
Method Article
Análisis de moléculas complejas y sus reacciones en superficies por medios de espectrometría de masas de desorción/ionización inducida por racimos
En este artículo
Resumen
Los grupos neutros SO2 de baja energía cinética (< 0,8 eV/constituyente) se utilizan para desorb moléculas superficiales complejas como péptidos o lípidos para su posterior análisis mediante espectrometría de masas utilizando un espectrómetro de masas trampa de iones. No se requiere ninguna preparación especial de la muestra, y es posible la observación en tiempo real de las reacciones.
Resumen
La desorción/ionización inducida por los cúmulos neutrales SO2 (DINeC) se emplea como una técnica de desorción/ionización muy suave y eficiente para espectrometría de masas (MS) de moléculas complejas y sus reacciones en superficies. DINeC se basa en un haz de clústeres SO2 que impactan en la superficie de la muestra a baja energía del clúster. Durante el impacto en la superficie del racimo, algunas de las moléculas superficiales se desorbidan e ionizan a través de la disolvación en el cúmulo que impacta; como resultado de este mecanismo de desorción mediado por la disresolución, la baja energía del racimo es suficiente y el proceso de desorción es extremadamente suave. Se pueden analizar tanto los adsorbatos superficiales como las moléculas de las que se compone la superficie. Se obtienen espectros claros y libres de fragmentación de moléculas complejas como péptidos y proteínas. DINeC no requiere ninguna preparación especial de la muestra, en particular no es necesario aplicar ninguna matriz. El método produce información cuantitativa sobre la composición de las muestras; moléculas con una cobertura superficial tan baja como el 0,1 % de una monocapa. Las reacciones superficiales como el intercambio de H/D o la descomposición térmica se pueden observar en tiempo real y se puede deducir la cinética de las reacciones. Utilizando una boquilla pulsada para la generación de haz de racimo, DINeC se puede combinar eficientemente con espectrometría de masas de trampa de iones. La naturaleza libre de matriz y suave del proceso DINeC en combinación con las capacidades MSn de la trampa iónica permite un análisis muy detallado e inequívoco de la composición química de muestras orgánicas complejas y adsorbatos orgánicos en superficies.
Introducción
Las técnicas de análisis sensibles a la superficie a menudo se basan en sondas de partículas como electrones de baja energía, átomos o iones que interactúan fuertemente con muestras sólidas. Como consecuencia, muestran una alta sensibilidad superficial y se puede obtener información detallada sobre la estructura de la superficie1. La información química, sin embargo, a menudo es limitada. Por ejemplo, la espectroscopia de fotoelectrón de rayos X puede proporcionar información cuantitativa sobre la composición atómica y sobre el entorno químico medio de una especie determinada (por ejemplo, los átomos de carbono de una molécula orgánica adsorbida en una superficie2). Sin embargo, es difícil obtener información más detallada sobre moléculas complejas y sacadas a la superficie, como su estructura detallada o sitios de unión, con técnicas estándar de análisis de superficie. Por otro lado, la necesidad de esta información está creciendo con el creciente interés en la funcionalización de la superficie por medio de moléculas orgánicas. Los campos en expansión de la síntesis en superficie3 o la funcionalización de la superficie por unión de biomoléculas4,5 son dos ejemplos prominentes. En todos estos campos, se investigan preguntas fundamentales sobre las interacciones de sento-adsorbato y adsorbato de sustrato con el fin de comprender mejor los sistemas. Para estas investigaciones, es deseable un máximo de información sobre las moléculas adsorbidas.
En parte, la espectrometría de masas ióniona secundaria (SIMS) puede proporcionar dicha información. En primer lugar, SIMS es altamente sensible a la superficie. En segundo lugar, a medida que los adsorbatos esputos y sus fragmentos se detectan por medio de la EP, se obtiene información mucho más allá de la composición atómica. Dependiendo de la naturaleza de la especie química adsorbida en la superficie, se puede identificar por su masa molecular y el patrón de fragmentos observado en el espectro de masa6. Los fragmentos inducidos por los iones primarios pueden ayudar a la identificación del material analizado. Por otro lado, si la modificación inducida por iones primarios (fragmentación, reacciones inducidas por iones, mezcla) de la muestra es demasiado fuerte, la mayor parte de la información sobre el estado original de la muestra se pierde. Así, se han realizado importantes esfuerzos para reducir la fragmentación en sims (por ejemplo, utilizando clústeres moleculares cargados como iones primarios7,8,9). Sin embargo, la fragmentación todavía domina los espectros SIMS de grandes macromoléculas y muestras biológicas10,limitando la aplicación de SIMS en varios campos.
Como alternativa, hemos demostrado que la desorción/ionización inducida por racimos neutros (DINeC) es un método de ionización suave y libre de matrices que se ha empleado con éxito para el análisis espectrométrico de masas de moléculas complejas11,12,13,14,15,16,17. DINeC se basa en un haz de racimos moleculares que consisten en 103 a 104 moléculas SO2 (Figura 1). Cuando los racimos impactan en la muestra, interactúan de varias maneras con las moléculas en y en la superficie: primero, una parte de la energía cinética del cúmulo se redistribuye y activa la desorción. Del mismo modo, la molécula de desorbing se disuelve en el cúmulo durante el impacto en la superficie del racimo11,18,19 (Figura 1 y Figura 2). En otras palabras, basado en el momento dipolo alto de SO2, los cúmulos sirven muy eficientemente como una matriz transitoria para los analitos polares. Como resultado, la desorción de las moléculas de analito tiene lugar en energías de racimo tan bajas como 1 eV/molécula y por debajo. La naturaleza suave del proceso de desorción se apoya aún más en el enfriamiento rápido del sistema cuando el clúster SO2 se rompe durante y después del impacto superficial11,19. Como consecuencia de estos diversos aspectos, la desorción inducida por racimos de moléculas complejas como péptidos, proteínas, lípidos y colorantes procede sin ninguna fragmentación de las moléculas desortonadas11,15; los espectros de masa típicos muestran el pico dominante en el valor m/z de la molécula intacta ([M+H]+ o [M-H]-, Figura 3). Dependiendo del número y la naturaleza de los grupos funcionales en la molécula, múltiples cationes cargados de la forma [M + n H]n+ se observan11,15,18. En el lugar de las biomoléculas, la ionización suele tener lugar mediante la aceptación o abstracción de un protón en un grupo funcional básico o ácido, respectivamente11. Si las moléculas de agua están presentes en la muestra, LAS moléculas SO2 del cúmulo pueden reaccionar con estas moléculas de agua formando ácido sulfuroso18. Este último puede actuar como una fuente de protones eficiente que promueve aún más el proceso de ionización en caso de ionización a través de la aceptación de protones (modo iónico positivo)13,18.

Figura 1: Ilustración esquemática de desorción/ionización inducida por clústeres y configuración experimental. La desorción/ionización inducida por racimos se realiza en un recipiente de alto vacío. Un haz de2 racimos SO (puntos amarillos) se produce a través de la expansión supersónica de una mezcla de gas SO2/He de una boquilla pulsada. Durante el impacto en la superficie del racimo, las moléculas superficiales se desorbidan e ionizan. Los iones moleculares (puntos rojos/naranjas) se transfieren a través de una cuadrícula sesgada, una entrada de embudo de iones doble y guías iónicas octopolares en la trampa iónica para espectrometría de masas. Los espectros de masa típicos muestran picos dominantes en los valores m/z de las moléculas intactas, aquí: M1 (naranja) y M2 (rojo) en modo iónico positivo. Explosión: Durante el impacto superficial del racimo, las moléculas desorbidas se disuelven en el cúmulo que impacta o en uno de sus fragmentos. La rotura y evaporación de las moléculas SO2 conducen entonces al ion molecular desnudo e intacto como se detecta en el espectrómetro de masas. Vea también la figura 2. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Instantáneas de simulaciones de dinámica molecular que ilustran la desorción inducida por clústeres a través de la disolvación. (A) Un grupo SO2 (300 moléculas) se acerca a la superficie con 1250 m/s perpendiculares a la superficie en la que se absorbe un dipéptido (ácido aspártico-arginina, ASP-ARG). (B) Durante el impacto en la superficie del clúster, el clúster se rompe. El dipéptido adsorbido interactúa con las moléculas SO2 circundantes que conducen a su diseminación en uno de los fragmentos del cúmulo. (C) Los fragmentos del clúster se repelin de la superficie. El fragmento etiquetado (círculo azul) lleva el dipéptido que se desorbia en este fragmento. Esta cifra ha sido modificada de la referencia 19. Haga clic aquí para ver una versión más grande de esta figura.
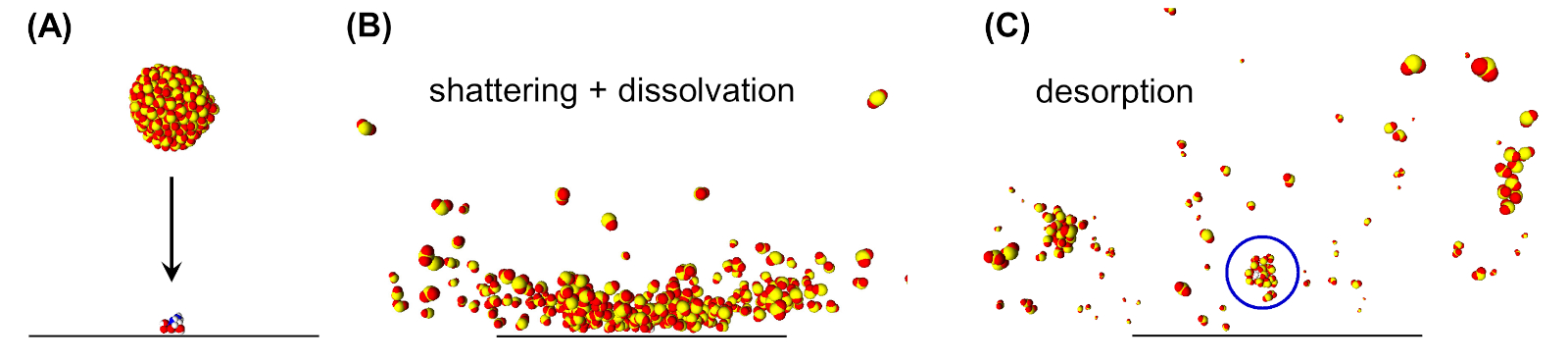{kind=link}

Figura 3: Espectro de masarepresentativo y modelo molecular de angiotensina II. (A) Espectros de masa (panel superior: modo iónico positivo, panel inferior: modo iónico negativo) obtenidos después de la desorción/ionización inducida por racimos de una muestra de angiotensina II. La muestra se preparó mediante la fundición de gotas de la solución respectiva en una oblea Si (cubierta por su óxido natural). Los picos principales se asignan a la biomolécula intacta, [M+H]+ y [M-H]- ; no se observan patrones de fragmentación. Dimers ([2M+H]+, flecha) indican además la naturaleza suave del proceso de desorción. La señal ion positiva es más intensa debido a la influencia de los clústeres SO2 18. (B) Modelo de llenado de espacio y secuencia de aminoácidos de angiotensina II. Las bolas blancas indican átomos de hidrógeno; negro: carbono; azul: nitrógeno; rojo: oxígeno. Haga clic aquí para ver una versión más grande de esta figura.
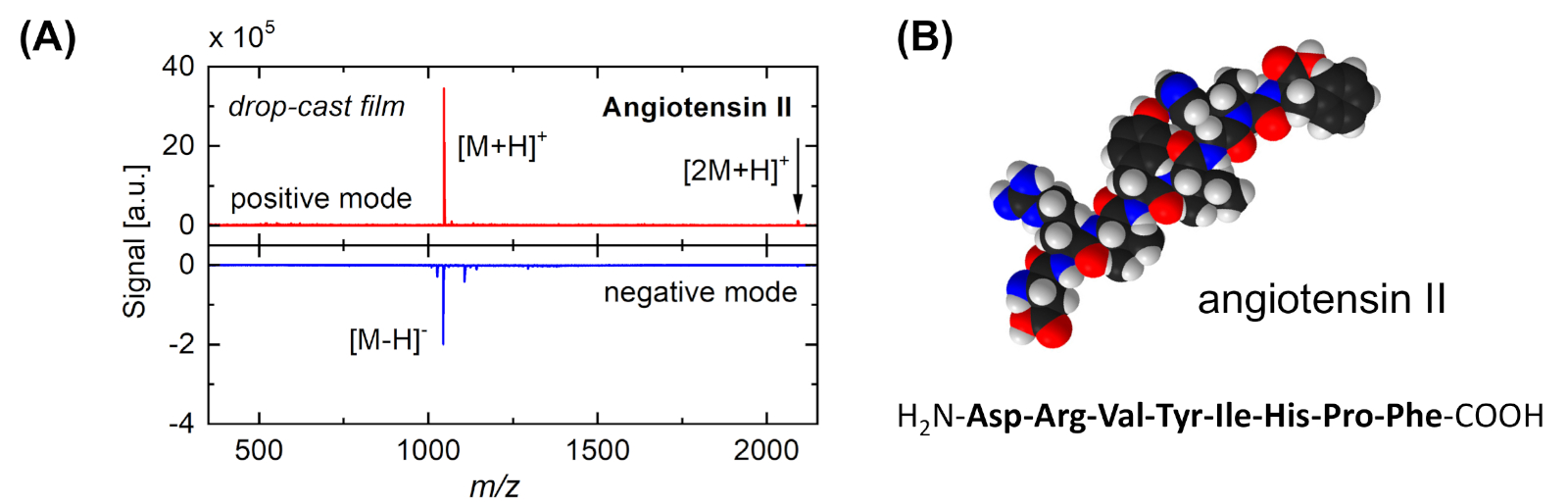{kind=link}
DINeC se puede aplicar a cualquier tipo de muestra sólida que sea compatible con condiciones de alto vacío. No es necesario realizar ninguna preparación especial de la muestra, en particular no es necesario aplicar ninguna matriz antes de las mediciones de DINeC-MS, a diferencia de la espectrometría de masas de desorción/ionización láser asistida por matriz (MALDI) y las técnicas conexas20,21. Esto permite mediciones en tiempo real de los cambios químicos de la muestra con diferentes condiciones experimentales, como la presión de fondo de las especies reactivas en la cámara de vacío22 o la temperatura de la muestra. Se ha demostrado que el límite de detección de DINeC-MS está en el rango de femtomole11. Cuando se aplica al análisis de biomoléculas adsorbidas en superficies sólidas en el régimen de submonocapa, se detectó una cobertura superficial tan baja como el 0,1% de una monocapa23. En este régimen de cobertura, la intensidad de la señal depende linealmente de la cobertura superficial y DINeC-MS se puede utilizar para el análisis cuantitativo de la composición superficial23. En el caso de muestras mixtas, es posible una evaluación cuantitativa de la composición de la muestra17,24, ya que no se observa ningún efecto importante del entorno químico sobre la probabilidad de ionización (por ejemplo, en el caso de muestras mixtas de lípidos/péptidos17). Esto contrasta claramente con los SIMS, para los que la probabilidad de ionización de una especie determinada suele estar fuertemente influenciada por la presencia de diferentes componentes químicos (el llamado "efecto matriz"25,26).
Además del análisis de superficie, la composición química en la región del subsuelo se puede sondear mediante perfiles de profundidad17. Con la configuración actual, las tasas típicas de desorción de la desorción inducida por racimos de biomoléculas son del orden 10-3 nm/s. Se ha observado una resolución de alta profundidad en el rango de 1 a 2 nm para muestras mixtas de lípidos/péptidos17.
Otro campo de aplicación es la combinación de DINeC-MS con cromatografía de capa fina (TLC). Las placas TLC convencionales se pueden analizar directamente mediante DINeC-MS. Los espectros de masa dependientes de la posición se pueden adquirir a partir de las placas TLC y, por lo tanto, los cromatogramas específicos de la masa se pueden obtener de las placas TLC27. No es necesaria la re-elución de los analitos separados, diferente a TLC en combinación con ESI28,29. Tampoco se necesita ninguna matriz para la combinación DINeC-MS + TLC, a diferencia del acoplamiento de TLC con MALDI28,29.
La ionización por electrospray de desorción (DESI) es también un método de desorción/ionización suave para aplicaciones MS30,31. Las diferencias más llamativas entre DINeC y DESI son: la naturaleza cuantitativa de DINeC23,su compatibilidad con condiciones de vacío ultra alto (UHV), en particular la posibilidad de investigar muestras preparadas y transferidas en condiciones UHV sin romper el vacío23,así como la posibilidad de desorb eficientemente moléculas no polares19.
En principio, DINeC como fuente de desorción/ionización se puede acoplar a cualquier tipo de espectrómetro de masas. Sin embargo, la combinación con espectrometría de masas de trampa iónial presenta dos ventajas principales: en primer lugar, la anchura del pulso y la tasa de repetición de un haz de racimo pulsado típico corresponden muy bien al tiempo de acumulación discontinua, así como a la velocidad espectral de la trampa ióniona15,32. En segundo lugar, la naturaleza blanda del proceso DINeC conduce a la desorción de moléculas intactas. En combinación con las capacidades MSn de espectrometría de masas de trampa iónial, esto permite un análisis más completo de las muestras investigadas15.
Protocolo
NOTA: El protocolo se puede pausar en cualquier momento.
1. Preparación de sustratos
- Para muestras estándar, corte los sustratos de las obleas de silicio (espesor de aprox. 0,5 a 1 mm) en trozos de 1 x 1 cm2.
- Limpie los sustratos de Si en un baño de ultrasonido de etanol y acetona durante 15 minutos cada uno.
- Secar los sustratos en una corriente de gas nitrógeno seco.
2. Preparación de muestras
-
Preparación de muestras estándar
- Para muestras estándar, prepare la solución que contiene las moléculas de analito de acuerdo con la pregunta científica que debe abordarse. La concentración del analito debe ser de al menos 1 x 10-10 mol/L.
- Lanzamiento de 5 a 30 l de la solución de muestra en el sustrato. Dependiendo de la presión de vapor del disolvente, deje que la muestra se seque en condiciones ambientales o en un desecador hasta que todo el disolvente se haya evaporado y se haya formado una película seca. Dependiendo de la cantidad de sustancia aplicada, el espesor de la película puede estar entre varios diez m (inspección visual posible) y el régimen de monocapa a subcapa (por lo tanto, no detectable por el ojo).
- Monte las muestras en el soporte de la muestra (por ejemplo, utilizando cinta adhesiva o abrazaderas apretadas por tornillos, dependiendo de las muestras y las condiciones de vacío requeridas).
- Si es posible, monte además una muestra de referencia, como una película de micrometro de espesor de angiotensina II, en el soporte de la muestra.
-
Preparación alternativa de muestras
- Utilice esquemas alternativos de preparación de muestras, como la deposición de haz iónico de electrospray (ES-IBD) en el recubrimiento al vacío o a la inmersión en una solución respectiva, si procede.
- Monte las muestras que se deben preparar y transferir al vacío en el soporte de muestra DINeC antes de los pasos de preparación.
- Asegúrese de que las muestras recubiertas de inmersión estén secas después del paso final de preparación.
- Considere el esquema de preparación más simple. Por ejemplo, para la investigación de la tinta del resaltador, simplemente dibuje un punto en la superficie del sustrato.
3. Transferencia de muestras al espectrómetro de masas DINeC
-
Transferencia de muestras de las condiciones ambientales a la cámara DINeC evacuada
- Ventile el sistema de bloqueo de carga.
- Abra el bloqueo de carga y monte el soporte de muestra.
- Cierre el bloqueo de carga y bombee la cámara de bloqueo de carga a una presión inferior a 2 x 10-5 mbar.
- Abra la válvula a la cámara DINeC y transfiera el soporte de la muestra con la varilla de transferencia al manipulador principal. Fije el soporte de la muestra al manipulador.
- Retirar la varilla de transferencia y cerrar la válvula entre el bloqueo de carga y la cámara DINeC.
-
Transferencia de muestras desde el vacío a la cámara DINeC evacuada
- Utilice un contenedor de vacío transportable, que se puede conectar a la brida CF40 de la cámara DINeC. Transfiera las muestras, que se prepararon al vacío, con este recipiente sin romper el vacío. Asegúrese de que las muestras estén montadas en un soporte de muestra compatible con el manipulador utilizado en el sistema DINeC.
- Fije el recipiente de vacío transportable a la brida CF40 y bombee el volumen entre el contenedor y la cámara DINeC.
- Una vez que la presión haya caído por debajo de 2 x 10-5 mbar, abra las válvulas de compuerta a la cámara DINeC y al contenedor de vacío transportable y transfiera la muestra a la cámara DINeC al manipulador utilizando un bastón de tambaleo u otro sistema de transferencia con más de 50 cm de movimiento lineal.
- Retirar el sistema de transferencia y cerrar las dos válvulas de compuerta.
4. Preparación de la mezcla de gas
- Preparar una mezcla de aprox. 3% SO2 en helio evacuando primero los cilindros de gas del sistema de mezcla de gas durante 10 min.
- Llene los cilindros con SO2 hasta alcanzar una presión de 1 bar.
- Llene aún más los cilindros con helio hasta alcanzar una presión total de 30 bar.
ADVERTENCIA: Cuando se utiliza SO2, las precauciones de seguridad respectivas, como el almacenamiento de2 cilindros SO en armarios de gas designados, siempre deben cumplirse.
5. Preparación del espectrómetro de masas DINeC
- Abra la válvula entre el cilindro de gas y la boquilla. Ajuste la presión de la mezcla de gas SO2/He en el contorno del sistema de mezcla de gas a 15 bar.
- Establezca la posición del manipulador en la posición de la muestra de referencia.
- Para medir espectros de masa catiónicos, establezca el sesgo de la muestra y la rejilla en +40 y +7 V, respectivamente.
- Para conducir la boquilla pulsada y el espectrómetro de masas de trampa de iones, ajuste el generador de funciones externas a 2 Hz. Con el generador de retardo, ajuste el retardo de tiempoentre la señal de trampa clara de la trampa iónca y la señal de disparo para la boquilla pulsada a 5 ms.
- En el software de control, ajuste los siguientes parámetros pulsando los botones respectivos o escribiendo los valores respectivos en la página Modo de la ventana de diálogo principal: Modo de escaneo:Resolución mejorada, Rango: m/z 50 – 3000, Accu-time: 0.1 ms, Promedio: 10 ciclos, Polaridad: positivo para la medición de espectros de masa catiónicos
NOTA: Para medir los espectros aniónicos, el sesgo de la muestra y la rejilla tiene que ser negativo con respecto al suelo, la polaridad tiene que cambiarse a Negativo en el software de control.
6. Medición de espectros de masa
- Una vez que se ha alcanzado una presión por debajo de 3 x 10-6 mbar en la cámara DINeC, se puede iniciar la medición. Inicie la medición pulsando primero Stand by y, a continuación, pulsando Operar en el software de control. Comience a grabar las medidas pulsando el botón Reproducir.
- Mida un espectro de prueba a partir de una muestra de referencia como angiotensina II durante unos 300 s. Siga la dependencia temporal de la señal utilizando el Cromatograma establecido en el valor m/z respectivo. Optimice la intensidad de la señal mediante el ajuste del retardo de tiempoentre la señal de trampa clara y la señal que activa la boquilla pulsada.
- Mueva el manipulador a la posición de la muestra que se va a medir. Optimice la intensidad de la señal mediante el ajuste de la posición de la muestra dentro del plano del soporte de la muestra.
- Adquiera espectros de masas durante el intervalo de tiempo de interés. Modifique los parámetros experimentales como la temperatura de la muestra o la presión de fondo en la cámara de acuerdo con los detalles del experimento. Continúe tomando espectros de masas al variar los parámetros experimentales.
7. Evaluación de datos
- Una vez finalizada la medición, cargue el conjunto de datos respectivo en el programa Análisis de datos. Seleccione el intervalo de tiempo de interés en el cromatograma con el botón derecho del ratón. El espectro promediado se mostrará en una ventana separada.
- Exporte el espectro como un archivo de datos para su posterior procesamiento en un programa de elección.
Resultados
A continuación, se presentan dos ejemplos para la aplicación en tiempo real de DINeC-MS. La Figura 4 muestra el cambio del espectro de masa obtenido a partir de la angiotensina II cuando la muestra se calienta a aprox. 140 oC. Cuando se alcanza la temperatura final(Figura 4B, Figura 4E), el espectro se caracteriza por un pico adicional que indica la pérdida de una entidad H2O (m/z a 1029). Al mantener la muestra a esa temperatura, se observa una mayor descomposición de las moléculas de angiotensina II (Figura 4C), incluida la pérdida de una de las unidades de aminoácidos terminales, ácido aspártico (pico en m/z a 932, Figura 4D). El análisis cuantitativo de los datos permite evaluar la cinética de reacción subyacente(Figura 4E). En particular, la Figura 4E demuestra que la entidad con m/z a 1029 es un intermedio que se descompone aún más en fragmentos más pequeños a medida que la intensidad aumenta primero y luego disminuye. Las constantes de velocidad concomitantes están en el mismo orden de magnitud.
Como segundo ejemplo, la investigación del intercambio de hidrógeno/deuterio en angiotensina II22 se ilustra en la Figura 5. Tras la exposición de la muestra de angiotensina a D2O en la cámara DINeC (pD2O a 10-4 mbar), el patrón isotópico de angiotensina II se amplía y se desplaza hacia valores m/z más altos que indican el intercambio de átomos H por D. El proceso es rápido durante los primeros 60 s, pero se ralentiza significativamente en el curso posterior del experimento: El patrón de isótopos en la Figura 5B cubre un amplio rango m/z (aproximadamente 15 unidades m/z). Cuando definimos el grado de deuterización d como el número de átomos H intercambiados en una molécula, los valores de d entre d a 0 a d a 13 se pueden extraer del espectro. En la Figura 5C, el patrón de isótopos se reduce de nuevo en anchura. Esta observación se puede atribuir a la intensidad fuertemente reducida de los picos que están asociados con los grados más bajos de deuteración. En la Figura 5D,el espectro se muestra para tiempos de reacción aún más largos. El rango m/z cubierto sigue siendo casi el mismo, pero el centro de masa del espectro todavía cambia lentamente hacia el aumento de los valores m/z. Para los largos tiempos de exposición, una parte de las moléculas alcanza el grado más alto de deuteración, dmáx. 17. Corresponde al número máximo de átomos H intercambiables, dado por el número de átomos H unidos a grupos funcionales como ácidos carboxílicos o grupos de aminas.
Ya a partir de la evolución temporal de los espectros, se puede deducir que el intercambio H/D tiene lugar con diferentes constantes de velocidad. Para una descripción cuantitativa de esta observación, el grado medio de deuteración de d á se traza en la Figura 5E en función del tiempo. La inspección de los resultados experimentales (símbolos) revela tres regímenes diferentes: un rápido aumento de d -operative para t < 50 s, un régimen intermedio para 50 s < t < 200 s, y un aumento lento pero casi continuo para t > 200 s. Los resultados experimentales fueron simulados por medio de simulaciones de Monte Carlo; se asumió la cinética de reacción de pseudo-primer orden con constantes de reacción ki para el intercambio H/D en los grupos funcionales de las moléculas investigadas22. Sólo se obtuvo un buen acuerdo entre las simulaciones y los resultados experimentales en los tres regímenes cuando se aplicaron al menos tres constantes de velocidad diferentes ki para el intercambio de H/D en moléculas de angiotensina II. En la Figura 5F,G, se muestra la cinética como se deduce de ajustar los patrones experimentales de isótopos por la suma de patrones de isótopos para diferentes grados de deuteración(Figura 5A a Figura 5D). Se observa claramente el buen acuerdo entre los datos experimentales y las simulaciones, así como los muy diferentes tipos de cambio para los grados bajos y altos de deuteración. En comparación con diferentes oligopéptidos como la hexaglicina, los tipos de cambio rápidos se atribuyeron a los grupos funcionales explícitos, mientras que los tipos de cambio lentos se asociaron con los grupos de amida de la columna vertebral del péptido22.
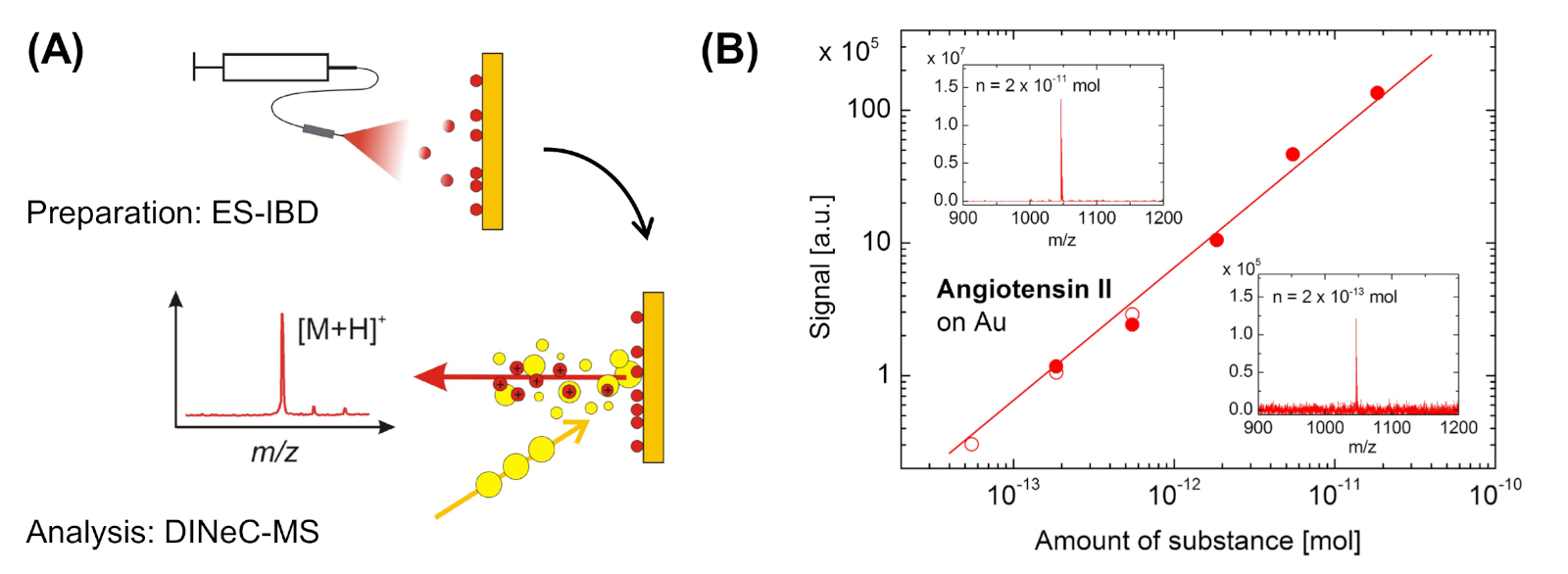
Mientras que estos dos primeros ejemplos se midieron con muestras de angiotensina II de espesor micrómetro, la Figura 6 muestra los resultados obtenidos de la cobertura de submonocapa de angiotensina II en muestras de oro preparadas mediante deposición de haz iónico de electrospray (ES-IBD)23. Se observa una dependencia lineal de la intensidad de la señal en la cantidad de sustancia en 3 órdenes de magnitud, la menor cantidad de sustancia detectada corresponde al 0,1% de una monocapa de moléculas de angiotensina II en la superficie de oro. Los experimentos de intercambio de H/D como se muestra en la Figura 5 también se realizaron con angiotensina II sobre oro en el régimen de submonocapa23.

Figura 4: Observación en tiempo real de la degradación térmica de la angiotensina II. (A-C) Espectros de masa obtenidos después de la desorción/ionización inducida por racimos de una muestra de angiotensina II. (A) Muestra fresca a RT. (B) Muestra calentada a aprox. 140 oC. Además del pico en m/z a 1047, que se asocia con la molécula intacta [M+H]+, aparecen picos en m/z a 932, 1012 y 1029 (indicados por flechas). (C) Estos últimos picos aumentan y el pico principal disminuye con el tiempo al mantener la muestra a temperatura elevada. (D) Fórmula estructural de la angiotensina II que indica el fragmento (corchetes marrones) que conduce a la aparición del pico en m/z a 932 por la pérdida de una unidad de aminoácidos (ácido aspártico). (E) Dependencia del tiempo de la temperatura de la muestra y la intensidad de los picos principales indicados en las parcelas (A) a (C). Las líneas sólidas son guías para el ojo. Haga clic aquí para ver una versión más grande de esta figura.
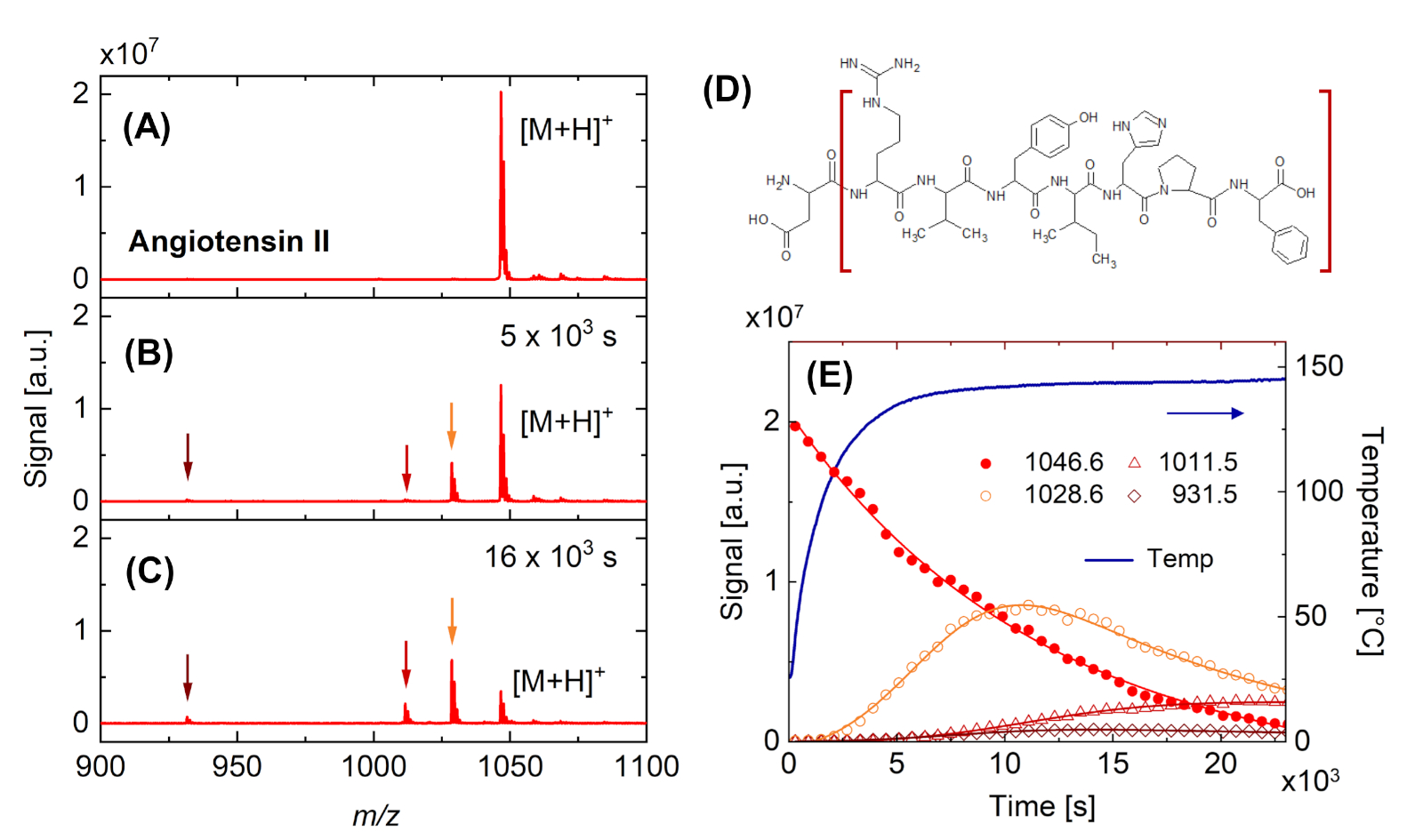{kind=link}

Figura 5: Observación en tiempo real del intercambio H/D en angiotensina II. (A-D) Debido al intercambio de H/D, el patrón de isótopos se ensancha y se desplaza hacia valores más altos de m/z en (B) a (D) en comparación con el patrón de isótopos de las especies no diuteadas que se muestra en (A). Las líneas rojas son datos, las líneas cian discontinuas se ajustan a los datos teniendo en cuenta diferentes grados de deuteración. (E) Grado medio de deuteración d - en función del tiempo, como se deduce de los experimentos (puntos abiertos). Además, se muestra la función de tiempo deducida por medio de simulaciones de Monte Carlo. Curva discontinua negra: simulaciones teniendo en cuenta una constante de tasa (k1); curva roja: teniendo en cuenta tres constantes de velocidad(k1, k2, k3). (F,G) Intensidades de señal relativas de determinados grados de deuteración de la angiotensina II (símbolos + líneas sólidas) en función del tiempo junto con los resultados correspondientes de las simulaciones de Monte Carlo (líneas discontinuas). Esta cifra se ha modificado a partir de la referencia 22. Haga clic aquí para ver una versión más grande de esta figura.
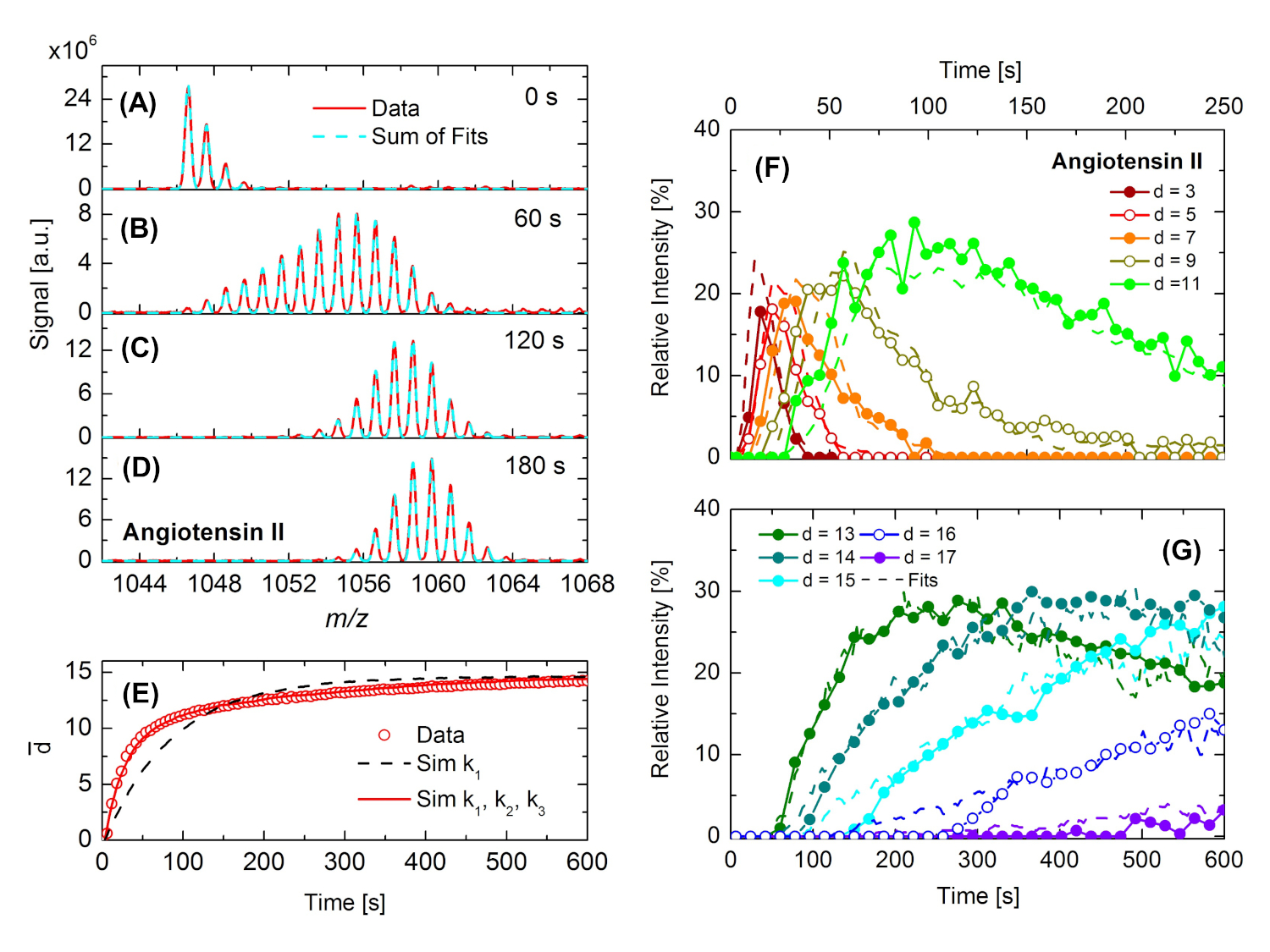{kind=link}

Figura 6: Aplicación de DINeC-MS a la angiotensina II sobre el oro en el régimen de submonocapa. (A) Representación esquemática de la combinación de ES-IBD para deposición y DINeC-MS para espectrometría de masas de moléculas aisladas de angiotensina II en el régimen de submonocapa. (B) Dependencia de la intensidad de la señal en la sustancia de importe depositada en la muestra obtenida a partir de dos conjuntos independientes de datos (símbolos llenos y abiertos). Inserciones: espectros de masa DINeC obtenidos a partir de muestras en las que se depositó una cantidad de sustancia como se indica. Esta cifra se ha modificado a partir de la referencia 23. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
En muchos estudios realizados hasta ahora, se ha demostrado una alta sensibilidad de DINeC-MS en diversas sustancias. De hecho, esto permite mediciones de analitos hasta una cantidad de sustancia en el régimen de femtomole11. Debido a esta alta sensibilidad, la preparación de la muestra, en particular la limpieza del sustrato, debe realizarse con productos químicos altamente puros para evitar la contaminación en los espectros de masa DINeC. Como es el caso de muchas técnicas de análisis, una medición de fondo adecuada de un sustrato en blanco ayuda a separar los picos del analito y los picos que tienen su origen en la preparación del sustrato/muestra.
Aunque hemos demostrado que la probabilidad de ionización de una molécula de analito determinada no está fuertemente influenciada por la presencia de co-adsorbatos o coconstituyentes en muestras mixtas17,24, la probabilidad de ionización puede variar de sustancia a sustancia13. Por lo tanto, es aún más importante trabajar en condiciones limpias, ya que los contaminantes, dependiendo de su probabilidad de ionización, pueden contribuir a la señal mucho más fuerte que el analito. Los iones preformados (por ejemplo, como se encuentran en el caso de muchas moléculas de tinte), o moléculas con grupos funcionales que muestran una clara tendencia hacia la aceptación de protones o la desprotonación (es decir, bases o ácidos), suelen mostrar una alta probabilidad de ionización en DINeC-MS. Si no hay tal grupo funcional presente en el analito, la probabilidad de ionización puede ser baja. Las muestras pueden ser tratadas por agentes ionizantes como el ácido trifluoro (por ejemplo, mediante la exposición de la muestra a la presión de vapor del agente ionizante).
Los resultados representativos examinados en la Figura 4 y en la Figura 5 demuestran la aplicabilidad de DINeC-MS para investigaciones en tiempo real de reacciones químicas mediante espectrometría de masas. La Figura 6 ilustra la sensibilidad de la submonocapa del método. Si se combinan las dos propiedades, las reacciones químicas en las superficies y sus productos se pueden seguir en tiempo real23. Esto puede ser en particular de interés para la llamada "síntesis en superficie" que conduce al montaje de estructuras macromoleculares en superficies3,33,34,35,36. En la configuración actual, la observación de tales reacciones superficiales es posible en superficies con menor reactividad como el oro23 y otros metales nobles; los experimentos son más difíciles de realizar en superficies altamente reactivas como superficies de silicio37,ya que la presión base en la cámara de desorción está en el rango de 10-7-mbar. Las actividades actuales abordan esta limitación y se está construyendo un aparato DINeC compatible con UHV. En el caso de superficies reactivas, la interacción entre SO2 y la superficie del sustrato debe probarse antes de las mediciones de adsorbatos superficiales y reacciones superficiales.
Como el haz de racimo es neutro, no se puede enfocar. De este modo, el tamaño del haz en la muestra se indica mediante la geometría de la configuración y el orificio del skimmer en uso; los valores típicos para el diámetro de la viga en la muestra son de uno a varios milímetros. Como resultado, la toma de imágenes mediante el escaneo de la muestra es posible sólo con muy baja resolución. Por otro lado, dada por la alta probabilidad de ionización13,DINeC hace uso eficiente mente de las moléculas desorbidas. Por lo tanto, una combinación de DINeC-MS y un detector de imágenes ióniales38 parece ser muy atractiva.
Divulgaciones
Los autores declaran que no tienen nada que revelar.
Agradecimientos
Los autores reconocen el apoyo financiero del Helmholtz International Center for FAIR (HICforFAIR) y de la Helmholtz Graduate School for Hadron and Ion Research (P.S.). Los autores agradecen al profesor Rauschenbach (Universidad de Oxford) y a su equipo por su fructífera colaboración en experimentos combinados ES-IBD/DINeC.
Materiales
| Name | Company | Catalog Number | Comments |
| Acetone rotisolv HPLC | Roth | 7328.2 | HPLC Gradient Grade |
| Copper tape | |||
| Ethanol rotisolv HPLC | Roth | p076.1 | HPLC Gradient Grade |
| Helium | Praxair | 4800086706 | Purity 99.9999% |
| Nitrogen | Praxair | 40728408 | Purity 99.5 - 100% |
| Silicon Wafers | Active Business Company GmbH | G60007 | |
| Sulfur dioxide | Air Liquide | P1734S10R0A001 | Purity 99.98% |
| Water rotisolv LC-MS | Roth | HN43.1 | Ultra LC-MS |
Referencias
- Vickerman, J. C., Gilmore, I. Surface Analysis: The Principal Techniques. , 2nd ed, John Wiley & Sons. New York. (2009).
- Reutzel, M., Münster, N., Lipponer, M. A., Länger, C., Höfer, U., Koert, U., Dürr, M. Chemoselective Reactivity of Bifunctional Cyclooctynes on Si(001). Journal of Physical Chemistry C. 120, 26284-26289 (2016).
- Grill, L., Dyer, M., Lafferentz, L., Persson, M., Peters, M., Hecht, S. Nano-architectures by covalent assembly of molecular building blocks. Nature Nanotechnol. 2, 687-691 (2007).
- Stutzmann, M., Garrido, J. A., Eickhoff, M., Brandt, M. S. Direct biofunctionalization of semiconductors: A survey. Physica Status Solidi A. 203, 3424-3437 (2006).
- Adler-Abramovich, L., Gazit, E. The physical properties of supramolecular peptide assemblies: from building block association to technological applications. Chemical Society Reviews. 43, 6881-6893 (2014).
- Vickerman, J. C., Briggs, D. TOF-SIMS: Materials Analysis by Mass Spectrometry, 2nd ed. , IM Publications. Chichester, UK. (2013).
- Winograd, N. The magic of cluster SIMS. Analytical Chemistry. 77, 142-149 (2005).
- Ichiki, K., Ninomiya, S., Nakata, Y., Honda, Y., Seki, T., Aoki, T., Matsuo, J. High Sputtering Yields of Organic Compounds by Large Gas Cluster Ions. Applied Surface Science. 255, 1148-1150 (2008).
- Mochiji, K., Hashinokuchi, M., Moritani, K., Toyoda, N. Matrix-free Detection of Intact Ions from Proteins in Argon-Cluster Secondary Ion Mass Spectrometry. Rapid Communications in Mass Spectrometry. 23, 648-652 (2009).
- Yokoyama, Y., Aoyagi, S., Fujii, M., Matsuo, J., Fletcher, J. S., Lockyer, N. P., Vickerman, J. C., Passarelli, M. K., Havelund, R., Seah, M. P. Peptide Fragmentation and Surface Structural Analysis by Means of ToF-SIMS Using Large Cluster Ion Sources. Analytical Chemistry. 88, 3592-3597 (2016).
- Gebhardt, C. R., Tomsic, A., Schröder, H., Durr, M., Kompa, K. L. Matrix-Free Formation of Gas-Phase Biomolecular Ions by Soft Cluster-Induced Desorption. Angewandte Chemie, International Edition. 48, 4162-4165 (2009).
- Baur, M., Lee, B. J., Gebhardt, C. R., Durr, M. Soft Clusterinduced Desorption and Ionization of Biomolecules - Influence of Surface Load and Morphology on Desorption Efficiency. Applied Physics Letters. 99, 234103(2011).
- Lee, B. J., Baur, M., Gebhardt, C. R., Durr, M. Quantification of the Ionization Probability During Desorption/Ionization of Oligopeptides Induced by Neutral Cluster Impact. Rapid Communications in Mass Spectrometry. 27, 1090-1094 (2013).
- Lee, B. J., Gebhardt, C. R., Schroder, H., Kompa, K. L., Durr, M. Observation of Ionic Desorption Channels in Cluster-induced Desorption of Alkali Halides - Influence of Surface Electronic Properties and Surface Configuration. Chemical Physics Letters. 556, 77-81 (2013).
- Baur, M., Gebhardt, C. R., Durr, M. Desorption/Ionization Induced by Neutral Cluster Impact as a Soft and Efficient Ionization Source for Ion Trap Mass Spectrometry of Biomolecules. Rapid Communications in Mass Spectrometry. 28, 290-296 (2014).
- Kley, C. S., Dette, C., Rinke, G., Patrick, C. E., Cechal, J., Jung, S. J., Baur, M., Durr, M., Rauschenbach, S., Giustino, F., Stepanow, S., Kern, K. Atomic-Scale Observation of Multiconformational Binding and Energy Level Alignment of Ruthenium-Based Photosensitizers on TiO2 Anatase. Nano Letters. 14, 563-569 (2014).
- Portz, A., Aoyagi, S., Durr, M. Soft depth-profiling of mixed peptide/lipid samples by means of cluster induced desorption/ionization mass spectrometry - high depth resolution and low matrix effect. Biointerphases. 13, 03B405(2018).
- Portz, A., Baur, M., Gebhardt, C. R., Frank, A. J., Neuderth, P., Eickhoff, M., Durr, M. Influence of the Cluster Constituents' Reactivity on the Desorption/Ionization Process Induced by Neutral SO2 Clusters. Journal of Chemical Physics. 146, 134705(2017).
- Schneider, P., Durr, M. Cluster-induced desorption investigated by means of molecular dynamics simulations - Microsolvation in clusters of polar and non-polar constituents. Journal of Chemical Physics. 150, 214301(2019).
- Karas, M., Hillenkamp, F. Laser Desorption Ionization of Proteins with Molecular Masses Exceeding 10 000 Daltons. Analytical Chemistry. 60, 2299-2301 (1988).
- Buchberger, A. R., DeLaney, K., Johnson, J., Li, L. Mass Spectrometry Imaging: A Review of Emerging Advancements and Future Insights. Analytical Chemistry. 90, 240-265 (2018).
- Portz, A., Gebhardt, C. R., Durr, M. Real-Time Investigation of the H/D Exchange Kinetics of Porphyrins and Oligopeptides by Means of Neutral Cluster-Induced Desorption/Ionization Mass Spectrometry. Journal of Physical Chemistry B. 121, 11031-11036 (2017).
- Portz, A., Baur, M., Rinke, G., Abb, S., Rauschenbach, S., Kern, K., Dürr, M. Chemical Analysis of Complex Surface-Adsorbed Molecules and Their Reactions by Means of Cluster-Induced Desorption/Ionization Mass Spectrometry. Analytical Chemistry. 90, 3328(2018).
- Portz, A., Baur, M., Gebhardt, C. R., Durr, M. Mass Spectrometry of Oligopeptides in the Presence of Large Amounts of Alkali Halides Using Desorption/Ionization Induced by Neutral Cluster Impact. Biointerphases. 11, 02A316(2016).
- Shard, A. G., Spencer, S. J., Smith, S. A., Havelund, R., Gilmore, I. S. International Journal of Mass Spectrometry. 377, 599-609 (2015).
- Nakano, S., Yamagishi, T., Aoyagi, S., Portz, A., Durr, M., Iwai, H., Kawashima, T. Evaluation of Matrix Effects on TOF-SIMS Data of Leu-enkephalin and DOPC Mixed Samples. Biointerphases. 13, 03B403(2018).
- Heep, J., Tuchecker, P. H. K., Gebhardt, C. R., Dürr, M. Coupling of planar chromatography to mass spectrometry. ACS Omega. 4, 22426-22430 (2019).
- Morlock, G., Schwack, W. Coupling of planar chromatography to mass spectrometry. Trends in Analytical Chemistry. 29, 1157-1171 (2010).
- Cheng, S. C., Huang, M. Z., Shiea, J. Thin layer chromatography/mass spectrometry. Journal of Chromatography A. 1218, 2700-2711 (2011).
- Takats, Z., Wiseman, J. M., Gologan, B., Cooks, R. G. Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science. 306, 471(2004).
- Cooks, R. G., Ouyang, Z., Takats, Z., Wiseman, J. M. Ambient mass spectrometry. Science. 311, 1566(2006).
- Dürr, M., Gebhardt, C. Ion generation in mass spectrometers by cluster bombardment. US Patent. , 926322 B2 (2019).
- Lindner, R., Kuhnle, A. Bottom-up Assembly of Molecular Wagons on a Surface. ChemPhysChem. 16, 1582-1592 (2015).
- Dong, L., Liu, P. N., Lin, N. Bottom-up Assembly of Molecular Wagons on a Surface. Accounts of Chemical Research. 48, 2765-2774 (2015).
- Björk, J. Reaction mechanisms for on-surface synthesis of covalent nanostructures. Journal of Physics: Condensed Matter. 28, 083002(2016).
- Rauschenbach, S., Rinke, G., Gutzler, R., Abb, S., Albarghash, A., Le, D., Rahman, T. S., Durr, M., Harnau, L., Kern, K. Two-Dimensional Folding of Polypeptides into Molecular Nanostructures at Surfaces. ACS Nano. 11, 2420-2427 (2017).
- Dürr, M., Höfer, U. Dissociative adsorption of molecular hydrogen on silicon surfaces. Surface Science Reports. 61, 465-526 (2006).
- Zhang, J., Franzreb, K., Aksyonov, S. A., Williams, P. Mass Spectra and Yields of Intact Charged Biomolecules Ejected by Massive Cluster Impact for Bioimaging in a Time-of-Flight Secondary Ion Microscope. Analytical Chemistry. 87, 10779-10784 (2015).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados