto fines de semana y festivos). Significa copias +/-desviaciones estándar para RET positivo control fueron 586 +-182. No muestran ninguna de la transcriptasa reversa y sin controles de plantilla que también se ejecutan cada día y eran todos negativos. Adaptado de la página web Biodesix con autorización. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4 : Solución de problemas de dPCR RT. Parcelas de 2D que representa dPCR óptimo resultados cuando hay contaminación (A) en el no control de la transcriptasa inversa, (B) contaminación en el no control de RNA, (C) corte y une de gotitas y (D ) mal optimizado las condiciones de la polimerización en cadena o inhibición de la polimerización en cadena. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5 : Tiempo de respuesta (TAT). TAT (en horas) fue compilado para pruebas solicitando una variante de RNA (n = 984). Datos excluyen fines de semana, fiestas y muestras destinadas > 24 h debido a la incompleta información clínica en el laboratorio de formas de solicitud de prueba. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Tabla 1: Preparación de los reactivos de reversa de la transcripción para controles de proceso.
| Componente | Volumen |
| 2 x dPCR supermix para sondas (no hay 2'-desoxiuridina 5'-trifosfato) | 10 ΜL |
| set de 20 x objetivo variante cartillas/sondas (cartillas de 450 nmol/L, sonda de FAM de 250 nmol/L) | 1 ΜL |
| 20 juego de primers/sonda control objetivo x (cartillas de 450 nmol/L, sonda HEX de 250 nmol/L) | 1 ΜL |
| agua libre de nucleasas | 1 ΜL |
| cDNA | 7 ΜL |
Tabla 2: Preparación de la mezcla principal de dPCR.
| Bicicleta paso | Temperatura | Tiempo | # Ciclos | Tasa de rampa |
| Activación enzimática | 95 ° C | 10 min | 1 | ~ 2 oC/s |
| Desnaturalización | 94 ° C | 30 s | 40 | |
| Recocido/extensión | 55 ° C | 1 min | ||
| Desactivación de la enzima | 98 ° C | 10 min | 1 | |
| Hold (opcional) | 4 ° C | infinito | 1 | ~ 1 oC/s |
Tabla 3: Condiciones de ciclo termal.
Discusión
Reordenamientos RET y ROS1 componen ~ 3% de las mutaciones de controlador en el CPCNP población18. Aunque sea rara, la detección de estas alteraciones genéticas es vital. Pacientes de CPCNP con estas alteraciones pueden beneficiarse de terapias específicas que inhiben específicamente la actividad de la cinasa aberrante que resulta de la onco-proteína13. Algunas de estas terapias ya son aprobados por la FDA para uso en el CPCNP positivo ROS1, mientras que otros se han demostrado para ser eficaz contra RET en ensayos clínicos19.
Tecnología PCR digital ofrece una sensibilidad que es ideal para aplicaciones de biopsia líquida20. Ha habido una adopción importante de esta tecnología para el uso con circulación libre de ADN para la determinación de mutaciones del tumor en pacientes con NSCLC4,6,21,22,23 . Además de cfDNA, hemos desarrollado un protocolo diseñado para la medida robusta de las variantes de fusión más frecuentes en pacientes con NSCLC de circulación tumor RNA (figura 1A)10.
Nuestro protocolo establecido permite límites analíticos de detección hasta 0.2% (figura 2). Mientras que la dPCR RT es muy específica y sensible, los ensayos se limitan al panel de variantes de fusión conocidos que se eligen y multiplexado para la detección en el análisis de la polimerización en cadena. Por lo tanto, fusiones en ensayos de multiplexado deben seleccionarse cuidadosamente para asegurar una cobertura adecuada en la población de pacientes con NSCLC. Hemos diseñado con éxito los ensayos para RET y ROS1 que simultáneamente detectar ocho resultantes de variantes de fusión de los cambios de los loci RET o ROS1 y cubrir el 99% y el 88% de la población positivo RET y ROS1, respectivamente (figura 1B-C )17.
El flujo de trabajo de final de la prueba como se describe en este estudio incluye controles de lote para asegurar la consistencia de los resultados. Estos controles incluyen un nivel analítico positivo, así como dos controles negativos, que juntos garantizan que no haya contaminación o inhibición de la polimerización en cadena que ocurren dentro del lote (figura 3). Para garantizar la robustez del ensayo, se realizó un estudio utilizando los controles de lote durante un período de 21 días (figura 3D, H). Estos datos demuestran la consistencia del proceso de RNA según lo establecido en este protocolo.
Buenas prácticas de laboratorio y manejo adecuado de la RNA son componentes clave de asegurar resultados robustos y exactos. Espacio de laboratorio y equipo dedicado a utilizar con el ARN, limpieza del equipo después de cada uso, utilizando consumibles y reactivos libres de RNasa y aplicar un spray de inactivación de Rnasa al espacio de trabajo ayudan a reducir contaminantes RNasas. Consciente manipulación de las muestras de RNA por técnicos, incluyendo una capa del laboratorio dedicado, frecuentes cambios de guante, trabajando rápidamente a través del procedimiento de extracción de RNA, y mantenimiento de las muestras en hielo es de suma importancia para preservar la integridad de la muestra. Una vez que la RNA ha sido revés transcrito a cDNA, la muestra es de una forma más estable que es menos propensa a la degradación. Además de las prácticas que apoyan la integridad del RNA, las muestras y los componentes de la PCR se deben mantener en áreas segregadas para evitar la contaminación cruzada que puedan conducir a resultados falsos positivos. Los valores reactivos de PCR y preparación de mezclas principales PCR deben ser apartadas del plantillas de PCR y grandes se debe a la segregación amplificada plantilla (post-PCR) de todos los materiales previamente amplificados incluyendo reactivos, RNA y las muestras de cDNA. Finalmente, la correcta generación y manejo de mezclas emulsionadas de PCR antes de la amplificación es central para mantener la integridad de la gotita y dPCR óptimas condiciones. Medidas como estas son fundamentales durante la ejecución de este protocolo para obtener resultados consistentes y exactos. Todos los datos deben ser examinados por personal capacitado antes de la publicación de los resultados para asegurarse que se cumplan los parámetros de control de calidad todos. En el caso de resultados subóptimos (figura 4), el lote debe ser revisado por personal técnico y el Director del laboratorio y requerir volver a procesar.
RT-dPCR resultados pueden obtenerse tan pronto como 24 horas desde la recepción de la muestra y 95% de los resultados de las muestras en el aparato utilizado en este estudio de prueba (n = 984) notificaron al pedido médico en menos de 72 horas desde el momento de la recepción (figura 5). Este tiempo brinda a los médicos mucho la información molecular en un marco de tiempo que permite la iniciación de la terapia adecuada que necesita. Estos resultados están típicamente disponibles antes que las obtenidas mediante una biopsia de tejido convencional. Adicional para NSCLC y otros tipos de cáncer podrían ser desarrollados utilizando métodos similares basados en RNA circulantes y se beneficiarían con el mismo tiempo-a-resultados rápidos. Por ejemplo, medición de la transcripción de mRNA de ligando de muerte programada 1 (PD-L1) mediante RT-dPCR podría informar a los médicos sobre las opciones de inmunoterapia. También hay un creciente interés en la utilidad de la biopsia líquida y dPCR en el seguimiento de la eficacia terapéutica. Las indicaciones anteriores de reaparición del tumor mediante pruebas específicas variantes genómicas podrían permiten a los médicos ajustar los regímenes de tratamiento antes de que los pacientes son sintomáticos por estándar de medidas de cuidado como la proyección de imagen24. Protocolos como el reportado en este estudio son ideales para monitoreo debido a su no invasividad, sensibilidad, tiempo de vuelta rápido y la rentabilidad. El ensayo descrito en el presente documento proporciona resultados dentro de las 72 horas desde la recepción de muestras, con tasas de detección mínima de falsos positivos, que facilita las decisiones de tratamiento rápido y evita algunas limitaciones con base de tejido de prueba4.
Nuestro protocolo y datos demuestran un sistema de prueba robusta para la identificación de variantes de RNA de baja abundancia así como la posibilidad de mutación de base de sangre de prueba en la práctica clínica. Para aquellos pacientes que no tienen un conductor accionable mutación identificada por biopsia líquida específica rápida acerca como éste, la adición de más extenso genoma y proteoma pruebas de tejido y sangre puede proporcionar información clínica más amplia apoyar la planificación del tratamiento.
Divulgaciones
H.M., L.J., K.A., G.A.P. son empleados de y tienen acciones en H.M. Biodesix, Inc., L.J. y G.A.P. son inventores Co en una solicitud de patente presentada por Biodesix, que abarca un sistema de prueba de diagnóstico para la detección de variantes genéticas en células no pequeñas en circulación cáncer de pulmón.
Agradecimientos
Agradecemos a nuestros colaboradores, Stephen Jones, Nia Charrington, Dr. Dianna Maar y el Dr. Samantha Cooper desde el centro Digital de la biología (Bio-Rad Inc. CA) por su diseño de ensayo; Nezar Rghei y el Dr. Moemen Abdalla (Norgen Biotek, Canadá) para asesoramiento crítico cuando optimizar el protocolo de extracción de RNA; y Shannon Campbell, Scott Thurston, Jeff Fensterer, Shannon Martello y Joellyn Enos ayuda con requisitos y seguimiento comercial.
Materiales
| Name | Company | Catalog Number | Comments |
| Ultrapure Distilled Water (DNAse, RNAse Free) (500 mL) | Life Technologies | 10977-015 | 1604071 |
| Ultrapure Distilled Water (DNAse, RNAse Free) (500 mL) | Life Technologies | 10977-015 | 1809353 |
| Nuclease-free water (molecular grade) | Ambion | AM9938 | 1604071 |
| Nuclease-free water (molecular grade) | Ambion | AM9938 | 1606077 |
| Phosphate Buffered Saline 1X, Sterile | Amresco | K812-500mL | 1446C189 |
| Phosphate Buffered Saline 1X, Sterile – 500 mL | Invitrogen | 10010023 | 1916C092 |
| RNase Zap (Life Tech) (250 mL) | Ambion | AM9780 | 353952 |
| Beta-Mercaptoethanol (BME) (250 mL) | CalbioChem | 6050 | W105B |
| OmniPur Ethyl Alcohol | CalbioChem | 4455-4L | 56054611 |
| OmniPur Ethyl Alcohol | CalbioChem | 4455-4L | 56238638 |
| Isopropyl Alcohol | VWR | 0918-4L | 2116C416 |
| TranscriptAid T7 High Yield Transcription Kit | Thermo Scientific | K0441 | 403648 |
| TranscriptAid T7 High Yield Transcription Kit | Thermo Scientific | K0441 | 288461 |
| DNase I | Thermo | K0441 | 371299 |
| QIAzol Lysis Reagent | Qiagen | 79306 | 54809699 |
| 20x TE buffer pH 8.0 | Alfa Aesar | J62388 | R13C548 |
| UltraPure Agarose | Invitrogen | 16500-100 | 552730 |
| 10x TBE buffer | Invitrogen | AM9863 | 353065 |
| Cell-Free RNA BCT | Streck | 218976 | 60110327 |
| Cell-Free RNA BCT | Streck | 218976 | 61900327 |
| Cell-Free RNA BCT | Streck | 218976 | 61480327 |
| Cell-Free RNA BCT | Streck | 218976 | 62320327 |
| Plasma/Serum Circulating and Exosomal RNA Purification Kit (Slurry Format) 50 preps | Norgen | 42800 | 585849 |
| Plasma/Serum Circulating and Exosomal RNA Purification Kit (Slurry Format) 50 preps | Norgen | 42800 | 588308 |
| Lysis Buffer | Norgen | 21205 | A5F61E |
| RNA Cleanup and Concentration Micro-Elute Kit (Norgen) 50 preps | Norgen | 61000 | 585848 |
| RNA Cleanup and Concentration Micro-Elute Kit (Norgen) 50 preps | Norgen | 61000 | 588309 |
| DNA Clean and ConcentratorTM- 5 200 preps (samples) | Zymo | D4014 | ZRC186976 |
| DNA Clean and ConcentratorTM- 5 200 preps (samples) | Zymo | D4014 | ZRC188077 |
| DNA Clean and ConcentratorTM- 5 200 preps (samples) | Zymo | D4014 | ZRC188413 |
| Collection Tubes 500 pack | Zymo | C1001-500 | N/A |
| SuperScript IV First Strand Synthesis System 200 rxn (samples) | Life Technologies | 18091200 | 391657 |
| SuperScript IV First Strand Synthesis System 200 rxn (samples) | Life Technologies | 18091200 | 392504 |
| SuperScript IV First Strand Synthesis System 200 rxn (samples) | Life Technologies | 18091200 | 448001 |
| SuperScript IV Reverse Transcriptase | Life Technologies | 18090200 | 451702 |
| Qubit HS RNA Assay Kit (500) | Life Technologies | Q32854 | 1745264 |
| Qubit assay tubes (500) | Life Technologies | Q32856 | 13416Q311 |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023 | 64031651 |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023 | 64063941 |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023 | 64065740 |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023 | 64065741 |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023 | 64079083 |
| ddPCR Buffer Control for Probes | Bio-Rad | 1863052 | 64025320 |
| ddPCR Buffer Control for Probes | Bio-Rad | 1863052 | 64052358 |
| gBlock KIF5B-RET K15:R12 | IDT | 151004172 | 4-Oct-16 |
| gBlock KIF5B-RET K16:R12 | IDT | 151004173 | 4-Oct-16 |
| gBlock KIF5B-RET K22:R12 | IDT | 151004174 | 4-Oct-16 |
| gBlock KIF5B-RET K23:R12 | IDT | 151004175 | 4-Oct-16 |
| gBlock KIF5B-RET K24:R11 | IDT | 151004176 | 4-Oct-16 |
| gBlock KIF5B-RET K24:R8 | IDT | 151004177 | 4-Oct-16 |
| gBlock CCDC6-RET C1:R12 | IDT | 151004178 | 4-Oct-16 |
| gBlock TRIM33-RET T14:R12 | IDT | 151004179 | 4-Oct-16 |
| RET exon 8 RT Gene Specific Primer | IDT | 150554385 | 28-Sep-16 |
| 5’-CTCCACTCACACCTG-3’ | IDT | 150554385 | 28-Sep-16 |
| RET exon 11 RT Gene Specific Primer | IDT | 150554384 | 28-Sep-16 |
| 5’-GCAAACTTGTGGTAGCAG-3’ | IDT | 150554384 | 28-Sep-16 |
| RET exon 12 RT Gene Specific Primer | IDT | 150554383 | 28-Sep-16 |
| 5’-CTGCCTTTCAGATGGAAG-3’ | IDT | 150554383 | 28-Sep-16 |
| gBlock CD74-ROS1 C6:R34 | IDT | 152324366 | 15-Nov-16 |
| gBlock CD74-ROS1 C6:R32 | IDT | 152324367 | 15-Nov-16 |
| gBlock SDC4-ROS1 S2:R32 | IDT | 152324368 | 15-Nov-16 |
| gBlock SDC4-ROS1 S2:R34 | IDT | 152324369 | 15-Nov-16 |
| gBlock S13del2046-ROS1 S13del2046:R32 | IDT | 152324370 | 15-Nov-16 |
| gBlock S13del2046-ROS1 S13del2046:R34 | IDT | 152324371 | 15-Nov-16 |
| gBlock EZR-ROS1 E10:R34 | IDT | 152324372 | 15-Nov-16 |
| gBlock TPM3-ROS1 T8:R35 | IDT | 152324373 | 15-Nov-16 |
| ROS1 exon 34 RT Gene Specific Primer | IDT | 152704983 | 21-Nov-16 |
| 5’-CCTTCCTTGGCACTTT-3’ | IDT | 152704983 | 21-Nov-16 |
| ROS1 exon 35 RT Gene Specific Primer | IDT | 152704985 | 21-Nov-16 |
| 5’-CTCTTGGGTTGGAAGAGTATG-3’ | IDT | 152704985 | 21-Nov-16 |
| ALK Gene Specific Primer | IDT | 140035422 | 26-Aug-16 |
| 5’-CAGTAGTTGGGGTTGTAGTCG-3’ | IDT | 140035422 | 26-Aug-16 |
| EML4-ALK Cell line pellet | Horizon Discovery | N/A | 11-Jun-15 |
| SLC34A2-ROS1 Cell line pellet | Horizon Discovery | N/A | 11-Jun-15 |
| CCDC6-RET Cell line pellet | Horizon Discovery | N/A | 11-Jun-15 |
| Human Brain Total RNA | Ambion | AM7962 | 1703548 |
| PrimePCR ddPCR Expert Design Assay: K15:R12 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: K16:R12 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: K22:R12 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: K23:R12 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: K24:R11 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: K24:R8 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: C1:R12 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: T14:R12 | Bio-Rad | N/A | 17-Aug-16 |
| PrimePCR ddPCR Expert Design Assay: C6:R34 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: C6:R32 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: S2:R32 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: S2:R34 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: S13del2046:R32 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: S13del2046:R34 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: E10:R34 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: T8:R35 | Bio-Rad /Biodesix | N/A | 6-Dec-16 |
| (version 2) | Bio-Rad | 12003909 | 213939881 |
| PrimePCR ddPCR Expert Design Assay: ROS1 Multiplex (version 3.2) | Bio-Rad | N/A | 13-Dec-16 |
| PrimePCR ddPCR Expert Design Assay: ROS1 Multiplex (version 3.2) | Bio-Rad | N/A | 20170112v3.2 |
| PrimePCR ddPCR Gene Expression Probe Assay: GUSB, Human | Bio-Rad | 10031257 | 212851151 |
| PrimePCR ddPCR Gene Expression Probe Assay: GUSB, Human | Bio-Rad | 10031257 | 207383915 |
| PrimePCR ddPCR Gene Expression Probe Assay: GUSB, Human | Bio-Rad | 10031257 | 195995635 |
| PrimePCR ddPCR Gene Expression Probe Assay: GUSB, Human | Bio-Rad | 10031257 | 212851152 |
| PrimePCR ddPCR Gene Expression Probe Assay: GUSB, Human | Bio-Rad | 10031257 | 213949301 |
| PrimePCR ddPCR Expert Design Assay: EML4-ALK | Bio-Rad | 12003909 | 20160914 |
| PrimePCR ddPCR Expert Design Assay: EML4-ALK | Bio-Rad | 12003909 | 211383227 |
| Droplet Generation Oil for Probes | Bio-Rad | 186-3005 | 1065C220 |
| Droplet Generation Oil for Probes | Bio-Rad | 186-3005 | 64052953 |
| Droplet Generation Oil for Probes | Bio-Rad | 186-3005 | 64052358 |
| Automated Droplet Generation Oil for Probes (20x96) | Bio-Rad | 186-4110 | 1065C320 |
| Automated Droplet Generation Oil for Probes (20x96) | Bio-Rad | 186-4110 | 64052952 |
| Automated Droplet Generation Oil for Probes (20x96) | Bio-Rad | 186-4110 | 64064127 |
| DG8 Cartridges for QX100/QX200 Droplet Generator | Bio-Rad | 186-4008 | C000065883 |
| DG8 Cartridges for QX100/QX200 Droplet Generator | Bio-Rad | 186-4008 | C000084276 |
| DG8 Cartridges for QX100/QX200 Droplet Generator | Bio-Rad | 186-4008 | C000079928 |
| DG8 Cartridges for QX100/QX200 Droplet Generator | Bio-Rad | 186-4008 | C000084395 |
| DG8 Cartridges for QX100/QX200 Droplet Generator | Bio-Rad | 186-4008 | C000084634 |
| Droplet Generator DG8 Gasket | Bio-Rad | 186-3009 | 20160627 |
| Droplet Generator DG8 Gasket | Bio-Rad | 186-3009 | 20161107 |
| Droplet Generator DG8 Gasket | Bio-Rad | 186-3009 | 20161206 |
| Droplet Generator DG8 Gasket | Bio-Rad | 186-3009 | 20161216 |
| Droplet Generator DG8 Gasket | Bio-Rad | 186-3009 | 20170125 |
| Pipet Tips for Automated Droplet Generator | Bio-Rad | 1864120 | PR125340 |
| DG32 Cartridge for Automated Droplet Generator (10-96 well plates) | Bio-Rad | 186-4108 | 206894 |
| DG32 Cartridge for Automated Droplet Generator (10-96 well plates) | Bio-Rad | 186-4108 | 206893 |
| Pierceable Foil Heat Seal | Bio-Rad | 1814040 | 1409850 |
| Pierceable Foil Heat Seal | Bio-Rad | 1814040 | 100402 |
| Pierceable Foil Heat Seal | Bio-Rad | 1814040 | 145851 |
| Microseal 'B' seals | Bio-Rad | MSB1001 | BR00428490 |
| ddPCR Droplet Reader Oil | Bio-Rad | 186-3004 | 64039089 |
| ddPCR Droplet Reader Oil | Bio-Rad | 186-3004 | 64049253 |
| ddPCR Droplet Reader Oil | Bio-Rad | 186-3004 | 64049255 |
| ddPCR Droplet Reader Oil | Bio-Rad | 186-3004 | 64081870 |
| DNA Lo Bind Tube 0.5 mL | Eppendorf | 22431005 | E1629620 |
| DNA Lo Bind Tube 1.5 mL | Eppendorf | 22431021 | F16698K |
| DNA Lo Bind Tube 2 mL | Eppendorf | 22431048 | E160610I |
| 50 mL Conicals, Polypropylene (25) | Thermo | 339652 | G5ZF5W8118 |
| TempAssure PCR 8-Strips, Optical Caps, Natural, polypropylene (120) | USA Scientific | 1402-4700 | 16202 |
| For Rainin LTS Pipettors 0.5-20 µL tips | Pipette.com | LF-20 | 40155-642C4-642C |
| For Rainin LTS Pipettors 5-200 µL tips | Pipette.com | LF-250 | 40154-642C4-642B |
| Tips LTS 200 ul Filter 960/10 RT-L200F (10 boxes) | Rainin | 17002927 | 1635 |
| Pipet Tips, 10 ul TipOne RPT ultra low retention filter tip refill cassette, sterile | USA Scientific | 1181-3710 | F1175551-1108 |
| Pipet Tips, 10 ul TipOne RPT ultra low retention filter tip refill cassette, sterile | USA Scientific | 1181-3710 | F118054L-1720 |
| Pipet Tips, 100 ul TipOne RPT ultra low retention filter tip refill cassette, sterile (10x96) | USA Scientific | 1180-1740 | 0014961Q-2501 |
| Pipet Tips, 200 ul TipOne RPT ultra low retention filter tip refill cassette, sterile | USA Scientific | 1180-8710 | E116684P-1540 |
| Pipet Tips, 1000 ul XL TipOne RPT ultra low retention filter tip refill cassette, sterile (10x96) | USA Scientific | 1182-1730 | F118815P |
| 5 mL Standard Racked Gilson-fit Reference Tips | Scientific Specialties | 4411-00 | 14312 |
| Combitips advanced, 0.1 mL Biopur | Eppendorf | 003 008 9618 | F165414H |
| Combitips advanced, 0.2 mL Biopur | Eppendorf | 0030 089.626 | F166689J |
| Combitips advanced, 5 mL Biopur | Eppendorf | 0030.089 669 | F166054J |
| Combitips advanced, 50 mL Biopur | Eppendorf | 003.008.9693 | F166055I |
| Reagent Reservoir | VWR | 89094-680 | 141500 |
| Twin tec PCR Plate 96, semi-skirted, Clear | Eppendorf | 951020303 | E163697P |
| Twin tec PCR Plate 96, semi-skirted, Clear | Eppendorf | 951020303 | F165029I |
| Twin tec PCR Plate 96, semi-skirted, Clear | Eppendorf | 951020303 | F165028G |
| Twin tec PCR Plate 96, semi-skirted, Clear | Eppendorf | 951020303 | E163697P |
| Twin tec PCR Plate 96, semi-skirted, Green | Eppendorf | 951020346 | F166183K |
| Equipment Type | Equipment ID | ||
| Analytical Balance | EQP0125 | ||
| Cryogenic Freezer 1, -80oC | EQP0095 | ||
| Refrigerator 6.1 cu ft GP06W1AREF | EQP0139 | ||
| -20oC Freezer | EQP0140 | ||
| Beckman Coulter Microfuge 22R | EQP0025 | ||
| Beckman Coulter Microfuge 22R | EQP0124 | ||
| Thermo Scientific Hereaus Megafuge 8 | EQP0104 | ||
| Mini Centrifuge | EQP0131 | ||
| Mini Centrifuge | EQP0136 | ||
| Mini Centrifuge | EQP0134 | ||
| Mini Centrifuge | EQP0235 | ||
| Mini Centrifuge | EQP0216 | ||
| Thermo Scientific HeraTherm Incubator | EQP0105 | ||
| Pipette 0.1 - 2.5 μL | EQP0182 | ||
| Pipette 0.1 - 2.5 μL | EQP0072 | ||
| Pipette 0.1 - 2.5 μL | EQP0070 | ||
| Pipette 0.5-10 μL | EQP0218 | ||
| Pipette 0.5-10 μL | EQP0075 | ||
| Pipette 0.5-10 μL | EQP0169 | ||
| Pipette 0.5-10 μL | EQP0074 | ||
| Pipette 0.5-10 μL | EQP0147 | ||
| Pipette 2 - 20 μL | EQP0128 | ||
| Pipette 2 - 20 μL | EQP0160 | ||
| Pipette 2 - 20 μL | EQP0018 | ||
| Pipette 2 - 20 μL | EQP0146 | ||
| Pipette 10 - 100 μL | EQP0079 | ||
| Pipette 10 - 100 μL | EQP0181 | ||
| Pipette 10 - 100 μL | EQP0085 | ||
| Pipette 10 - 100 μL | EQP0077 | ||
| Pipette 20 - 200 μL | EQP0088 | ||
| Pipette 20 - 200 μL | EQP0087 | ||
| Pipette 20 - 200 μL | EQP0231 | ||
| Pipette 100 - 1000 μL | EQP0050 | ||
| Pipette 100 - 1000 μL | EQP0158 | ||
| Pipette 100 - 1000 μL | EQP0217 | ||
| Pipette 100 - 1000 μL | EQP0082 | ||
| Pipette 100 - 1000 μL | EQP0183 | ||
| Pipette 100 - 1000 μL | EQP0083 | ||
| Pipette 5 mL | EQP0153 | ||
| Timer | S/N 140623950 | ||
| Hamilton SafeAire VAV Fume Hood | EQP0206 | ||
| Biosafety Cabinet | EQP0205 | ||
| Biosafety Cabinet | EQP0204 | ||
| Qubit 3.0 | EQP0102 | ||
| Benchmark Digital Heat Block | EQP0108 | ||
| Benchmark Digital Heat Block | EQP0231 | ||
| Polaroid Z2300 Instant Print Digital Gel Camera with WiFi and 16GB SDHC memory card | EQP0111 | ||
| Electrophoresis Power Unit | EQP0113 | ||
| Electrophoresis Small Gel Box | EQP0116 | ||
| Maestro Transilluminator | EQP0118 | ||
| Microwave | EQP0215 | ||
| Multichannel 8-well Pipette 2 - 20 μL | EQP0207 | ||
| Multichannel 8-well Pipette 10 - 100 μL | EQP0090 | ||
| Rainin Multichannel 8-well Pipette 50 μL | EQP0094 | ||
| Rainin Multichannel 8-well Pipette 50 μL | EQP0161 | ||
| Rainin Multichannel 8-well Pipette 50 μL | EQP0162 | ||
| Rainin Multichannel 8-well Pipette 50 μL | EQP0163 | ||
| Vortex Genie 2 | EQP0052 | ||
| Vortex Genie 2 | EQP0007 | ||
| Vortex Genie 2 | EQP0132 | ||
| Vortex Genie 2 | EQP0137 | ||
| Vortex Genie 2 | EQP0135 | ||
| Air Clean PCR Workstation | EQP0203 | ||
| Air Clean PCR Workstation | EQP0096 | ||
| Air Clean PCR Workstation | EQP0148 | ||
| Air Clean PCR Workstation | EQP0097 | ||
| QX200 Droplet Generator | EQP0202 | ||
| QX200 Droplet Generator | EQP0121 | ||
| Automated Droplet Generator | EQP0179 | ||
| PX1 PCR Plate Sealer | EQP0123 | ||
| PX1 PCR Plate Sealer | EQP0186 | ||
| C1000 Touch Cycler w/96W FS RM | EQP0120 | ||
| S1000 Cycler w/96W FS RM | EQP0174 | ||
| S1000 Cycler w/96W FS RM | EQP0173 | ||
| T100 Thermal Cycler | EQP0180 | ||
| T100 Thermal Cycler | EQP0175 | ||
| QX200 Droplet Reader | EQP0194 | ||
| QX200 Droplet Reader | EQP0122 |
Referencias
- Ignatiadis, M., Lee, M., Jeffrey, S. S. Circulating Tumor Cells and Circulating Tumor DNA: Challenges and Opportunities on the Path to Clinical Utility. Clin Cancer Res. 21 (21), 4786-4800 (2015).
- Alix-Panabieres, C., Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. , (2016).
- Paxton, A. Is Molecular AP testing in sync with guidelines. CAP Today. , (2014).
- Sacher, A. G., et al. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol. , (2016).
- Sozzi, G., et al. Quantification of free circulating DNA as a diagnostic marker in lung cancer. J Clin Oncol. 21 (21), 3902-3908 (2003).
- Oxnard, G. R., et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 20 (6), 1698-1705 (2014).
- Best, M. G., et al. RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell. 28 (5), 666-676 (2015).
- Rodriguez, M., et al. Different exosome cargo from plasma/bronchoalveolar lavage in non-small-cell lung cancer. Genes Chromosomes Cancer. 53 (9), 713-724 (2014).
- Kalluri, R. The biology and function of exosomes in cancer. J Clin Invest. 126 (4), 1208-1215 (2016).
- Mellert, H., et al. Development and Clinical Utility of a Blood-Based Test Service for the Rapid Identification of Actionable Mutations in Non-Small Cell Lung Carcinoma. J Mol Diagn. 19 (3), 404-416 (2017).
- Qin, J., Williams, T. L., Fernando, M. R. A novel blood collection device stabilizes cell-free RNA in blood during sample shipping and storage. BMC Res Notes. 6, 380 (2013).
- Chomczynski, P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques. 15 (3), 532-537 (1993).
- Kohno, T., et al. Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl Lung Cancer Res. 4 (2), 156-164 (2015).
- Takeuchi, K., et al. ROS1 and ALK fusions in lung cancer. Nat Med. 18 (3), 378-381 (2012).
- Rimkunas, V. M., et al. Analysis of receptor tyrosine kinase ROS1-positive tumors in non-small cell lung cancer: identification of a FIG-ROS1 fusion. Clin Cancer Res. 18 (16), 4449-4457 (2012).
- Tsuta, K., et al. RET-rearranged non-small-cell lung carcinoma: a clinicopathological and molecular analysis. Br J Cancer. 110 (6), 1571-1578 (2014).
- Forbes, S. A., et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45 (1), 777-783 (2017).
- Salgia, R. Diagnostic challenges in non-small-cell lung cancer: an integrated medicine approach. Future Oncol. 11 (3), 489-500 (2015).
- Cagle, P. T., Raparia, K., Portier, B. P. Emerging Biomarkers in Personalized Therapy of Lung Cancer. Adv Exp Med Biol. 890, 25-36 (2016).
- Vogelstein, B., Kinzler, K. W. Digital PCR. Proc Natl Acad Sci U S A. 96 (16), 9236-9241 (1999).
- Oxnard, G. R., et al. Association Between Plasma Genotyping and Outcomes of Treatment With Osimertinib (AZD9291) in Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 34 (28), 3375-3382 (2016).
- Reckamp, K. L., et al. A Highly Sensitive and Quantitative Test Platform for Detection of NSCLC EGFR Mutations in Urine and Plasma. J Thorac Oncol. 11 (10), 1690-1700 (2016).
- Yanagita, M., et al. A prospective evaluation of circulating tumor cells and cell-free DNA in EGFR mutant non-small cell lung cancer patients treated with erlotinib on a phase II trial. Clin Cancer Res. , (2016).
- Abbosh, C., et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 545 (7655), 446-451 (2017).
- Chomczynski, P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. BioTechniques. 15 (3), 532-534 (1993).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados