
Method Article
Secuenciación de próxima generación de ARN y una tubería bioinformática para identificar los LINE-1s expresados en el nivel específico de locus
En este artículo
Resumen
Aquí presentamos un enfoque y análisis bioinformáticos para identificar la expresión LINE-1 en el nivel específico del locus.
Resumen
Los elementos intercalados largos-1 (LINEs/L1s) son elementos repetitivos que pueden copiarse e insertarse aleatoriamente en el genoma, resultando en inestabilidad genómica y mutagénesis. La comprensión de los patrones de expresión de los loci L1 a nivel individual prestará a la comprensión de la biología de este elemento mutagénico. Este elemento autónomo conforma una porción significativa del genoma humano con más de 500.000 ejemplares, aunque el 99% están truncados y defectuosos. Sin embargo, su abundancia y el número dominante de copias defectuosas hacen que sea difícil identificar auténticamente expresado L1s de secuencias relacionadas con L1 expresadas como parte de otros genes. También es difícil identificar qué locus L1 específico se expresa debido a la naturaleza repetitiva de los elementos. Superando estos desafíos, presentamos un enfoque Bioinformático de ARN-SEQ para identificar la expresión L1 en el nivel específico del locus. En Resumen, recogemos el ARN citoplasmático, seleccionamos para las transcripciones poliadeniladas, y utilizamos análisis de ARN-SEQ específicos de la hebra para mapear lecturas de forma única a loci L1 en el genoma de referencia humano. Se curan visualmente cada locus L1 con lecturas asignadas de forma única para confirmar la transcripción de su propio promotor y ajustar las lecturas de transcripción asignadas para tener en cuenta la capacidad de asignabilidad de cada locus L1 individual. Este enfoque se aplicó a una línea celular de tumor de próstata, DU145, para demostrar la capacidad de este protocolo para detectar la expresión a partir de un pequeño número de elementos L1 de longitud completa.
Introducción
Los retrotransposones son elementos de ADN repetitivos que pueden "saltar" en el genoma en un mecanismo de copiado y pegado a través de los intermedios de ARN. Un subconjunto de retrotransposones se conoce como Long INterspersed Elements-1 (LINEs/L1s) y constituye una sexta parte del genoma humano con más de 500, 0000 copias1. A pesar de su abundancia, la mayoría de estas copias son defectuosas y truncadas con sólo un estimado 80-120 elementos L1 que se cree que son activos2. Una L1 de longitud completa tiene una longitud de aproximadamente 6 KB con regiones no traducidas de 5 ' y 3 ', un promotor interno y un promotor anti-sentido asociado, dos marcos de lectura abierta no solapados (ORFs) y una cola de señal y Polya3,4,5 . En los seres humanos, L1s se componen de subfamilias distinguidas por la edad evolutiva con las familias mayores que han acumulado más mutaciones de secuencia únicas en el tiempo en comparación con la subfamilia más joven, L1HS6,7. L1s son los únicos retrotransposones autónomos y humanos y sus ORFs codifican una transcriptasa inversa, endonuclease, y RNPs con las actividades de enlace de ARN y chaperona requeridas para retrotransponer e insertar en el genoma en un proceso referido como objetivo-cebado transcripción inversa8, 9,10,11,12.
Se ha notificado que la retrotransposición de L1s causó enfermedades de la línea germinal humana por una variedad de mecanismos que incluyen mutagénesis insercional, deleciones en el lugar de destino y rearreglos13,14,15, 16. recientemente se ha presumido que el L1s puede desempeñar un papel en la oncogénesis y/o progresión tumoral, ya que se han observado mayores manifestaciones y acontecimientos de inserción de este elemento mutagénico en una variedad de cánceres epiteliales17,18 . Se estima que hay una nueva inserción L1 en cada 200 nacimientos19. Por lo tanto, es imperativo entender mejor la biología de la expresión activa L1s. La naturaleza repetitiva y la abundancia de copias defectuosas encontradas en las transcripciones de otros genes han hecho que este nivel de análisis sea desafiante.
Afortunadamente, con el advenimiento de las tecnologías de secuenciación de alto rendimiento, se han hecho progresos para analizar e identificar auténticamente expresar L1s en el nivel específico del locus. Existen diferentes filosofías sobre cómo identificar mejor el L1s expresado usando la secuenciación de próxima generación de ARN. Sólo se han sugerido dos enfoques razonables para mapear transcripciones L1 en el nivel específico de locus. Uno se centra sólo en la transcripción potencial que lee a través de la señal de poliadenilación L1 y en las secuencias flanqueante20. Nuestro enfoque aprovecha las pequeñas diferencias de secuencia entre los elementos L1 y solo asigna las lecturas de ARN-SEQ que se asignan de forma única a un locus21. Ambos métodos tienen limitaciones en términos de cuantificación de los niveles de transcripción. La cuantificación se puede mejorar potencialmente añadiendo una corrección para la "asignabilidad única" de cada locus21L1, o usando algoritmos más complejos que redistribuyan las lecturas de múltiples mapeados que no podrían asignarse de forma única a un locus específico22. Aquí, detallaremos paso a paso la extracción de ARN y la secuenciación de última generación y el protocolo Bioinformático para identificar los elementos expresados en L1 en el nivel específico del locus. Nuestro enfoque aprovecha al máximo nuestro conocimiento de la biología de los elementos funcionales L1. Esto incluye saber que los elementos L1 funcionales deben generarse a partir del promotor L1, iniciado al principio del elemento L1, debe traducirse en el citoplasma y que sus transcripciones deben ser co-lineales con el genoma. Brevemente, recolectamos ARN citoplásmico fresco, seleccionamos para las transcripciones poliadeniladas, y utilizamos análisis de ARN-SEQ específicos de la hebra para mapear lecturas de forma única a loci L1 en el genoma de referencia humano. Estas lecturas alineadas todavía requieren una extensa curación manual para determinar si las lecturas de transcripción provienen del promotor L1 antes de designar un locus como un L1 auténticamente expresado. Aplicamos este enfoque en la muestra de la línea celular del tumor de próstata DU145 para demostrar cómo identifica a unos pocos miembros de L1 transcritos activamente de la masa de copias inactivas.
Protocolo
1. extracción del ARN citoplasmático
- Obtenga las celdas a través de los siguientes métodos.
- Recoge células vivas de 2,75% – 100% confluentes, frascos T-75.
- Lavar el matraz 2 veces en 5 mL de PBS frío, y en el último lavado raspar las células y transferir a un tubo cónico de 15 mL. Centrifugar durante 2 min a 1.000 x g y 4 ° c, y retirar y desechar con cuidado el sobrenadante (tabla de materiales).
- Recoja las células de los especímenes de tejido.
- Prepare el tejido para la extracción del ARN citoplásmico dentro de una hora de ser diseccionado y siempre mantener en el hielo. Para el almacenamiento a largo plazo, utilice soluciones de inhibidor de ARN para almacenar el tejido durante un máximo de 72 horas después de la disección siguiendo el protocolo del fabricante (tabla de materiales).
- Dados una muestra de 10 μm3 y homogeneizar la muestra fresca con 5 ml de PBS frío en un homogeneizador de Dounce estéril, transferir a un tubo cónico de 15 ml, centrifugar durante 2 min a 1.000 x g a 4 ° c, y retirar y desechar cuidadosamente el sobrenadante (tabla de materiales < /C8 >).
- Recoge células vivas de 2,75% – 100% confluentes, frascos T-75.
- Añadir 2 mL de tampón de lisis a la mezcla de pellets celulares e incubar sobre hielo durante 5 min.
- Preparar tampón de lisis fresca con 150 mM NaCl, 50 mM HEPES (pH 7,4) y 25 μg/mL digitonina (tabla de materiales).
- Como la concentración mínima de digitonina en el tampón de lisis necesaria para penetrar la membrana plasmática puede variar según el tipo de célula, se confirma microscópicamente que las células tratadas con tampón de lisis pierden la membrana plasmática y retienen la membrana nuclear intacta.
- Justo antes de su uso, añadir 1.000 U/mL inhibidor de la RNase (tabla de materiales).
- Centrifugar durante 1 minuto a 1.000 x g y 4 ° c, y recoger el sobrenadante.
- Añadir sobrenadante a 7,5 mL de TRIzol pre-enfriado y 1,5 mL de cloroformo. Todos los pasos que requieren el cloroformo deben realizarse dentro de una campana química limpia (tabla de materiales).
- Centrifugar por 35 min a 3.220 x g y 4 ° c.
- Transfiera la porción acuosa (capa superior) a un tubo fresco pre-enfriado de 15 mL.
- Añadir 4,5 mL de cloroformo y vórtice.
- Centrifugar durante 10 min a 3.220 x g y 4 ° c.
- Transfiera la porción acuosa al tubo pre-enfriado fresco.
- Añadir 4,5 mL de isopropanol, agitar bien e incubar a-80 ° c durante la noche (tabla de materiales).
- Centrifugar a 3.220 x g y 4 ° c durante 45 minutos.
- Extraer isopropanol, añadir 15 mL de etanol al 100% (tabla de materiales).
- Centrifugar a 3.220 x g durante 10 min.
- Retire el etanol, escurrir y secar durante aproximadamente 1 h.
- Utilice un bastoncillo de algodón estéril para borrar cualquier etanol restante (tabla de materiales).
- Vuelva a suspender la muestra en 100 a 200 μL de agua libre de RNase dependiendo del tamañodel pellet (tabla de materiales).
- Muestras fraccionadas utilizando tecnología de electroforesis para determinar la calidad y concentración de las muestras según las intrucciones del fabricante23 (tabla de materiales).
- Las muestras califican para el análisis de ARN-SEQ si RIN > 824.
2. secuenciación de próxima generación
- Presentar muestras de ARN citoplásmico para secuenciar utilizando la plataforma de secuenciación de próxima generación destinada a generar al menos 50 millones lecturas de 100 BP de final emparejado.
- Seleccione para RNAs poli-adeniladas y secuenciación específica de la hebra.
3. crear anotaciones (opcional si uno tiene una anotación existente)
- Cree una anotación L1 de longitud completa o descargue la anotación L1 de longitud completa (archivo suplementario 1A-b).
- Descargue las anotaciones de REPEAT Masker para los elementos LINE-1 desde el navegador genoma UCSC con la herramienta de navegador de tablas (https://genome.ucsc.edu/cgi-bin/hgTables). Especifique el clado mamífero, el genoma humano, el conjunto hg19 (o hg38 para un genoma más actualizado) y filtre por "LINE1" en nombre de clase. Descárguelo como archivo. GTF y etiquégalo como FL-L1-BLAST. GTF.
- Ejecute una búsqueda local de BLAST de la primera 300 BP del elemento L1 de longitud completa L 1.3 que abarca la región promotora en el genoma humano y agregue 6.000 BP aguas abajo para crear un final de las coordenadas L1 al archivo de anotación. Guardar en un archivo GTF y etiquetar como FL-L1-RM. GTF.
- Interseca la anotación RepeatMasker y la anotación L1 basada en el promotor utilizando bedtools y Label como FL-L1-BLAST_RM. txt (paquetes de software).
- Utilice este comando en el terminal Linux: bedtools Intersect-a FL-L1-Blast. GTF-b FL-L1-RM. gtf > FL-L1-BLAST_RM. txt.
- Separe la anotación FL-L1 intersectada por la hebra superior e inferior.
- Copiar sobre el FL-L1-BLAST_RM. txt en el software de hoja de cálculo y ordenar por el "menos" y "más" Strand y luego ordenar por la ubicación del cromosoma.
- Cree dos nuevos documentos de hoja de cálculo, uno con las coordenadas intersectadas para la longitud completa L1s en la hebra menos y otra en la hebra inferior, y guárdela como FL-L1-BLAST_RM_minus. xls y FL-L1-BLAST_RM_plus. xls.
- Guarde los dos nuevos documentos como archivos. txt.
- Utilice el programa mac2unix para convertir los archivos. txt a los archivos de anotación correctos (paquetes de software).
- Utilice este comando en el terminal: Mac2unix.sh FL-L1-BLAST_RM_minus. GFF.
- Utilice este comando en el terminal: Mac2unix.sh FL-L1-BLAST_RM_plus. GFF.
- Guarde los nuevos archivos con la extensión. GFF.
- Como alternativa, utilice AWK para filtrar filas asociadas con la hebra + y –.
- Utilice el siguiente comando para obtener el + Strand: awk '/+/' FL-L1_BLAST_RM. gtf > FL-L1_BLAST_RM_plus. GTF.
- Utilice la siguiente línea de comandos para obtener el-Strand: awk '/-/' FL-L1_BLAST_RM. gtf > FL-L1_BLAST_RM_minus. GTF.
4. leer canalización de alineación para identificar expresado L1s
| extra | descripción |
| – p | Esto detalla el número de subprocesos que el equipo debe usar para ejecutar la alineación. Mayor memoria del ordenador permitirá más hilos y debe ser empíricamente d. |
| – m 1 | Esto le dice al programa que sólo acepte lecturas que tienen una coincidencia en el genoma que es mejor que cualquier otro fósforo del genoma. |
| – y | Este es el interruptor de tryhard que hace la búsqueda de mapeo para todas las coincidencias posibles y no permitir que se salga después de que se alcance un número fijo de coincidencias. |
| – v 3 | Esto sólo permite que el programa utilice la memoria para las lecturas asignadas con 3 o menos discrepancias al genoma. |
| – X 600 | Esto sólo permite las lecturas emparejadas que se asignan dentro de 600 bases entre sí. Esto asegura que los pares leídos son co-lineales en el genoma y se seleccionan contra s que involucran moléculas de ARN procesado. |
| – chunkmbs 8184 | Este comando asigna memoria adicional para controlar la gran cantidad de alineaciones posibles para cada lectura relacionada con L1. |
Tabla 1: opciones de línea de comandos para Bowtie.
- Ejecute los archivos fastq de secuenciación de final emparejado de alineación con la muestra de ARN-SEQ de interés utilizando Bowtie.
Nota: Bowtie1 debe ser utilizado y no Bowtie2 porque los parámetros requeridos para la alineación única se encuentran específicamente sólo en esta versión de bowtie (paquetes de software). Bowtie se utiliza sobre alineadores conscientes del empalme como STAR para evaluar concordantes, las lecturas contiguas más relevantes para la biología y la expresión L1.- Utilice esta línea de mandatos en el terminal Linux: bowtie-p 10-m 1-S-y-v 3-X 600--chunkmbs 8184 hg_X_Y_M_index-1 hg_sample_1. FQ-2 hg_sample_2. FQ | samtools View-hbuS-| samtools Sort – hg_sample_sorted. BAM. Consulte la tabla 1 para obtener una descripción de las opciones de línea de comandos de Bowtie.
- Strand separa el archivo de BAM de salida mediante samtools (paquetes de software) y los siguientes comandos de Linux. Tenga en cuenta que los valores de indicador reales pueden variar si uno no está utilizando protocolos de secuenciación de próxima generación estándar.
- Utilice esta línea de comandos para seleccionar el hilo superior: samtools View-h hg_sample_sorted. BAM | awk ' substr ($ 0,1, 1) = = "@" | | $2 = = 83 | | $2 = = 163 {print} ' | samtools ver-BS-> hg_sample_sorted_topstrand. BAM.
- Utilice esta línea de comandos para seleccionar el Strand inferior: samtools View-h hg_sample_sorted. BAM | awk ' substr ($ 0,1, 1) = = "@" | | $2 = = 99 | | $2 = = 147 {print} ' | samtools ver-BS-> hg_sample_sorted_bottomstrand. BAM.
- Genere recuentos de lectura contra anotaciones para loci L1 mediante bedtools (paquetes de software).
- Utilice esta línea de mandatos para generar recuentos de lectura para L1s en la dirección de sentido del hilo superior: cobertura de bedtools-abam FL-L1-BLAST_RM_plus. GFF-b hg_sample_sorted_topstrand. bam > hg_sample_sorted_bowtie_tryhard_plus_top. txt.
- Utilice esta línea de mandatos para generar recuentos de lectura para L1s en la dirección de sentido de la hebra inferior: cobertura de bedtools-abam FL-L1-BLAST_RM_minus. GFF-b hg_sample_sorted_bottomstrand. bam > hg_sample_sorted_bowtie_tryhard_minus_bottom. txt.
- Archivo de BAM de índice del paso 5.1.1 para que sea visible en el Inteative Genomics Viewer (IGV)25 (paquetes de software).
- Utilice esta línea de mandatos: samtools index hg_sample_sorted. BAM
- Para utilizar un modo por lotes para aumentar el número de muestras de ARN-SEQ canalizado a la vez, utilice un script supercomputadora para completar el paso 4,1 llamado human_bowtie. sh, un script para completar los pasos 4.2-4.3 se ha creado llamado human_L1_pipeline. sh, y un script para completar se ha creado el paso 4,4 llamado bam_index. sh. Estos scripts se pueden encontrar en el archivo suplementario 2 con comandos de superordenador asociados para ejecutar los scripts.
5. la curación manual
- Cree una hoja de cálculo para lecturas asignadas a cada locus L1 anotado.
- Copie sobre hg_sample_sorted_bowtie_tryhard_minus_bottom. txt creado en el paso 4.3.2 y la página de la etiqueta como "menos-abajo."
- Ordene todas las columnas según el número de lecturas más alto y más bajo que se encuentra en la columna J.
- Copia sobre hg_sample_sorted_bowtie_tryhard_plus_top. txt creado en el paso 4.3.1 y etiqueta como "Top-Plus" en otra hoja de cálculo.
- Ordene todas las columnas según el número de lecturas más alto y más bajo que se encuentra en la columna J.
- Cree una tercera página etiquetada como "combinada" y añada todos los loci con diez o más lecturas de las páginas "menos-Bottom" y "Plus-Top".
- Ordene todas las columnas según el número de lecturas más alto y más bajo que se encuentra en la columna J.
- Cargue los siguientes archivos en IGV25 (paquetes de software): 1) genoma de referencia de interés para visualizar genes anotados, 2) FL-L1-BLAST_RM. GFF para visualizar la anotación L1, 3) hg_sample_sorted. BAM para visualizar transcripciones asignadas desde muestra de interés, y 4) hg_genomicDNA_sorted. BAM para evaluar la asignabilidad de las regiones genómicas.
- Quite las filas de cobertura y de cruce asociadas a cada archivo de BAM.
- Comprima hg_sample_sorted. BAM y hg_genomicDNA_sorted. BAM para que todas las pistas de IGV quepan en una pantalla.
- Copie sobre hg_sample_sorted_bowtie_tryhard_minus_bottom. txt creado en el paso 4.3.2 y la página de la etiqueta como "menos-abajo."
- Curar manualmente.
- Utilizando las coordenadas de loci enumeradas en la hoja de cálculo "combinado" página, vista llamada loci en IGV25 (paquetes de software).
- Curar un locus para ser auténticamente expresado de su propia si no hay lecturas aguas arriba en la dirección L1 hasta 5 KB.
- Etiquete la fila en color verde y observe por qué es una L1 auténticamente expresada.
Nota: existe una excepción a esta regla si la región de arriba de la L1 no es asignable. Si este es el caso, etiquete la fila en color rojo y observe que la expresión de la región que precede al promotor L1 no se puede evaluar y, por lo tanto, la expresión L1's no es capaz de determinarse con confianza.
- Etiquete la fila en color verde y observe por qué es una L1 auténticamente expresada.
- Cura un locus para que no se exprese auténticamente de su propio promotor si hay lecturas ascendentes de hasta 5 KB.
- Etiquete la fila en color rojo y observe por qué no es una L1 auténticamente expresada.
- Curar un locus como falso si se expresa dentro de un intrón de un gen expresado en la misma dirección con lecturas aguas arriba de la L1, si está aguas abajo de un gen expresado en la misma dirección con lecturas aguas arriba de la L1, o para patrones de expresión no anotadas con re anuncios en sentido ascendente de la L1.
Nota: una excepción a esta regla se aplica cuando hay lecturas mínimas superpuestas directamente al sitio de inicio del promotor L1, pero ligeramente aguas arriba de la L1. Si no hay otras lecturas en sentido ascendente de un caso L1 como este, considere que esta L1 se exprese auténticamente. Etiquete la fila de color verde y observe por qué es una L1 auténticamente expresada.
- Curar un locus L1 como probable que sea falso si el patrón de las lecturas asignadas al locus no se correlacionan con las regiones L1's específicas de la asignabilidad.
Nota: por ejemplo, si una L1 es altamente asignable pero sólo tiene una pila de lecturas en una región condensada dentro de la L1, es menos probable que esté relacionada con la expresión L1 de su propio promotor y más probable que sea de fuentes no anotadas como exones o LTRs. En casos como este, se curan los loci como naranja y se nota por qué el locus es sospechoso. Verifique las fuentes de los apilamientos sospechosos comprobando la ubicación L1 en el navegador del genoma UCSC. - Cura un locus para que no se exprese auténticamente si se encuentra dentro de un entorno genómico de regiones sin anotar esporádicamente expresadas
Nota: por ejemplo, las lecturas se pueden expresar 10 KB en sentido ascendente de la L1, pero cada 10 KB o así hay lecturas asignadas y algunas de esas lecturas se alinean con la L1. Estos L1s tienen menos probabilidades de ser expresados en su propio promotor, y es más probable que hayan mapeado lecturas debido a patrones no anotados de expresión genómica. En casos como este, se curan los loci como naranja y se nota por qué el locus es sospechoso.
6. Lea la estrategia de alineación para evaluar la asignabilidad en el genoma de referencia (opcional si uno tiene un DataSet de ADN genómico alineado existente)
- Descargue archivos de secuencias de ADN del genoma completo y conviértalo a archivos. FQ
- Vaya al sitio web de NCBI que se encuentra aquí: https://www.ncbi.nlm.nih.gov/sra
- Escriba en WGS HeLa final emparejado.
- Seleccionar para Homo sapiens bajo resultados por taxón.
- Seleccione un ejemplo que esté emparejado final y tenga lecturas con 100 o más BP como el ejemplo siguiente: https://www.ncbi.nlm.nih.gov/sra/ERX457838 [accn]
- Confirme la longitud de lectura seleccionando Ejecutar y luego metadatos como se muestra aquí: https://Trace.ncbi.nlm.nih.gov/Traces/Sra/?Run=ERR492384
- Para descargar los datos enteros de la secuencia de ADN del genoma, ingrese este comando en el terminal de Linux: sratoolkit. 2.9.2-mac64/bin/Prefetch-X 100G ERR492384
Nota: la función de captación previa de la SRA Toolkit descarga el número de accesión "ERR492384" que se encuentra en el sitio NCBI (paquetes de software). El "100G" limita la cantidad de datos descargados a 100 gigabytes. - Ingrese este comando en el terminal de Linux: fastq-dump--Split-files ERR492384
Nota: esto divide el DataSet de ADN genómico descargado en dos archivos fastq.
- Ejecuta la alineación usando Bowtie.
- Utilice este comando en Linux para la alineación: bowtie-p 10-m 1-S-y-v 3-X 600--chunkmbs 8184 hg_X_Y_M_index-1 hg_genomicDNA_1. FQ-2 hg_genomicDNA_2. FQ | samtools vista-hbuS-| samtools Sort – hg_genomicDNA_sorted. BAM.
- Consulte el paso 4,1 para conocer los parámetros utilizados en la alineación de bowtie (paquetes de software).
- Descargue el archivo BAM alineado genómicamente para evaluar la asignabilidad disponible tras la solicitud del autor.
- Utilice este comando en Linux para la alineación: bowtie-p 10-m 1-S-y-v 3-X 600--chunkmbs 8184 hg_X_Y_M_index-1 hg_genomicDNA_1. FQ-2 hg_genomicDNA_2. FQ | samtools vista-hbuS-| samtools Sort – hg_genomicDNA_sorted. BAM.
- Archivo de BAM de índice del paso 4.2.1 utilizando samtools para que sea visible en IGV25 (paquetes de software) para informar aún más de la curación manual.
- Utilice esta línea de comandos en Linux: Índice samtools hg_genomicDNA_sorted. BAM
- Evalúe la capacidad de asignabilidad de cada loci L1
- Determine el número de lecturas asignadas de forma exclusiva a loci L1 mediante el programa bedtools, la anotación FL-L1 y los datos de secuencia genómica alineados (paquetes de software).
- Utilice esta línea de comandos en Linux: cobertura bedtools-abam FL-L1-BLAST_RM. GTF – b hg_genomicDNA_sorted. bam ≫ L1_Mappability_hg_genomicDNA. txt.
- Designe un locus L1 para que tenga una capacidad de asignabilidad completa cuando se alinean 400 lecturas únicas.
- Determine el factor necesario para escalar hacia arriba o hacia abajo las lecturas de ADN genómica alineadas a 400 para cada L1 individual.
- Para tener una medida de expresión a escala según la capacidad de asignabilidad del locus L1 individual, multiplique el factor determinado en el paso 6.4.3 al número de lecturas de transcripción de ARN que se alinean con L1s expresadas auténticamente en las secciones 4 – 5.
- Determine el número de lecturas asignadas de forma exclusiva a loci L1 mediante el programa bedtools, la anotación FL-L1 y los datos de secuencia genómica alineados (paquetes de software).
Resultados
Los pasos descritos anteriormente y descritos gráficamente en la figura 1 se aplicaron a una línea celular del tumor de próstata humano DU145. La muestra de ARN se preparó de forma citoplásmicamente y fue secuenciada de próxima generación en un protocolo poli-A seleccionado, específico de la hebra, del extremo emparejado. Con bowtie, los archivos de secuenciación de extremo emparejado se alinearon permitiendo solo coincidencias únicas en las que la lectura del extremo emparejado coincido mejor con una ubicación genómica en comparación con cualquier otra ubicación genómica. Los archivos de secuencia DU145 se alinearon con el genoma de referencia humano creando un archivo BAM, que está disponible a petición del autor. Utilizando bedtools, los datos se extrajeron de los archivos BAM separados por hebras DU145 en el número de lecturas que se asignaron a L1s de longitud completa. Esas lecturas se clasificaron en una hoja de cálculo de mayor a menor y se curaron manualmente examinando el entorno genómico alrededor de cada locus L1 en IGV para confirmar su autenticidad (tabla 1 suplementaria). Si se seleccionó una muestra para que se expresaba auténticamente, se codificó en color verde con una explicación para su aceptación en la columna de la derecha. En la figura 2a-bse muestran ejemplos de loci L1 aceptados para ser expresados auténticamente siguiendo las pautas descritas en la sección de métodos. Si se rechazó una muestra para que se expresara auténticamente, se codificó en color como rojo con el motivo del rechazo en la columna de la derecha. Los ejemplos de loci L1 rechazados debido a la expresión de un promotor que no sean sus propias pautas descritas en la sección de métodos se detallan en la figura 2c-e.
Aquí, solo se estudiaron L1s de longitud completa con una región promotora intacta. Si no se hace esta distinción, se introduce una gran fuente de ruido transcripcional procedente de la L1s truncada. Los ejemplos de L1s truncados en DU145 se muestran en la figura 3a-b , donde se identificaron como que tienen lecturas de ARN-SEQ asignadas de forma única. En IGV, sin embargo, es evidente que esas transcripciones no se iniciaron a partir de la L1 truncada, sino de la inclusión de la secuencia L1 en un gen o aguas abajo de un gen expresado.
En general, en DU145, el porcentaje de loci L1 de longitud completa y lecturas que se rechazan como L1s expresa auténticamente después de la curación manual es aproximadamente 50% (tabla complementaria 2) que demuestra el alto nivel de las lecturas de transcripción asignadas L1 que de lo contrario se registrará como falsos positivos sin la curación manual. Concretamente, en DU145 había 114 loci totales de L1 de longitud total para que las lecturas estuvieran asignadas de forma única en la dirección del sentido con un total de 3.152 lecturas, pero sólo había 60 loci identificados para ser expresados en su propio promotor después de la curación manual con 1.879 lecturas ( Tabla complementaria 1). Este es el caso incluso cuando se tomaron medidas para reducir la expresión irrelevante para la biología L1 mediante la selección de mRNA citoplasmática. Tenga en cuenta que el locus con el nivel más alto de transcripciones asignadas en DU145 fue rechazado porque no era un L1 auténticamente expresado (figura 4). En general, el número de transcripciones asignadas a rangos de loci L1 específicos se extiende de manera similar entre los loci L1 aceptados y rechazados como se expresan auténticamente después de la curación manual (figura 4).
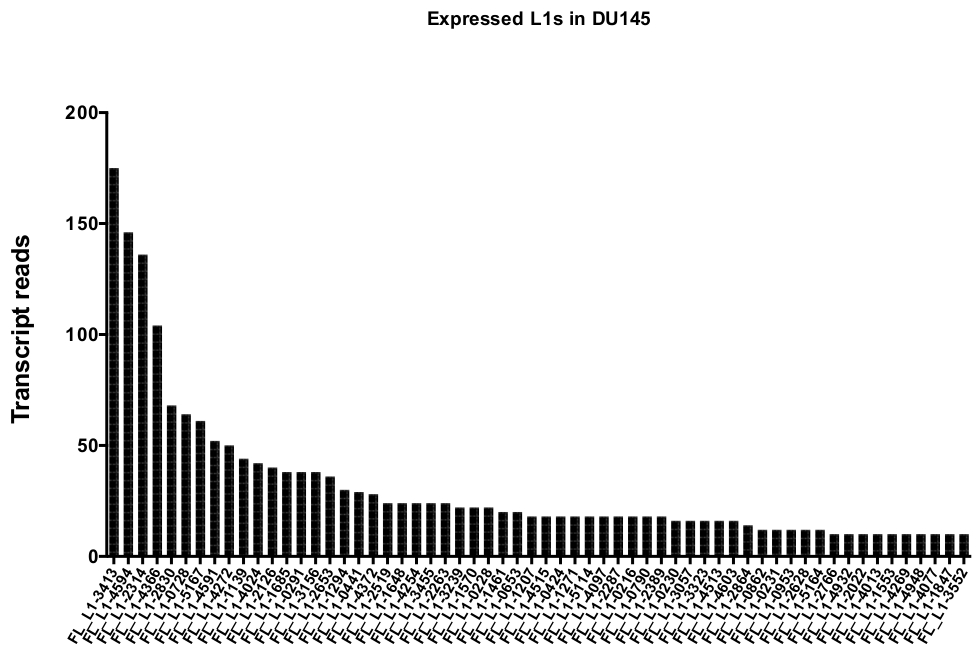
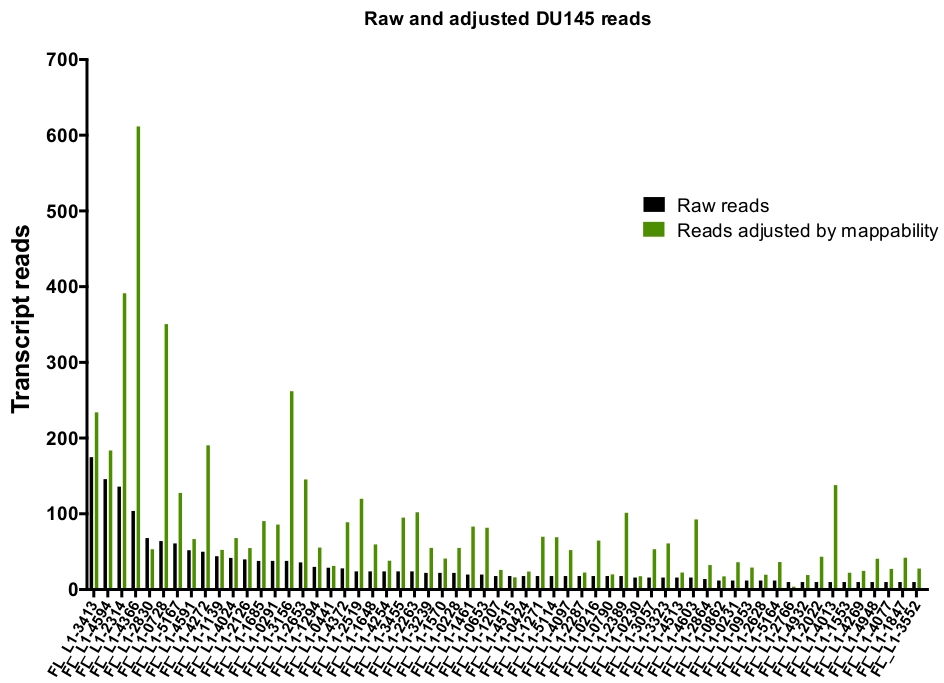
Después de la curación manual, el número de lecturas que se asignan únicamente a loci L1 específicos expresados auténticamente en DU145 oscilan entre 175 lecturas y un recorte mínimo elegido arbitrariamente de 10 lecturas (figura 5). Este enfoque de identificar las lecturas de transcripción asignadas de forma exclusiva a L1s limita la capacidad de cuantificar con precisión la expresión. Para tener en cuenta esto, se creó un factor de corrección para cada locus basado en su capacidad de asignabilidad. Para crear este factor de corrección, se utilizaron las primeras herramientas para extraer el número de lecturas asignadas de forma exclusiva del archivo HeLa genomic de BAM que se alinearon con todos los loci de L1 de longitud completa y grafiaron esos loci de lecturas de transcripción asignadas de mayor a menor correlación (suplementario Figura 1). Se designó arbitrariamente que L1s con 400 lecturas tenían una capacidad de asignabilidad completa. El número de lecturas que se pueden asignar a un locus L1 en la muestra de secuenciación genómica HeLa se ha escalado en relación con 400 lecturas y ese número escalado se multiplicó entonces por el número de lecturas que se asignaban a cada loci L1 auténticamente expresado en DU145 (tabla complementaria 2) . Como era de esperar, los elementos L1 que tenían puntuaciones de corrección más grandes para la asignabilidad provenían de subfamilias más jóvenes como L1PA2 (tabla complementaria 2). Una vez que se ajustaron las lecturas para las puntuaciones de la asignabilidad en cada locus, aumentó la cuantificación de la expresión para la mayoría de los loci (figura 6). El número de lecturas que se asignaron de forma exclusiva a los loci L1 específicos auténticamente expresados con correcciones de mappability en DU145 varió de 612 a 4 lecturas y hubo una reorganización de loci de mayor a menor expresión (figura 6).

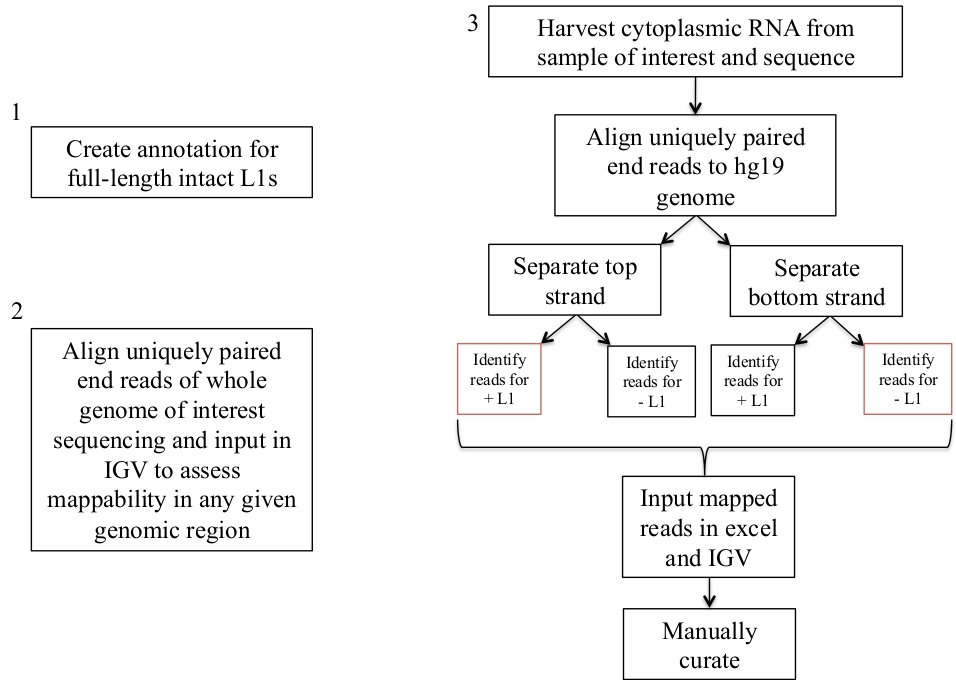
Figura 1: esquema de flujo de trabajo.
Se describen gráficamente los pasos para identificar el L1s expresado en una muestra humana. Tenga en cuenta que los pasos 1 y 2 no necesitan repetirse si los archivos apropiados ya están disponibles. Estos archivos apropiados se pueden descargar de suplemento archivo 1A-b y el suplemento de archivo 2. Las casillas en rojo indican los pasos donde el programa de cobertura de bedtools se utiliza para contar el número de lecturas de mapeo a L1s en la misma dirección de sentido. Estos loci con mapas orientados al sentido son los L1s que deben ser seleccionados manualmente. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

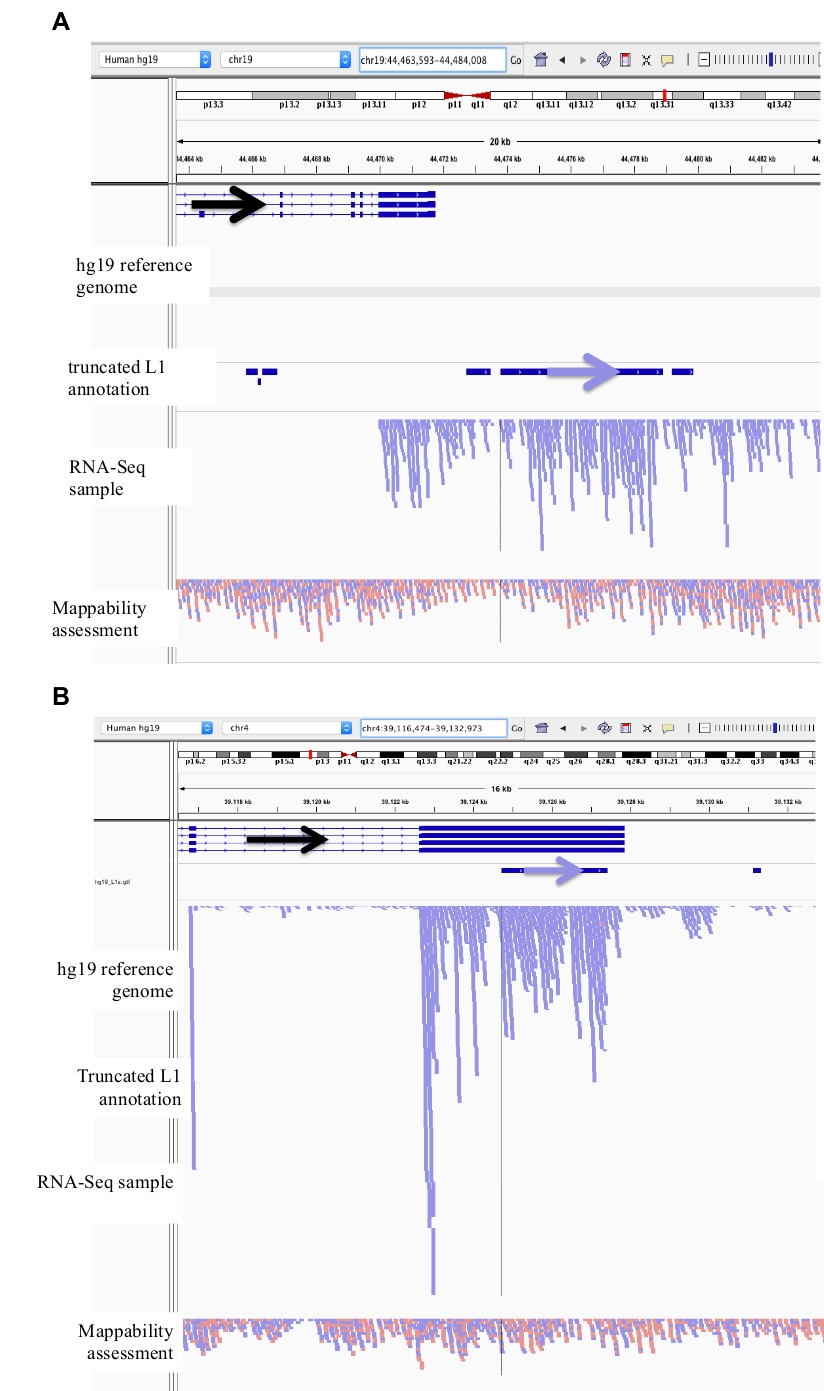
Figura 2: ejemplos de loci L1 comisariadas en DU145.
Cargados en IGV son el genoma de referencia, el archivo de anotación L1 GFF de longitud completa que coincide con la versión del genoma de referencia (suplemento de archivo 1), el archivo BAM DU145 y, por último, el archivo de BAM de Hela para evaluar la asignabilidad, que están disponibles sobre el autor pedido. Se han añadido flechas para ayudar en la visualización de la dirección de la L1 anotada. Las flechas y las lecturas en rojo están orientadas en secuencia de derecha a izquierda. Las flechas y las lecturas en azul están orientadas en secuencia de izquierda a derecha. a) en IGV, este locus L1 parece estar expresado en su propio promotor, ya que no hay lecturas aguas arriba de la L1 en la orientación del sentido para más de 5 KB. Este L1 tiene baja capacidad de asignabilidad, no está en un gen, y tiene evidencia de la actividad de promotor de antisentido esperada26. b) en IGV, este locus L1 parece estar expresado en su propio promotor, ya que no hay lecturas aguas arriba de la L1 en la orientación del sentido para más de 5 KB. Esta L1 tiene una baja capacidad de asignabilidad y está dentro de un gen de dirección opuesta. c) en IGV, este locus L1 fue rechazado como un L1 expresado ya que hay lecturas ascendentes en la misma orientación dentro de 5 KB. Esta L1 está dentro de un gen de la misma dirección por lo que las lecturas de transcripción son más probables originadas por el promotor del gen expresado. d) en IGV, este locus L1 fue rechazado como un L1 expresado ya que hay lecturas ascendentes en la misma orientación dentro de 5 KB. Esta L1 está aguas abajo de un gen altamente expresado en la misma dirección por lo que las lecturas de transcripción son más probables originadas por el promotor de ese gen expresado y extendiéndose más allá del terminador genético normal. e) en IGV, este locus L1 fue rechazado como un L1 expresado ya que hay lecturas ascendentes en la misma orientación dentro de 5 KB. Esta L1 no está dentro ni cerca de un gen anotado en el gen de referencia, por lo que el origen de estas transcripciones dentro y arriba del elemento L1 sugiere un promotor no anotado. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: el ruido de fondo se origina de L1s truncados también.
Nuestra anotación L1 no incluye L1s truncados, ya que son una fuente importante de ruido de fondo. Se han añadido flechas para ayudar en la visualización de la dirección de la L1 anotada. Las flechas y las lecturas en azul están orientadas en secuencia de izquierda a derecha. a) demostrado es un ejemplo de una L1 truncada en la SUFAMILIA L1MB5 que es 2706 bps. En la IGV es evidente que las lecturas proceden de la extensión descendente de un gen expresado. b) se muestra otro ejemplo de una L1 truncada. Este L1 es un L1PA11 que es 4767 bps de largo. En IGV es evidente que el mapeo de lecturas de forma exclusiva a la L1 se origina en el exón expresado, que el L1 está dentro. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: la transcripción lee ese mapa de forma exclusiva para todos los L1s intactos de longitud completa en el genoma humano expresado en DU145 línea celular del tumor de próstata.
En negro son los loci específicos que se identifican como auténticamente expresados después de la curación manual y en rojo son los loci específicos que se rechazarán como lecturas auténticamente expresadas después de la curación manual. En gris son loci con menos de diez lecturas de mapeo a cada uno. Como estos loci representan una pequeña fracción de las lecturas de transcripción, no fueron curadas manualmente. Las marcas de graduación del eje x denotan cada 100 de longitud completa, intacto L1s. aproximadamente 4.500 loci no se muestran gráficamente ya que tenían cero lecturas asignadas. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: la transcripción lee ese mapa de forma exclusiva para expresar auténticamente la longitud completa L1s intacta en DU145 línea celular del tumor prostático.
Se muestra el número de lecturas de transcripción que se asignan a loci específicos en DU145 celdas después de la curación manual. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 6: Lee la asignación a L1 auténticamente expresada cuando se ajusta por asignabilidad.
Se muestra el número de lecturas de transcripción ajustadas por puntuaciones de asignabilidad específicas de loci que se asignan a loci L1 seleccionados manualmente en celdas DU145. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Archivo suplementario 1: anotaciones para la L1s humana intacta de longitud completa según la orientación. a) FL-L1-BLAST_RM_minus. GFF. b) FL-L1-BLAST_RM_plus. GFF. Por favor haga clic aquí para descargar este archivo.
Archivo suplementario 2: guiones Superinformáticos utilizados para automatizar la canalización bioinformática detallada en la sección 4. Por favor haga clic aquí para descargar este archivo.
Figura complementaria 1: muestra de ADN genómica utilizada para determinar la asignabilidad L1.
Se muestra el número de lecturas de transcripción genómica de la muestra de línea de células HeLa que se asignan exclusivamente a todos los 5.000 loci L1 de longitud completa en el genoma. Se designó que una L1 tiene una capacidad de asignación de cobertura completa cuando 400 lee el mapa a la L1. Por favor haga clic aquí para descargar esta figura.
Tabla complementaria 1: curación manual de L1s en DU145. Por favor haga clic aquí para descargar esta tabla.
Tabla complementaria 2: comisariada L1s en DU145 con ajuste de asignabilidad. Por favor haga clic aquí para descargar esta tabla.
Discusión
Se ha demostrado que la actividad L1 causa daño genético e inestabilidad que contribuyen a la enfermedad27,28,29. De las aproximadamente 5.000 copias completas de L1, sólo unas cuantas docenas de jóvenes evolutivamente L1s representan la mayoría de la actividad de retrotransposición2. Sin embargo, hay evidencia de que incluso algunos más antiguos, retrotransposicionalmente-incompentent L1s todavía son capaces de producir ADN perjudicial proteínas30. Para apreciar plenamente el papel de la L1s en la inestabilidad genómica y la enfermedad, se debe entender la expresión L1 en el nivel específico del locus. Sin embargo, el alto fondo de las secuencias relacionadas con L1 incorporadas en otros RNAs no relacionados con la retrotransposición L1 plantea un desafío significativo en la interpretación de la expresión L1 auténtica. Otro desafío en la identificación y por lo tanto la comprensión de los patrones de expresión de los loci L1 individuales se produce debido a su naturaleza repetitiva que no permite que muchas secuencias de lectura cortas se mapear a un único locus único. Para superar estos desafíos, desarrollamos el enfoque descrito anteriormente para identificar la expresión de loci L1 individuales utilizando datos de ARN-seq.
Nuestro enfoque filtra el nivel alto (más del 99%) de ruido transcripcional generado a partir de secuencias L1 que no están relacionadas con la retrotransposición L1 mediante la toma de una serie de pasos. El primer paso consiste en la preparación del ARN citoplásmico. Al seleccionar el ARN citoplásmico, las lecturas relacionadas con L1 encontradas dentro del mRNA intrónico expresado en el núcleo se agotan significativamente. En la preparación de la biblioteca de secuenciación, otro paso tomado para reducir el ruido transcripcional no relacionado con el L1s incluyen la selección de transcripciones poliadeniladas. Esto elimina el ruido de transcripción relacionado con L1 que se encuentra en especies que no son mRNA. Otro paso incluye la secuenciación específica de la hebra para identificar y eliminar las transcripciones relacionadas con el antisentido L1. El uso de una anotación para L1s de longitud completa con regiones promotoras funcionales al identificar el número de transcripciones de ARN-SEQ que se asignan a L1s también elimina el ruido de fondo que de otro modo se originan a partir de L1s truncados. Por último, el último paso crítico para eliminar el ruido transcripcional de las secuencias L1 no relacionadas con la retrotransposición de L1 es la curación manual de la longitud total de L1s identificada para haber mapeado transcripciones de ARN-seq. La curación manual implica la visualización de cada locus L1 identificado de manera bioinformáticamente en el contexto de su entorno genómico circundante para confirmar que la expresión proviene del promotor L1. Este enfoque se aplicó a DU145, una línea celular de tumor de próstata. Incluso con todas las medidas relacionadas con la preparación adoptadas para reducir el ruido de fondo, aproximadamente el 50% de los loci L1 identificados de manera bioinformáticamente en DU145 fueron rechazados como ruido de fondo L1 procedente de otras fuentes transcripcionales (figura 4), enfatizando el rigor necesario para producir resultados fiables. Este enfoque mediante la curación manual es laborioso, pero necesario en el desarrollo de esta tubería para evaluar y comprender el entorno genómico que rodea a un L1 de longitud completa. Los siguientes pasos incluyen la reducción de la cantidad de curación manual necesaria automatizando algunas de las reglas de curación, aunque debido a la naturaleza todavía no completamente conocida de la expresión genómica, las fuentes de expresión no anotadas en el genoma de referencia, las regiones de baja asignabilidad, e incluso factores que complican la construcción de un genoma de referencia, no es posible automatizar completamente la curación L1 en este momento.
El segundo desafío en la identificación de la expresión de loci L1 individuales con la secuenciación se relaciona con el mapeo de transcripciones L1 repetitivas. En esta estrategia de alineación, se requiere que una transcripción deba alinearse única y colinealmente con el genoma de referencia para que se asigne. Al seleccionar las secuencias de final emparejado que se asignan concordantemente, aumenta la cantidad de transcripciones que se alinean de forma única a los loci L1 que se encuentran en el genoma de referencia. Esta estrategia de mapeo único proporciona confianza en la llamada de lecturas que se mapear específicamente a un solo locus L1, aunque potencialmente subestima la cantidad de expresión de cada L1, repetitivo, expresado, a ser auténticamente, repetido. Para corregir aproximadamente esta infravaloración, se desarrolló una puntuación de "asignabilidad" para cada locus L1 basado en su capacidad de asignabilidad y se aplicó al número de lecturas de transcripción asignadas de forma exclusiva (figura 6). Es de notar que idealmente, la asignabilidad se debe anotar a las lecturas de la cobertura completa a través del L1 de longitud completa según la muestra emparejada WGS. Aquí, utilizamos WGS de células HeLa para determinar las puntuaciones de la asignación de cada loci L1 con el fin de inflar o DEFLATE lecturas de mapeo para L1 loci en DU145 líneas celulares de tumor de próstata. Este cálculo de la asignabilidad es una puntuación de corrección bruta, pero la "asignabilidad completa de la cobertura" de 400 lecturas se determinó con la naturaleza dinámica de las líneas celulares tumorales en mente. Puede observarse en la figura 1 suplementaria, que hay algunos loci L1 con Hela WGS con un número extremadamente alto de lecturas asignadas. Estos probablemente provienen de secuencias cromosómicas duplicadas dentro de HeLa que no están dentro del genoma de referencia, razón por la cual esos loci no fueron elegidos para ser representativos de la cobertura completa de la asignabilidad. En su lugar, se determinó que el promedio de 100% de cobertura de lectura se produce alrededor de 400 lecturas de acuerdo con la figura 1 suplementaria y luego se asumió que este promedio se aplica a la línea de la célula de próstata del tumor DU145, así.
Esta estrategia de alineación con 100-200 BP Lee de la tecnología RNA-SEQ también selecciona preferentemente para los L1s evolutivamente más antiguos dentro del genoma de referencia como mayores L1s han acumulado con el tiempo mutaciones únicas que los hacen más asignables. Este enfoque, por lo tanto, tiene una sensibilidad limitada a la hora de identificar al más joven de L1s, así como de no referencia, polimórfico L1s. Para identificar al más joven de L1s, sugerimos usar la selección 5 ' RACE de transcripciones L1 y tecnología de secuenciación como PacBio que hacen uso de lecturas más largas21. Esto permite un mapeo más singular y, por lo tanto, una identificación segura de los L1s expresados, jóvenes. el uso de ARN-SEQ y PacBio enfoques juntos puede conducir a una lista más completa de L1s expresa auténticamente. Para identificar el polimórfico L1s auténticamente expresado, los primeros pasos siguientes incluyen la construcción e inserción de secuencias polimórficas en el genoma de referencia.
Los desafíos biológicos y técnicos en el estudio de las secuencias repetidas son grandes, aunque con el procedimiento riguroso anterior para eliminar el ruido transcripcional de las secuencias L1 no relacionadas con la retrotransposición utilizando la tecnología de secuenciación de ARN, comenzamos a tamizar a través los grandes niveles de ruido de fondo transcripcional y la identificación segura y rigurosa de los patrones de expresión L1 y la cantidad a nivel de locus individual.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Nos gustaría agradecer al Dr. Yan dong por las células del tumor de próstata DU145. Nos gustaría agradecer al Dr. Nathan Ungerleider por su guía y Consejo en la creación de guiones de supercomputadoras. Parte de este trabajo fue financiado por NIH Grants r01 GM121812 a PD, r01 AG057597 a VPB, y 5TL1TR001418 a TK. También nos gustaría reconocer el apoyo de los cruzados del cáncer y el núcleo Bioinformático del centro oncológico Tulane.
Materiales
| Name | Company | Catalog Number | Comments |
| 1 M HEPES | Affymetrix | AAJ16924AE | |
| 5 M NaCl | Invitrogen | AM9760G | |
| Agilent bioanalyzer 2100 | Agilent technologies | ||
| Agilent RNA 6000 Nano Kit | Agilent technologies | 5067-1511 | |
| bedtools.26.0 | https://bedtools.readthedocs.io/en/latest/content/installation.html | ||
| bowtie-0.12.8 | https://sourceforge.net/projects/bowtie-bio/files/bowtie/0.12.8/ | ||
| Cell scraper | Olympus plastics | 25-270 | |
| Chloroform | Fisher | C298-500 | |
| Digitonin | Research Products International Corp | 50-488-644 | |
| Ethanol | Fisher | A4094 | |
| Gibco (Phosphate Buffered Saline) | Invitrogen | 10-010-049 | |
| Homogenizer | Thomas Scientific | BBI-8541906 | |
| IGV 2.4 | https://software.broadinstitute.org/software/igv/download | ||
| Isopropanol | Fisher | A416-500 | |
| mac2unix | https://sourceforge.net/projects/cs-cmdtools/files/mac2unix/ | ||
| Q-tips | Fisher | 23-400-122 | |
| RNAse later solution | Invitrogen | AM7022 | |
| RNaseZap RNase Decontamination Solution | Invitrogen | AM9780 | |
| samtools-1.3 | https://sourceforge.net/projects/samtools/files/ | ||
| sratoolkit.2.9.2 | https://github.com/ncbi/sra-tools/wiki/Downloads | ||
| SUPERase·In RNase Inhibitor | Invitrogen | AM2694 | |
| Trizol | Invitrogen | 15-596-018 | |
| Water (DNASE, RNASE free) | Fisher | BP2484100 |
Referencias
- International Human Genome Sequencing. Initial sequencing and analysis of the human genome. Nature. 409, 860 (2001).
- Brouha, B., et al. Hot L1s account for the bulk of retrotransposition in the human population. Proceedings of the National Academy of Sciences of the United States of America. 100 (9), 5280-5285 (2003).
- Dombroski, B. A., Mathias, S. L., Nanthakumar, E., Scott, A. F., Kazazian, H. H. Isolation of an active human transposable element. Science. 254 (5039), 1805 (1991).
- Swergold, G. D. Identification, characterization, and cell specificity of a human LINE-1 promoter. Molecular and Cellular Biology. 10 (12), 6718-6729 (1990).
- Speek, M. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Molecular and Cellular Biology. 21 (6), 1973-1985 (2001).
- Deininger, L., Batzer, M. A., Hutchison, C. A., Edgell, M. H. Master genes in mammalian repetitive DNA amplification. Trends in Genetics. 8 (9), 307-311 (1992).
- Boissinot, S., Chevret, P., Furano, A. L1 (LINE-1) Retrotransposon Evolution and Amplification in Recent Human History. Molecular Biology and Evolution. 17 (6), 915-918 (2000).
- Khazina, E., Weichenrieder, O. Non-LTR retrotransposons encode noncanonical RRM domains in their first open reading frame. Proceedings of the National Academy of Sciences of the United States of America. 106 (3), 731-736 (2009).
- Martin, S. L., Bushman, F. D. Nucleic acid chaperone activity of the ORF1 protein from the mouse LINE-1 retrotransposon. Molecular and Cellular Biology. 21 (2), 467-475 (2001).
- Feng, Q., Moran, M. H., Kazazian, H. H., Boeke, J. D. Human L1 Retrotransposon Encodes a Conserved Endonuclease Required for Retrotransposition. Cell. 87 (5), 905-916 (1996).
- Mathias, S. L., Scott, A. F., Kazazian, H. H., Boeke, J. D., Gabriel, A. Reverse transcriptase encoded by a human transposable element. Science. 254 (5039), 1808 (1991).
- Luan, D. D., Korman, M. H., Jakubczak, J. L., Eickbush, T. H. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: A mechanism for non-LTR retrotransposition. Cell. 72 (4), 595-605 (1993).
- van den Hurk, J. A. J. M., et al. Novel types of mutation in the choroideremia (CHM) gene: a full-length L1 insertion and an intronic mutation activating a cryptic exon. Human Genetics. 113 (3), 268-275 (2003).
- Miné, M., et al. A large genomic deletion in the PDHX gene caused by the retrotranspositional insertion of a full-length LINE-1 element. Human Mutation. 28 (2), 137-142 (2007).
- Solyom, S., et al. Pathogenic orphan transduction created by a nonreference LINE-1 retrotransposon. Human Mutation. 33 (2), 369-371 (2012).
- Hancks, D. C., Kazazian, H. H. Roles for retrotransposon insertions in human disease. Mobile DNA. Mobile DNA. 7, 9-9 (2016).
- Tubio, J. M. C., et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 345 (6196), 1251343-1251343 (2014).
- Ewing, A. D., et al. Widespread somatic L1 retrotransposition occurs early during gastrointestinal cancer evolution. Genome Research. 25 (10), 1536-1545 (2015).
- Beck, C. R., Garcia-Perez, J. L., Badge, R. M., Moran, J. V. LINE-1 elements in structural variation and disease. Annual Review of Genomics and Human Genetics. 12, 187-215 (2011).
- Philippe, C., et al. Activation of individual L1 retrotransposon instances is restricted to cell-type dependent permissive loci. eLife. 5, e13926 (2016).
- Deininger, P., et al. A comprehensive approach to expression of L1 loci. Nucleic Acids Research. 45 (5), e31-e31 (2017).
- Jin, Y., Tam, O. H., Paniagua, E., Hammell, M. TEtranscripts: a package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics. 31 (22), 3593-3599 (2015).
- . . Agilent RNA 6000 Nano Kit Guide. , (2017).
- Mueller, O. L., Schroeder, A. . RNA Integrity Number (RIN) –Standardization of RNA Quality Control. , (2016).
- Robinson, J. T., et al. Integrative genomics viewer. Nature Biotechnology. 29, 24 (2011).
- Speek, M. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Molecular Cellular Biology. 21 (6), 1973-1985 (2001).
- Belancio, V. P., Deininger, L., Roy-Engel, A. M. LINE dancing in the human genome: transposable elements and disease. Genome Medicine. 1 (10), 97-97 (2009).
- Iskow, R. C., et al. Natural Mutagenesis of Human Genomes by Endogenous Retrotransposons. Cell. 141 (7), 1253-1261 (2010).
- Scott, E. C., et al. A hot L1 retrotransposon evades somatic repression and initiates human colorectal cancer. Genome Research. 26 (6), 745-755 (2016).
- Kines, K. J., Sokolowski, M., deHaro, D. L., Christian, C. M., Belancio, V. P. Potential for genomic instability associated with retrotranspositionally-incompetent L1 loci. Nucleic Acids Research. 42 (16), 10488-10502 (2014).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados