
Method Article
Flujo de trabajo de espectrometría de masas de inmunoprecipitación sin etiquetas para la generación de perfiles de interactome nuclear a gran escala
En este artículo
Resumen
Descrito es un flujo de trabajo proteómico para identificar socios de interacción de proteínas a partir de una fracción subcelular nuclear utilizando el enriquecimiento de inmunoafinidad de una proteína dada de interés y espectrometría de masas sin etiquetas. El flujo de trabajo incluye fraccionamiento subcelular, inmunoprecipitación, preparación de muestras asistidas por filtro, limpieza fuera de línea, espectrometría de masas y una canalización bioinformática aguas abajo.
Resumen
La espectrometría de masas de purificación de inmunoafinidad (IP-MS) ha surgido como un método cuantitativo robusto para identificar interacciones proteína-proteína. Esta publicación presenta un flujo de trabajo proteómico de interacción completo diseñado para identificar interacciones proteína-proteína de baja abundancia desde el núcleo que también podrían aplicarse a otros compartimentos subcelulares. Este flujo de trabajo incluye fraccionamiento subcelular, inmunoprecipitación, preparación de muestras, limpieza fuera de línea, espectrometría de masas sin etiquetas de un solo disparo y análisis computacional descendente y visualización de datos. Nuestro protocolo está optimizado para detectar interacciones compartimentadas y de baja abundancia que son difíciles de identificar a partir de lisados de células enteras (por ejemplo, interacciones con factores de transcripción en el núcleo) por inmunoprecipitación de proteínas endógenas compartimentos subcelulares fraccionados. La tubería de preparación de muestras que se describe aquí proporciona instrucciones detalladas para la preparación del extracto nuclear de células de HeLa, la purificación de inmunoafinidad de proteína de cebo endógeno y el análisis cuantitativo de espectrometría de masas. También discutimos consideraciones metodológicas para realizar inmunoprecipitaciones a gran escala en experimentos de perfilado de interacciones basados en espectrometría de masas y proporcionamos pautas para evaluar la calidad de los datos para distinguir las proteínas positivas verdaderas interacciones de interacciones inespecíficas. Este enfoque se demuestra aquí investigando el interactome nuclear de la quinasa CMGC, DYRK1A, una proteína quinasa de baja abundancia con interacciones mal definidas dentro del núcleo.
Introducción
El proteome humano exhibe una vasta diversidad estructural y bioquímica a través de la formación de complejos multisubunidades estables e interacciones proteicas-proteínas transitorias. En consecuencia, la identificación de los socios de interacción para una proteína de interés es comúnmente necesaria en las investigaciones para desentrañar el mecanismo molecular. Los recientes avances en los protocolos de purificación de afinidad y la llegada de la instrumentación de espectrometría de masas de escaneo rápido de alta resolución han permitido un mapeo fácil de los paisajes de interacción con proteínas en un solo experimento imparcial.
Los protocolos de interacción con proteínas suelen emplear sistemas de expresión ectópicos con construcciones de fusión etiquetadas con afinidad para identificar interacciones proteicas sin necesidad de anticuerpos de alta calidad que reconozcan una proteína de interés1,2. Sin embargo, los métodos basados en etiquetas de epítopos tienen varios inconvenientes. Las interacciones físicas con el epítopo pueden conducir a la detección de proteínas corepeantes no específicas3. Además, la fusión de estas etiquetas de epítopos en el Terminal N o C de una proteína puede bloquear las interacciones proteínas-proteínas nativas, o alterar el plegado de proteínas para promover las conformaciones no fisiológicas4. Además, los sistemas de expresión ectópica suelen sobreexpresar la proteína de cebo a concentraciones suprafisiológicas, lo que puede resultar en la identificación de interacciones de proteínas artifreales, particularmente para genes sensibles a la dosis5. Para eludir estos problemas, la proteína de cebo endógena puede ser inmunopreciada junto con las proteínas de presa que interactúan asociadas, asumiendo la disponibilidad de un anticuerpo de alta calidad que reconoce la proteína nativa.
Aquí se proporciona un flujo de trabajo proteómico de interacción para detectar el interactome nuclear de una proteína endógena utilizando la proteína cCM kinasa DYRK1A como ejemplo. La interrupción del número de copia, el nivel de actividad o la expresión de DYRK1A puede causar una discapacidad intelectual grave en los seres humanos, y la letalidad embrionaria en ratones6,7,8,9. DYRK1A exhibe regulación espaciotemporal dinámica10,e interacciones proteicas compartimentadas11,12, que requieren enfoques capaces de detectar socios de interacción de baja abundancia específicos de diferentes compartimentos subcelulares.
Este protocolo emplea el fraccionamiento celular de células HeLa humanas en fracciones de citosol y nucleoplasmo, inmunoprecipitación, preparación de muestras para espectrometría de masas y una visión general de una tubería bioinformática para evaluar la calidad de los datos y visualizar los resultados, con scripts r proporcionados para el análisis y visualización (Figura 1). Los paquetes de software de Proteomics utilizados en este flujo de trabajo están disponibles gratuitamente para su descarga o se puede acceder a ellos a través de una interfaz web. Para obtener información adicional sobre software y métodos computacionales, los tutoriales en profundidad y la instrucción están disponibles en los enlaces proporcionados.
Protocolo
NOTA: Todas las composiciones tampón y las mezclas de proteasa se describen en el Cuadro 1.
1. Preparación de células
NOTA: Se desea un material de partida de 1 a 10 mg de lisato nuclear por réplica para este enfoque de espectrometría de masas de inmunoprecipitación (IP-MS). Se administrarán cantidades celulares para 1 mg de inmunoprecipitaciones nucleares en triplicado más controles triplicados.
- Si utiliza una línea celular adherente, crezca las células a 90% de confluencia en platos de 3 x 15 cm por réplica antes de la cosecha.
NOTA: Se recomienda realizar un mínimo de tres inmunoprecipitaciones replicadas para las condiciones de cebo y control. Este protocolo asumirá el uso de controles de 'solo perlas', que controlan para abundantes interacciones inespecíficas con las cuentas, comenzando en la sección 4. Otros tipos de controles pueden ser útiles. Estos se describen en profundidad en la sección de discusión.- Lavar las placas 2x con solución salina tamponada de fosfato (PBS) y trippsinizar las células usando 3 ml de 0.25% de trippsina por placa de 15 cm. Gire las células a 1.200 x g durante 5 min y decant la trippsina.
- Para las células de suspensión, crecer a una escala / densidad similar para lograr 70 x 80 mg de proteína total.
- Células de pelet a 1.200 x g y 4oC durante 5 min. Decantar el medio con cuidado.
NOTA: Los pellets se pueden combinar durante este paso para permitir un procesamiento eficiente utilizando el fraccionamiento subcelular a gran escala descrito en la sección 2.
- Células de pelet a 1.200 x g y 4oC durante 5 min. Decantar el medio con cuidado.
- Lavar el pellet celular 2x con PBS + 5 mM MgCl2 complementado con inhibidores de la proteasa (PE) e inhibidores de la fosfatasa (PH) (ver Tabla 1).
NOTA: Los gránulos celulares pueden congelarse en nitrógeno líquido y almacenarse a -80 oC hasta que estén listos para el fraccionamiento.
2. Preparación del extracto nuclear
NOTA: Los inhibidores de la proteasa y la fosfatasa deben añadirse a los búferes de fraccionamiento dentro de los 30 minutos de uso.
- Si se congela, descongelar los gránulos celulares durante 15 minutos en 1 volumen de pellets de búfer frío A + PIs / PH. Coloque el gránulo celular sobre un nutator a 4 oC para ayudar a la resuspensión durante la descongelación. De lo contrario, resuspenda el pellet celular del paso 1.3 en 1 volumen de pellets Buffer A + PIs /PhIs.
- Pellet a 2.000 x g y 4oC durante 10 min. Decantar el tampón.
- Suspenda las células con 5 veces el volumen de celda saqueado con el tampón A e incubar en hielo durante 20 minutos.
- Pellet a 2.000 x g y 4oC durante 10 min. Decanta el buffer y resuspende con 2x volumen de celda empaquetado original Buffer A + PIs/PH y sounce 7x con "A"/loose pestle.
- Centrifugar el lisado durante 10 min a 2.000 x g y 4oC.
- Pipetear cuidadosamente el sobrenadante y congelar el flash con nitrógeno líquido. Conservar el lisado a -80oC. El sobrenadante de este paso es la fracción subcelular citosólica.
NOTA: El pellet nuclear se puede guardar durante este paso mediante la congelación del flash con nitrógeno líquido y el almacenamiento a -80 oC - Resuspende el pellet con un volumen de pellets de 0,9x de Tampón B + PI/FI y mezcle en un nutator durante 5 min a 4oC.
- Para cargar los núcleos, sounce 20x con un pestillo más apretado "B".
- Mezclar el lisado nuclear en un nutator durante 30 min a 4 oC para que sea homogéneo.
- Centrifugar el lisado nuclear durante 30 min a 21.000 x g a 4oC. Quita el sobrenadante y guárdalo como extracto de proteína nuclear soluble.
NOTA: El tratamiento de nucleasa del pellet nuclear resultante permite la recuperación de una fracción proteica asociada a la cromatina. - Dializar el extracto nuclear soluble contra Buffer C + PIs durante 3 h a 4 oC.
- Corte una longitud apropiada de tubo de diálisis de 24 mm de ancho con un peso molecular de 8 kDa cortado. Sujete un lado del tubo y cargue el nucleoplasmo en el tubo. Después de cargar el lisado, sujeta el otro extremo y sumérgete en un recipiente de vidrio limpio que contenga Buffer C + PIs.
- Centrifugar el extracto nuclear dializado/nucleoplasmo a 21.000 x g a 4 oC durante 30 min. Aliquot 3x 20 l volúmenes de extracto nuclear para validación de fraccionamiento por mancha occidental. El extracto nuclear utilizado para el análisis ip-MS puede ser alícuota y flash congelado en nitrógeno líquido y almacenado a -80 oC, si es necesario.
3. Validación del fraccionamiento subcelular
- Complete un ensayo proteico para determinar la concentración proteica del lisato nuclear. Un ensayo de proteína de ácido bicincino proporciona suficiente sensibilidad para la aplicación aguas abajo.
- Cargue 20 g de las fracciones citosólicas y nucleares en un gel SDS-PAGE para el análisis de la mancha occidental como se describió anteriormente13. Omita los carriles al cargar para evitar la caracterización incorrecta de una muestra.
- Sondear la mancha occidental para p84 (THOC1) como un marcador nuclear, y GAPDH como un marcador citosólico. Determinar el grado de fraccionamiento por la relación del marcador citosólico en la fracción nuclear y viceversa.
NOTA: Se pueden utilizar anticuerpos para otros marcadores nucleares y citosólicos.
4. Inmunoprecipitación de la proteína de cebo nuclear endógena
NOTA: Se recomienda utilizar tubos de baja retención a partir de este punto. Esto reducirá la unión inespecífica a los tubos durante la manipulación de la muestra y evitará la pérdida innecesaria de la muestra. Además, asegúrese de que el grado H2O de LCMS se utilice para preparar los búferes para los pasos restantes.
- Preparar una mezcla de perlas A/G de proteína para cada réplica combinando 12,5 ml de volumen de perlas tanto para proteínaS A como para proteínas G en tubos de microcentrífuga. Almacene las existencias de cuentas como una suspensión que contenga un 20% de etanol. Determinar la concentración de perlas dentro de la suspensión %(v/v) y pipetear el volumen necesario utilizando una punta de pipeta que se ha cortado en la punta para asegurarse de que las perlas pueden entrar en la punta.
- Lave la mezcla de perlas de proteína A/G 2veces con 300 ml de tampón IP 1. Gire las perlas a 1.500 x g a 4 oC durante 1 min y el búfer de cant.
- Preparar las perlas a los anticuerpos-proteína A/G: Para unir el anticuerpo a las perlas, añadir 300 éL de búfer IP 1 y 10 g del anticuerpo deseado. Deje que la mezcla de perlas/anticuerpos se mece en un nutator a 4 oC durante la noche. Para los controles de cuentas, no agregue ningún anticuerpo.
NOTA: Se puede utilizar un total de 10 g de anticuerpos por réplica como punto de partida, pero la cantidad exacta tendrá que optimizarse para cada anticuerpo individual y la escala del lisado utilizado en el experimento - Descongelar los lysates nucleares del paso 2.10 en un baño de agua y alícuota volúmenes apropiados en tubos de microcentrífuga de baja retención para 1 mg de entrada de proteína por réplica.
- Gire el lisado a 16.000 x g durante 30 minutos y transfiera el sobrenadante a un tubo nuevo.
- Añadir 1 l de benzonasa (250 unidades/L) por 1 mg del lisado nuclear y roca en un nutator a 4 oC durante 10 x 15 min.
- Preparen cuentas para prelimpiar el lisado. Añadir 12,5 ml de cada proteína A y perlas g a tubos de baja retención de 1,5 ml como en el paso 4.1. Lave 2x con IP Wash Buffer 1 + PIs y decant el búfer.
- Añadir 1 mg del lisado nuclear a las cuentas del paso 4.5. Incubar mientras se balancea sobre un nutator durante 1 h a 4oC.
- Centrifugar lisiado preaclarado a 1.500 x g y 4oC durante 1 min.
- Mientras que las lisades nucleares se están incubando con perlas en el paso 4.5.1, lave las perlas a anticuerpos-proteínas A/G 2x con IP Buffer 1 + PIs. Centrifugar a 1.500 x g y 4oC durante 1 min y decantar el tampón.
- Transfiera el lisado nuclear preclearizado del paso 4.6.1 a las cuentas A/G de anticuerpos y proteínas. Incubar mientras se balancea sobre un nutator a 4oC durante 4 h. Centrífuga después de la incubación a 1.500 x g y 4oC durante 1 min.
- Transfiera el sobrenadante en tubos etiquetados como el flujo a través de cada réplica.
- Lave las perlas a los anticuerpos y proteínas A/G con 1 ml de IP Buffer 2 + PIs. Centrifugar a 1.500 x g y 4oC durante 1 min, tampón de canto, y repetir para un total de 3x.
- Lave las perlas 2x con 1 ml de centrifugación IP Buffer 1+ PIs como en el paso anterior. Asegúrese de que todo el búfer se elimina después del último lavado.
- Elute 2x con 20 ml de glicina de 0,1 M (pH 2,75) durante 30 min cada uno. Asegúrese de que los tubos se balancean durante la incubación con el tampón de elución. Girar a 750 x g y 4 oC durante 1 min y quitar la pipeta del sobrenadante después de cada incubación de 30 minutos.
NOTA: Mientras que el método de glicina de bajo pH descrito aquí eluye la mayoría de las proteínas de cebo, algunas interacciones anticuerpo-antígeno requieren condiciones de amortiguación más estrictas. - La congelación del flash eluye en nitrógeno líquido y almacenar a -80 oC.
5. Preparación de muestras
NOTA: La insulina que se introdujo en las muestras de elución de inmunoprecipitación ayuda en la recuperación de proteínas durante la precipitación del ácido tricloroacético (TCA) y el procesamiento de muestras, que es importante para las proteínas de cebo endógeno de baja abundancia.
- Descongelar los eludos a temperatura ambiente si están congelados.
- Colocar las muestras sobre hielo y añadir 10 ml de insulina de 1,0 mg/ml por cada 100 ml de eluido. Vórtice y luego añadir inmediatamente 10 éL de 1% de desoxicolato de sodio. Vortex la muestra de nuevo y añadir 30 s de 20% TCA seguido de un vórtice final.
- Incubar las muestras sobre hielo durante 20 min, luego centrifugar a 21.000 x g a 4oC durante 30 min.
- Aspirar el sobrenadante y añadir 0,5 ml de acetona que ha sido pretalado a -20 oC. Vórtice y luego girar a 21.000 x g y 4 oC durante 30 min. Repita este paso.
- Aspirar el sobrenadante y secar al aire el pellet restante en la parte inferior del tubo.
- Prepare la muestra para espectrometría de masas utilizando un método modificado de preparación de muestras asistidas por filtro (FASP), optimizado para reducir el manejo de la muestra, como se describe a continuación14.
- Resuspenda el pellet proteico del paso 5.1.4 con 30 ml de tampón de alquilación SDS (ver Tabla 1). Incubar la muestra en un bloque de calor de 95 oC durante 5 min. Dejar enfriar a temperatura ambiente durante 15 minutos antes de preceder al siguiente paso.
- Añadir 300 sl de solución de UA y 30 l de 100 mM TCEP a cada muestra. Cargue esta solución en un filtro centrífugo de 30k. Gire el filtro centrífugo a 21.000 x g a temperatura ambiente durante 10 min.
NOTA: La proteína de cebo y sus interactores putativos deben estar unidos al filtro en este punto. Sin embargo, el flujo a través se puede mantener, en caso de que haya un problema con el filtro. - Lavar el filtro con 250 s l de UA y centrifugar a 21.000 x g durante 10 minutos. Decantar el flujo a través y repetir para un total de 3x.
- Lavar el filtro con 100 ml de 100 mM Tris pH 8.5 y centrifugar a 21.000 x g durante 10 min. Decantar el flujo y repetirlo durante un total de 3x.
- Añadir 3 sl de 1 g/l de Lys C resuspendido en 0,1 M Tris pH 8,5. Llenar los filtros hasta la marca de 100 l y dejar digerir durante 1 h a 37 oC mientras se balancea sobre un nutator.
- Añadir 1 l de 1 g/L de trippsina de grado MS. Mezcle suavemente y deje que la trippsina se incuba con la muestra durante la noche a 37 oC mientras se balancea sobre un nutator.
- Centrífuga a 21.000 x g durante 20 minutos para eluir el péptido del filtro.
NOTA: Es posible que se necesiten múltiples rondas de centrifugación para recuperar todo eluso. Si esto no se hace, hay un potencial de pérdida grave de la muestra.
- Desalar los péptidos usando columnas de giro C18. Siga el protocolo proporcionado por el fabricante.
- Resuspenda el péptido liofilizado en 7 l de 0,1% TFA en 5% acetonitrilo. Sonicar la muestra durante 3 minutos para asegurarse de que los péptidos han sido resuspendidos. Gire hacia abajo a 14.000 x g durante 10 min.
- Transfiera el péptido resuspendido a un vial de muestra adecuado para cargarlo en el sistema de cromatografía líquida y espectrometría de masas (LC/MS).
6. Adecuabilidad del sistema LC/MS
NOTA: Debido a la pequeña escala y generalmente a la menor abundancia de proteínas de muestras purificadas por afinidad, es fundamental que la plataforma LC/MS funcione con una sensibilidad y robustez máximas.
- Agregue 1 ml de ácido fórmico de grado LC/MS a 1 L de agua de grado LC/MS para la fase móvil A, y agregue 1 ml de ácido fórmico de grado LC/MS a 1 L de acetonitrilo de grado LC/MS para la fase móvil B.
- Preparar o instalar una columna capilar de sílice fundida de 75 m llena de resina C18 de fase invertida de 2 m que tenga una longitud de 250 mm. Los mejores resultados se obtendrán con una inyección directa de muestras en la columna.
- Purgue el sistema ultra performance Liquid Chromatography (UPLC) con nuevas fases móviles. Con una columna C18 instalada, establezca un caudal estable y electrospray con un emisor adecuado (es decir, 20 ém id x 360 m od tirados a una punta de 10 m). Mantenga la columna a 40 o 60 oC.
- Pruebe el rendimiento general del sistema LC/MS inyectando un estándar de control de calidad complejo, como 100 a 200 ng de un digerido tríptico de lisato de células enteras de HeLa. Elute con un gradiente adecuado para una muestra compleja (es decir, tiempo de elución de gradiente de 2 x 3 h). Establecer un rendimiento del sistema de referencia de las identificaciones de péptidos y proteínas.
NOTA: Para obtener mejores resultados, entre 3.000 y 5.000 o más identificaciones de proteínas de 20.000 a 35.000 péptidos únicos proporcionarán un rendimiento óptimo para muestras experimentales. - Para la idoneidad rutinaria del sistema LC/MS, inyecte 100 a 200 fmol o menos de un único estándar de digestión proteica, como la albúmina de suero bovino (BSA). Elute con un gradiente corto (es decir, de 20 a 30 min).
NOTA: Las inyecciones múltiples de un digerido proteico ayudarán a establecer el rendimiento basal del sistema LC/MS, y la inyección repetida después de cada muestra IP-MS proporciona una medida del rendimiento del sistema a lo largo del experimento y permite la detección de la deriva del instrumento, lo que puede sesgar los experimentos sin etiquetas. Una línea base de las intensidades pico individuales seleccionadas y las formas de pico informará sobre el rendimiento de mS, LC y columna. - Para evitar sobrecargar la columna analítica, cargue una pequeña porción (15-30% del total) de una muestra experimental en la columna y separe utilizando un degradado adecuado para muestras complejas (es decir, 2-3 h). Si el número de identificaciones proteicas es insatisfactorio, cargue toda la muestra en la columna.
- Ejecute un único estándar de digerido de proteína entre muestras para supervisar el rendimiento del sistema LC/MS y el arrastre de la muestra. Es posible que se requieran varios estándares para reducir el arrastre de la muestra dependiendo de sus muestras.
7. Procesamiento de datos
- Descargue el paquete de software de proteómica que MaxQuant se encuentra en https://www.maxquant.org/.
NOTA: Esto se utilizará para procesar el archivo de datos RAW MS del paso 6.6 en tablas de datos de ID de proteínas, nombres de genes y valores cuantitativos asociados con la identificación de estos para el análisis posterior.- Seleccione Cargar en el subencabezado Datos de entrada de la pestaña Datos sin procesar.
- Haga clic en la pestaña Específico del grupo y seleccione Digestión. Dentro de la lista de enzimas seleccione LysC y haga clic en la flecha derecha para agregar esta enzima a la lista que se utilizará en la búsqueda. A continuación, seleccione Instrumento y asegúrese de que el tipo de instrumento correcto aparece en la lista desplegable en la parte superior de la pantalla. Deje otros parámetros de búsqueda específicos del grupo en la configuración estándar.
- Haga clic en la pestaña Parámetros globales y seleccione Secuencias. Agregue el archivo FASTA adecuado para la taxonomía que se utilizará en esta búsqueda. Los péptidos no se asignarán correctamente si esto no se hace. Para el proteome humano, descargue el archivo FASTA de UniProt en https://www.uniprot.org/help/human_proteome.
- En la pestaña Parámetros globales, haga clic en Cuantificación de proteínas. En el menú desplegable Péptidos para cuantificación, seleccione Único + Razor.
NOTA: MaxQuant ofrece una cuantificación alternativa de proteínas a través de la cuantificación absoluta basada en intensidad (iBAQ) y la cuantificación sin etiquetas (LFQ). Sin embargo, la información del recuento de péptidos es suficiente para el análisis descendente en este protocolo15. - En la parte inferior izquierda de la interfaz MaxQuant, seleccione el número de procesadores que se utilizarán para la búsqueda. Esto afectará directamente al tiempo requerido para la ejecución, así que seleccione tantos como sea posible para esto). Haga clic en Inicio en la parte inferior izquierda de la pantalla para iniciar la ejecución. Seleccione la pestaña Rendimiento en la parte superior de la pantalla para ver el progreso de la búsqueda.
- Una vez completada la ejecución, abra el archivo proteingroups.txt en Perseus, una plataforma de cálculo proteómica u otro programa de hoja de cálculo para ver los datos16.
- Utilice Perseus para eliminar contaminantes comunes y golpes a secuencias de proteínas invertidas. Siga la documentación detallada de Perseus en http://www.coxdocs.org/doku.php?id=perseus:user:use_cases:interactions.
NOTA: Abrir el archivo proteingroups.txt en el software de análisis (por ejemplo, Excel) dañará automáticamente ciertos nombres de genes y proteínas.
- Utilice Perseus para eliminar contaminantes comunes y golpes a secuencias de proteínas invertidas. Siga la documentación detallada de Perseus en http://www.coxdocs.org/doku.php?id=perseus:user:use_cases:interactions.
- Analice los datos experimentales utilizando el Repositorio de Contaminantes para la Purificación de Afinidad (CRAPome). Registre una cuenta en este repositorio http://crapome.org/ y siga el tutorial según sea necesario17,18.
- Utilice el flujo de trabajo Analizar sus datos que se encuentra en la página principal de CRAPome. Seleccione controles externos que correspondan al sistema de purificación de afinidad utilizado en este experimento de interacción.
NOTA: Estos controles se pueden utilizar para calcular un segundo enriquecimiento de cambio de plegado que es útil para detectar contaminantes comunes. - Genere un archivo de entrada a partir de la salida proteingroups.txt de MaxQuant mediante Perseus o una aplicación de hoja de cálculo adecuada. Los detalles sobre el formato manual se pueden encontrar en http://crapome.org/?q=fileformatting. Como alternativa, utilice el script de R proporcionado "export_CRAPomeSAINT_Input_File.R" para generar el archivo de entrada SAINT/CRAPome. Consulte README.txt en Archivos de codificación suplementarios.
- Ejecuta un análisis para determinar el enriquecimiento de plegado y la probabilidad de SAINT (Análisis de significancia de INTeractome) para cada proteína de cebo en la inmunoprecipitación. Asegúrese de que 'Controlesde usuario' esté seleccionado en el menú desplegable bajo FC-A, 'Controles CRAPome' o 'Todos los controles' estén seleccionados en el menú desplegable FC-B y la Puntuación de probabilidad esté seleccionada para generar probabilidades SAINT. Cuando la ejecución haya concluido, vea la salida que está disponible en "Resultados de análisis" junto con un identificador de trabajo. Descargue la matriz de datos de los 'Resultados de análisis' para el trazado futuro y la visualización de datos.
- Utilice el flujo de trabajo Analizar sus datos que se encuentra en la página principal de CRAPome. Seleccione controles externos que correspondan al sistema de purificación de afinidad utilizado en este experimento de interacción.
- Trazar proteínas en función de FC-A (IPs frente a controles de usuario) y probabilidad SAINT siguiendo los R-Scripts según lo dispuesto en Archivos de codificación suplementarios.
NOTA: Se proporciona un conjunto de scripts R para producir gráficas de probabilidad FC-A frente a SAINT y iBAQ frente al registro2 (abundancia de proteínas), coloreadas por el rango de valor p ajustado a partir del análisis empírico de Bayes de las intensidades sin etiquetas. Los detalles del análisis estadístico diferencial y el trazado se encuentran en README.txt y en el script de R "main_differential_analysis. R" en los archivos de codificación suplementarios. - Evaluar dónde las proteínas interactuantes conocidas de la proteína de cebo se clasifican por FC-A y SAINT. Haga un corte de FC-A > 3.00 y SAINT > 0.7 para experimentos de cebo único en triplicado como punto de partida.
NOTA: La selección de los límites para un interactor de "alta confianza" y un interactor de "baja confianza" debe estar informada por información biológica.
8. Visualización de datos
NOTA: Hay muchos programas que pueden visualizar eficazmente los datos proteómicos (por ejemplo, R, Perseus, Cytoscape, STRING-DB). Analizar la conectividad entre los hits de alta confianza y el enriquecimiento funcional de estos interelementoes puede ser una estrategia útil para priorizar los hits para una mayor validación y caracterización funcional.
- Descargue Cytoscape, una herramienta de visualización de red de código abierto al https://cytoscape.org/download.html19.
- Prepare un archivo de entrada para los datos de interacción como un archivo delimitado por tabulaciones formateado con tres columnas: cebo (nodo de origen), presa (nodo de destino), tipo de interacción (tipo de borde). Esto se puede hacer en Perseo o cualquier programa de hoja de cálculo de su elección.
- Seleccione el icono Importar tabla desde archivo hacia la parte superior izquierda del programa (designado por una flecha hacia abajo y una matriz en el icono). Cytoscape rellenará automáticamente los datos de interacción en una red lista para el formato y el diseño personalizados.
- Seleccione la pestaña Estilo en el panel de control de Cytoscape y haga clic en los cuadrados de la columna Def para ajustar el atributo de toda la red. Seleccione nodos o aristas específicos en la red y, a continuación, seleccione el cuadrado dentro de la columna Byp. del menú de estilo para omitir la configuración predeterminada y ajustar solo los objetos seleccionados. Alternativamente, haga clic en el menú desplegable en la parte superior del menú de estilo para ver los formatos de red predefinidos.
NOTA: Los datos de interacción proteína-proteína STRING-db pueden integrarse en esta red en este momento, ya sea manualmente a través del archivo de entrada o a través de varias herramientas de enriquecimiento disponibles como plug-ins en Cytoscape, http://apps.cytoscape.org/20. Un plug-in de citopaisaje recomendado para el análisis de enriquecimiento se encuentra en http://apps.cytoscape.org/apps/cluego21. - Para aumentar la confianza en el conjunto de datos generado en este flujo de trabajo, realice experimentos recíprocos de IP-MS o IP-western dirigidos a las proteínas de presa de interés como cebo.
Resultados
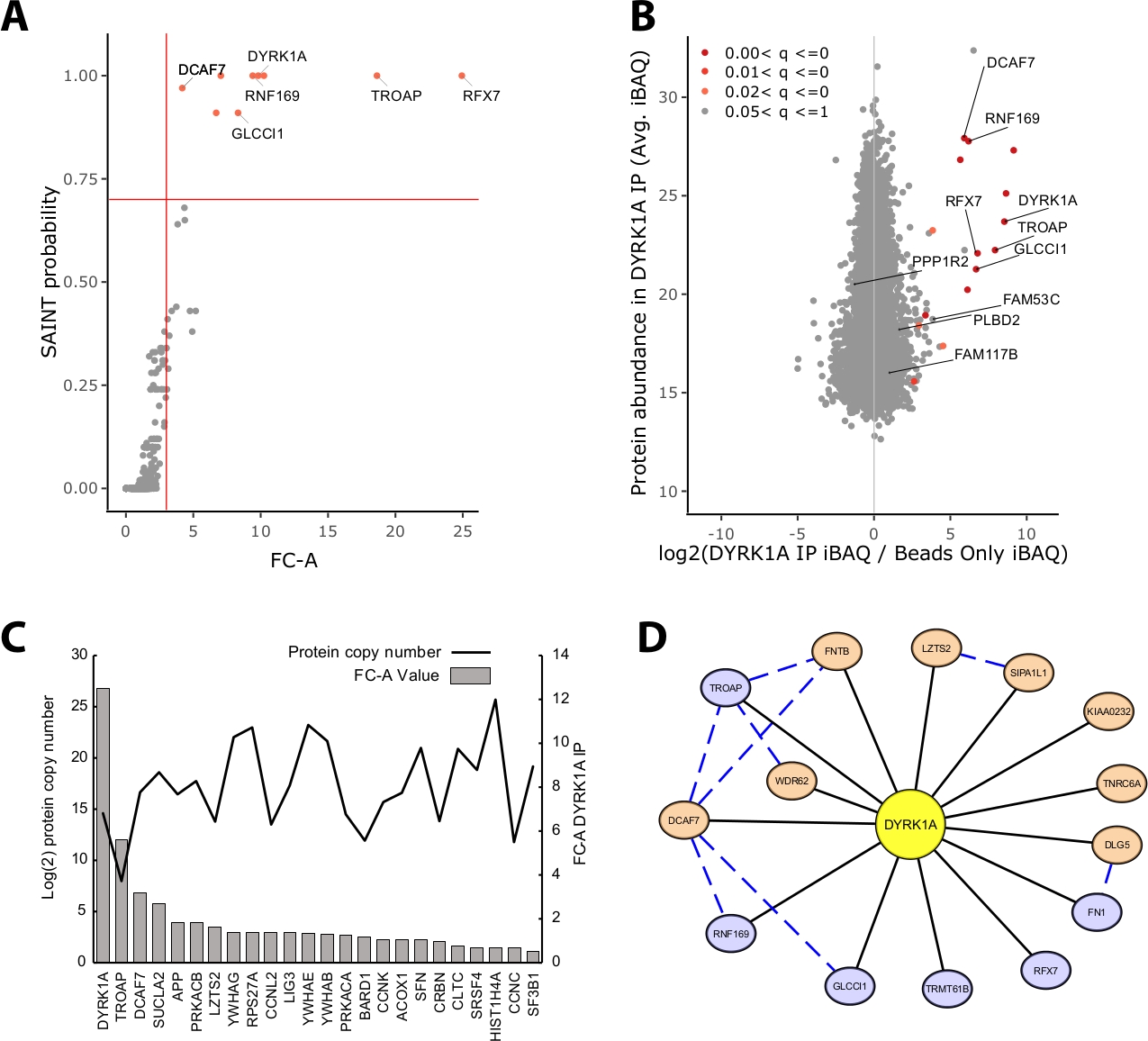
La mayor parte de la masa proteica identificada en un experimento IP-MS consiste en proteínas no específicas. Por lo tanto, uno de los desafíos clave de un experimento IP-MS es la interpretación de qué proteínas son los interactores de alta confianza frente a los interactores inespecíficos. Para demostrar los parámetros cruciales utilizados en la evaluación de la calidad de los datos, el estudio analizó las inmunoprecipitaciones triplicadas a partir de 5 mg de extracto nuclear de HeLa utilizando un control de perlas solamente. La primera comprobación interna para garantizar que un experimento IP-MS sea fiable es si la proteína de cebo se clasifica como una de las proteínas enriquecidas más altas identificadas por el cambio plegado y la probabilidad de SAINT. En este caso, el cebo DYRK1A se ubicó entre las tres principales proteínas enriquecidas sobre el control(Figura 2A,B). En un estudio de interactome nuclear de DYRK1A utilizando cuatro anticuerpos independientes, un corte FC-A de >3.00 y el corte de probabilidad SAINT >0.7 proporcionaron un estricto límite para la identificación de los interquenistas novedosos y previamente validados22. Cuando se aplica a este experimento, se puede ver una separación clara entre los interactores de alta confianza y >95% de las proteínas copurificadas identificadas como inespecíficas (Figura 2A,B). La aplicación de un límite de enriquecimiento y probabilidad de cambio de pliegue aumenta la rigensión al requerir un enriquecimiento constante de los documentos de identificación proteica a través de réplicas biológicas.
Además de la puntuación estadística, el flujo de trabajo de análisis CRAPome también asigna interacciones notificadas anteriormente en los datos de presa de cebo23. Si bien esta asignación puede ser útil para umbraldear interacciones de alta y baja confianza, las interacciones notificadas anteriormente pueden puntuar seriamente según las probabilidades FC-A y SAINT, lo que puede indicar que muchas interacciones conocidas de un cebo determinado pueden existir solo en tipos de células, contextos u orgánulos específicos. Para el conjunto de datos DYRK1A de ejemplo, los valores FC-A del interactor iREF eran tan bajos como 0,45, lo que representa un enriquecimiento muy bajo sobre el control(Figura 2C). Para evitar la inflación de falsos positivos, el umbral estadístico debe realizarse de una manera que priorice la rigásca sobre la reducción de falsos negativos. Cabe señalar que la detección de estas interacciones fue independiente de la abundancia de proteínas(Figura 2C). El número de copia absoluta calculado de cada interacción iREF dentro de las celdas de HeLa no mostró correlación con los niveles de detección de un socio de interacción por IP-MS24.
Cytoscape sirve como una herramienta eficaz para visualizar múltiples capas de datos de interacción19. En el experimento de inmunoprecipitación DYRK1A descrito aquí, el uso combinado de FC-A > 3.0 y SAINT > 0.9 redujo la lista de interactores de alta confianza a seis proteínas(Figura 2D). Sin embargo, al aplicar un corte FC-A de > 3.0 de forma aislada, se agregaron ocho proteínas adicionales a la red. Estos interactores de proteínas adicionales tienen una alta conectividad con los interactores que ya están en la red, lo que sugiere que están asociados en complejos similares o roles funcionales. Con este fin, la evidencia de la CADENA-DB de interacciones proteína-proteína se integró en esta red como líneas azules discontinuas20. Si bien este experimento de un solo cebo y triplicado proporciona una muestra limitada de la red de interacción DYRK1A completa, el uso de cebos adicionales, réplicas e integración de grandes conjuntos de datos públicos se puede utilizar para ampliar la red de interacciones de alta confianza. Por lo tanto, los recortes estadísticos serán específicos de cada experimento individual y deberán evaluarse a fondo.

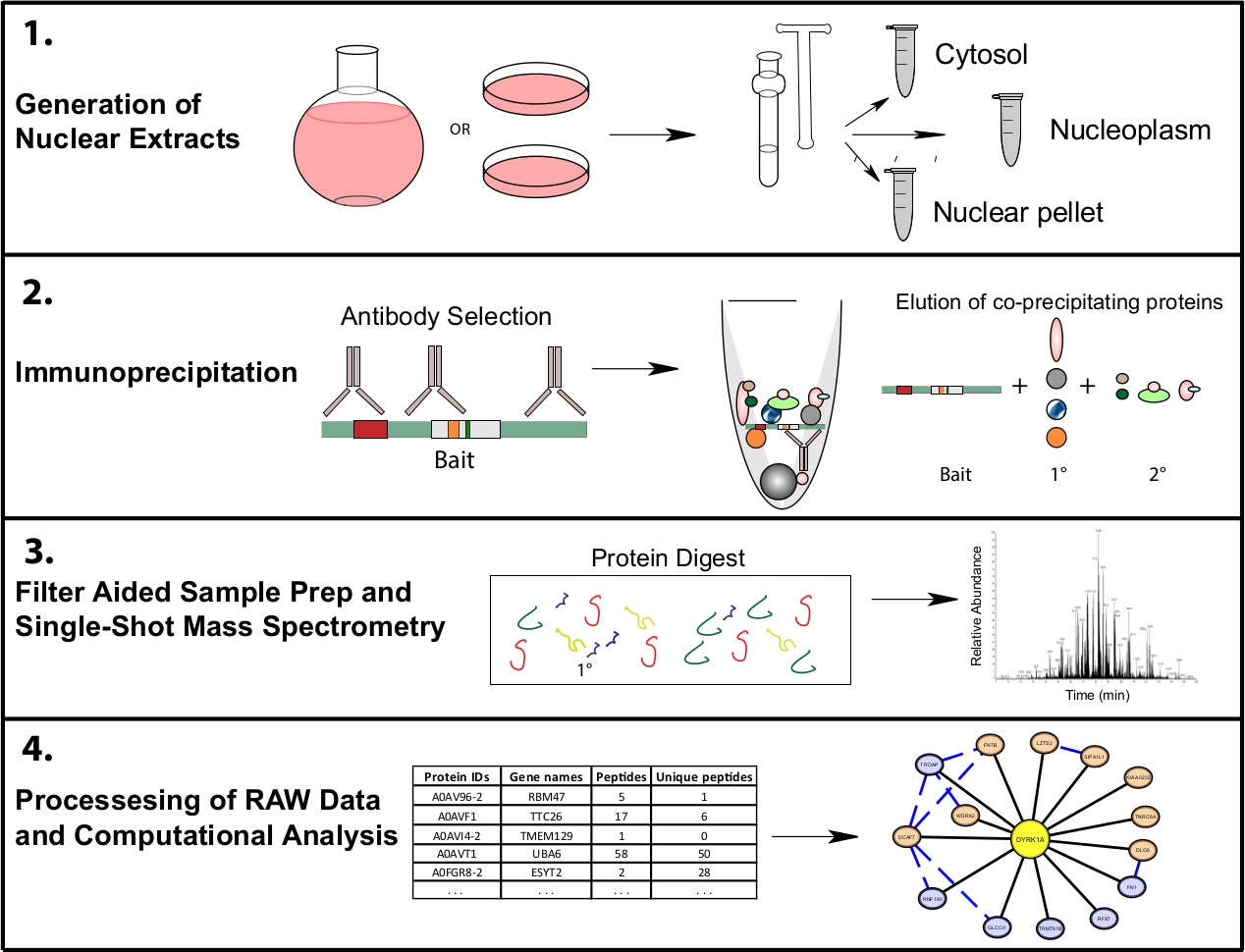
Figura 1: Flujo de trabajo proteómico representativo para IP-MS subcelular. Las células se cultivan en matraces de fondo redondo de 4 L o en platos de cultivo de tejido de 15 cm y se cosechan al mismo tiempo para el fraccionamiento subcelular. Las células se fraccionan en un citosólico, nuclear, y un pellet nuclear, y las inmunoprecipitaciones se realizan de 1 a 10 mg de lisado nuclear utilizando uno o varios anticuerpos que reconocen el mismo cebo. La preparación de muestras asistida por filtro (FASP) y la limpieza de muestras sin conexión se realizan antes de la espectrometría de masas de una sola toma. Una canalización computacional de nivel inferior se utiliza para procesar datos en datos de interacción interpretables. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Datos representativos para un experimento IP-MS de un solo cebo. (A) salida de probabilidad FC-A y SAINT del flujo de trabajo de análisis CRAPome para un experimento óptimo utilizando un solo anticuerpo para la quinasa DYRK1A (n.o 3). Se utilizaron controles de solo cordones para la comparación. Las líneas sólidas rojas representan los cortes establecidos en FC-A > 3.00 y SAINT > 0.7. (B) MaxQuant estima la producción de la abundancia de proteínas (iBAQ) frente a la relación log2 de abundancia de proteínas en DYRK1A IP para controlar, coloreada por el rango de valor p ajustado a partir del análisis empírico de Bayes de las intensidades sin etiquetas. (C) FC-A y el número estimado de proteínas enumeradas como proteínas que interactúan en la base de datos iRef23,24. (D) Visualización en red Cytoscape de los interactores DYRK1A. Nodos azules: FC-A > 3.00, SAINT > 0.7. Nodos naranjas: FC-A > 3.00. Bordes negros: proteínas identificadas como interactuadores en el experimento IPMS. Borde de corte azul: interacción de SAINT entre la proteína de presa (confianza > .150). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Mezcla de inhibidores de la proteasa (PI) | |
| Reactivo | Concentración final |
| Metabisulfita sódica | 1 mM |
| Benzamidina | 1 mM |
| Ditiothreitol (TDT) | 1 mM |
| Fluoruro de feniloquesulfonil (PMSF) | 0,25 mM |
| Aditivo de fosfatasa (PhI) mezcla | |
| Reactivo | Concentración final |
| Microcistina LR | 1 M |
| Ortovanato de sodio | 0,1 mM |
| Fluoruro de sodio | 5 mM |
| Búferes de fraccionamiento subcelular: | |
| Zona de influencia A pH 7,9 | |
| Reactivo | Concentración final |
| HEPES | 10 mM |
| MgCl2 | 1,5 mM |
| Kcl | 10 mM |
| Buffer B pH 7.9 | |
| Reactivo | Concentración final |
| HEPES | 20 mM |
| MgCl2 | 1,5 mM |
| Nacl | 420 mM |
| Acido etilendiaminetetraacético (EDTA) | 0,4 mM |
| Glicerol | 25% (v/v) |
| Buffer C pH 7.9 | |
| Reactivo | Concentración final |
| HEPES | 20 mM |
| MgCl2 | 2 mM |
| Kcl | 100 mM |
| Acido etilendiaminetetraacético (EDTA) | 0,4 mM |
| Glicerol | 20% (v/v) |
| Búferes de inmunoprecipitación: | |
| Búfer IP 1 | |
| Reactivo | Concentración final |
| HEPES | 20 mM |
| Kcl | 150 mM |
| Edta | 0,1 mM |
| NP-40 | 0,1% (v/v) |
| Glicerol | 10% (v/v) |
| Búfer IP 2 | |
| Reactivo | Concentración final |
| HEPES | 20 mM |
| Kcl | 500 mM |
| Edta | 0,1 mM |
| NP-40 | 0,1% (v/v) |
| Glicerol | 10% (v/v) |
| Tampón de alquilación SDS pH 8.5 | |
| Reactivo | Concentración final |
| Sds | 4% (v/v) |
| Cloroacetamida | 40 mM |
| TCEP | 10 mM |
| Tris | 100 mM |
| PH UA 8.5 | |
| Reactivo | Concentración final |
| Urea | 8 M |
| Tris | 0,1 M |
| * Utilizar H2O grado HPLC | |
Tabla 1: Composiciones de búfer
Archivos de codificación suplementarios. Haga clic aquí para descargar este archivo.
Discusión
El flujo de trabajo proteómico descrito aquí proporciona un método eficaz para identificar a los interactores de proteínas de alta confianza para una proteína de interés. Este enfoque reduce la complejidad de la muestra a través de la fracción subcelular y se centra en aumentar los socios de interacción de identificación a través de una preparación sólida de la muestra, la limpieza de muestras fuera de línea y el estricto control de calidad del sistema LC-MS. El análisis de datos aguas abajo descrito aquí permite una evaluación estadística simple de las proteínas identificadas como corependatas con el cebo. Sin embargo, debido a un alto número de variables experimentales (escala, línea celular, elección de anticuerpos), cada experimento requiere diferentes cortes y consideraciones con respecto a la visualización y el enriquecimiento de datos.
La primera consideración del diseño en un experimento IP-MS es la selección de anticuerpos que se utilizarán para la copurificación de la proteína de interés junto con sus socios que interactúan. Si bien la disponibilidad de anticuerpos comerciales se ha expandido para cubrir porciones más grandes del proteome humano en las últimas décadas, todavía hay muchas proteínas para las que los reactivos son limitados. Además, los anticuerpos que han sido validados para aplicaciones como la detección de manchas occidentales pueden ser incapaces de enriquecimiento selectivo de la proteína diana en un experimento de inmunoprecipitación. Antes de llevar a cabo un experimento proteómico de interacción a gran escala, se sugiere completar una IP a partir de un plato de 10 cm de confluente del 90%, o número de celda equivalente, y sondear la proteína objetivo de interés por la hincha occidental. Si hay más de un solo anticuerpo disponible para la inmunoprecipitación, además se sugiere seleccionar múltiples anticuerpos que reconozcan los epítopos dentro de diferentes porciones de la proteína. La unión de un anticuerpo a una proteína de cebo puede ocluir la interfaz de unión necesaria para los socios que interactúan putativamente. La selección de un epítopo secundario para la proteína de cebo aumentará la cobertura del perfil de interacción identificado por un experimento basado en espectrometría de masas.
Una segunda consideración importante radica en la selección del control adecuado para distinguir las interacciones de alta confianza de las interacciones de baja confianza o inespecíficos de las identificadas como copurificadas con el cebo. El control más estricto para un experimento IP-MS es completar la inmunoprecipitación de una línea celular CRISPR KO del cebo. Tal control permite identificar y filtrar proteínas inespecíficas que se unen directamente al anticuerpo en lugar de a la proteína de cebo. En los casos en que no es factible generar una línea celular CRISPR KO de cada proteína de cebo, se puede utilizar un control de perlas IgG del mismo isotipo del anticuerpo de cebo. En experimentos que emplean un panel de anticuerpos que representan múltiples especies, el uso de un control de cuentas sólo puede ser apropiado, pero aumentará la tasa de falsos positivos identificados como interactores de alta confianza.
La selección de la línea de celda utilizada en un experimento IP-MS depende de varios factores clave. La expresión y localización de proteínas dependen en gran medida del tipo de célula. Mientras que las estimaciones de la expresión de ARN se pueden encontrar para la mayoría de los genes en muchas líneas celulares de uso común, la expresión de proteína está mal correlacionada con la expresión de ARN y debe determinarse experimentalmente25. Las líneas celulares en las que una proteína de cebo se expresa en un número de copia muy bajo deben evitarse para eludir los problemas asociados con aumentos drásticos en la escala de cultivo celular que pueden ser necesarios. Cabe señalar, sin embargo, que la preparación de la muestra se puede optimizar para la detección de proteínas de muy baja abundancia. El método de preparación de muestras asistida por filtro (FASP), aunque robusto, puede causar una pérdida de péptido de más del 50% en una muestra. La preparación de muestras mejorada por fase sólida de una sola olla (SP3) es un método eficaz de generación de muestras para el análisis de espectrometría de masas que minimiza la pérdida de muestra26. El aumento de la recuperación habilitado por el método SP3 de preparación de muestras puede ser una alternativa útil en este flujo de trabajo para la cuantificación de proteínas que están cerca del límite de detección.
Este flujo de trabajo proteómico se ha aplicado a través de muchos cebos nucleares, incluyendo quinasas, ligasas de ubiquitina E3 y miembros de andamios de complejos multisubunidades. Suponiendo la validación adecuada de los reactivos de anticuerpos, la ejecución exitosa de este flujo de trabajo dará lugar a la detección de socios de interacción nuclear de proteínas de alta confianza para una proteína de interés.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Este trabajo fue apoyado por una beca Grand Challenge a W.M.O. del Linda Crnic Institute for Down syndrome y por un acuerdo de cooperación DARPA 13-34-RTA-FP-007. Nos gustaría agradecer a Jesse Kurland y Kira Cozzolino por sus contribuciones en la lectura y comentarios sobre el manuscrito.
Materiales
| Name | Company | Catalog Number | Comments |
| 0.25% Trypsin, 0.1% EDTA | Thermo Fisher Scientific | 25200056 | |
| 1.5 ml low-rention microcentrifuge tubes | Fisher Scientific | 02-681-320 | |
| 4-20% Mini PROTEAN TGX Precast Protein Gels | Bio-Rad | 4561096 | |
| acetone (HPLC) | Thermo Fisher Scientific | A949SK-4 | |
| Amicon Ultra 0.5 ml 30k filter column | Millipore Sigma | UFC503096 | |
| Benzamidine | Sigma-Aldrich | 12072 | |
| benzonase | Sigma-Aldrich | E1014 | |
| Chloroacetamide | Sigma-Aldrich | C0267 | |
| Dialysis tubing closure | Caroline Biological Supply Company | 684239 | |
| DTT | Sigma-Aldrich | 10197777001 | |
| EDTA | Sigma-Aldrich | EDS | |
| GAPDH antibody | Santa Cruz Biotechnology | Sc-47724 | |
| Glycerol | Fisher Scientific | 887845 | |
| Glycine | Sigma-Aldrich | G8898 | |
| HeLa QC tryptic digest | Pierce | 88329 | |
| HEPES | Fisher Scientific | AAJ1692630 | |
| insulin | Thermo Fisher Scientific | 12585014 | |
| iodoacetamide | Sigma-Aldrich | I1149 | |
| KONTES Dounce homogenizer 7 ml | VWR | KT885300-0007 | |
| Large Clearance pestle 7ml | VWR | KT885301-0007 | |
| Lysyl endopeptidase C | VWR | 125-05061 | |
| Magnesium Chloride | Sigma-Aldrich | 208337 | |
| Microcystin | enzo life sciences | ALX-350-012-C100 | |
| Nonidet P 40 Substitute solution | Sigma-Aldrich | 98379 | |
| p84 antibody | GeneTex | GTX70220 | |
| Phosphate Buffered Saline | |||
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | 23227 | |
| Pierce BSA Protein Digest, MS grade | Thermo Fisher Scientific | 88341 | LCMS QC |
| Pierce C18 spin columns | Thermo Fisher Scientific | PI-89873 | |
| Pierce Trypsin Protease, MS Grade | Thermo Fisher Scientific | 90057 | For mass spectrometry sample prep |
| PMSF | Sigma-Aldrich | P7626 | |
| Potassium Chloride | Sigma-Aldrich | P9541 | |
| Protein A Sepharose CL-4B | GE Healthcare Bio-Sciences | 17-0780-01 | |
| Protein G Sepharose 4 Fast Flow | GE Healthcare Bio-Sciences | 17-0618-01 | |
| SDS | Sigma-Aldrich | L3771 | |
| Silica emitter tip | Pico TIP | FS360-20-10 | |
| Small Clearance pestle 7ml | VWR | KT885302-0007 | |
| Sodium Chloride | Sigma-Aldrich | S3014 | |
| Sodium Fluoride | Sigma-Aldrich | 201154 | |
| Sodium metabisulfite | Sigma-Aldrich | 31448 | |
| Sodium orthovanadate | Sigma-Aldrich | S6508 | |
| Spectra/ Por 8 kDa 24 mm dialysis tubing | Thomas Scientific | 3787K17 | |
| TC Dish 150, Standard | Sarstedt | 83.3903 | Tissue culture dish for adherent cells |
| TCA | Sigma-Aldrich | T9159 | |
| TCEP | Thermo Scientific | PG82080 | |
| TFA | Thermo Fisher Scientific | 28904 | |
| Thermo Scientific Orbitrap Fusion MS | Thermo Fisher Scientific | ||
| Trizma Base | Sigma-Aldrich | T6066 | |
| Urea | Thermo Fisher Scientific | 29700 | |
| Waters ACQUITY M-Class UPLC | Waters | ||
| Waters ACQUITY UPLC M-Class Column Reversed-Phase 1.7µm Spherical Hybrid (1.7 µm, 75 µm x 250 mm) | Waters | 186007484 | nanoflow C18 column |
Referencias
- Varjosalo, M., et al. The protein interaction landscape of the human CMGC kinase group. Cell Reports. 3, 1306-1320 (2013).
- Kimple, M. E., Brill, A. L., Pasker, R. L. Overview of Affinity Tags for Protein Purification. Current Protocols in Protein Science. 73, (2013).
- Mahmood, N., Xie, J. An endogenous 'nonspecific' protein detected by a His-tag antibody is human transcription regulator YY1. Data in Brief. 2, 52 (2015).
- Zordan, R. E., Beliveau, B. J., Trow, J. A., Craig, N. L., Cormack, B. P. Avoiding the ends: internal epitope tagging of proteins using transposon Tn7. Genetics. 200, 47-59 (2015).
- Gibson, T. J., Seiler, M., Veitia, R. A. The transience of transient overexpression. Nature Methods. 10, 715-721 (2013).
- Bronicki, L. M., et al. Ten new cases further delineate the syndromic intellectual disability phenotype caused by mutations in DYRK1A. European Journal of Human Genetics. 23, 1482-1487 (2015).
- Antonarakis, S. E. Down syndrome and the complexity of genome dosage imbalance. Nature Reviews Genetics. , (2016).
- Dowjat, W. K., et al. Trisomy-driven overexpression of DYRK1A kinase in the brain of subjects with Down syndrome. Neuroscience Letters. 413, 77-81 (2007).
- Fotaki, V., et al. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Molecular and Cellular Biology. 22, 6636-6647 (2002).
- Hämmerle, B., Elizalde, C., Tejedor, F. J. The spatio-temporal and subcellular expression of the candidate Down syndrome gene Mnb/Dyrk1A in the developing mouse brain suggests distinct sequential roles in neuronal development. European Journal of Neuroscience. 27, 1061-1074 (2008).
- Funakoshi, E., et al. Overexpression of the human MNB/DYRK1A gene induces formation of multinucleate cells through overduplication of the centrosome. BMC Molecular and Cell Biology. 4, 12 (2003).
- Yu, D., Cattoglio, C., Xue, Y., Zhou, Q. A complex between DYRK1A and DCAF7 phosphorylates the C-terminal domain of RNA polymerase II to promote myogenesis. Nucleic Acids Research. , 1-14 (2019).
- Towbin, H., Staehelin, T., Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proceedings of the National Academy of Sciences of the United States of America. 76, 4350-4354 (1979).
- Wiśniewski, J. R., Zougman, A., Nagaraj, N., Mann, M. Universal sample preparation method for proteome analysis. Nature Methods. 6, 359-362 (2009).
- Tyanova, S., Temu, T., Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature Protocols. 11, 2301-2319 (2016).
- Tyanova, S., et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods. 13, 731-740 (2016).
- Mellacheruvu, D., et al. The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nature Methods. 10, 730-736 (2013).
- Choi, H., et al. SAINT: Probabilistic scoring of affinity purificationg-mass spectrometry data. Nature Methods. 8, 70-73 (2011).
- Shannon, P., et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research. 13, 2498-2504 (2003).
- Szklarczyk, D., et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Research. 45, 362-368 (2017).
- Bindea, G., et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 25, 1091-1093 (2009).
- Guard, S. E., et al. The nuclear interactome of DYRK1A reveals a functional role in DNA damage repair. Scientific Reports. 9, 6539 (2019).
- Razick, S., Magklaras, G., Donaldson, I. M. iRefIndex: A consolidated protein interaction database with provenance. BMC Bioinformatics. 9, 405 (2008).
- Kulak, N. A., Pichler, G., Paron, I., Nagaraj, N., Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nature Methods. 11, 319-324 (2014).
- Liu, Y., Beyer, A., Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell. 165, 535-550 (2016).
- Hughes, C. S., et al. Ultrasensitive proteome analysis using paramagnetic bead technology. Molecular Systems Biology. 10, 757 (2014).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados