
Method Article
Preparación de muestras TMT para la presentación de instalaciones proteómicas y análisis de datos posteriores
En este artículo
Resumen
Presentamos un protocolo de etiquetado optimizado de etiquetas de masa en tándem (TMT) que incluye información detallada para cada uno de los siguientes pasos: extracción de proteínas, cuantificación, precipitación, digestión, etiquetado, envío a una instalación proteómica y análisis de datos.
Resumen
Las tecnologías proteómicas son metodologías poderosas que pueden ayudar a nuestra comprensión de los mecanismos de acción en los sistemas biológicos al proporcionar una visión global del impacto de una enfermedad, tratamiento u otra condición en el proteomo en su conjunto. Este informe proporciona un protocolo detallado para la extracción, cuantificación, precipitación, digestión, etiquetado y posterior análisis de datos de muestras de proteínas. Nuestro protocolo de etiquetado TMT optimizado requiere una menor concentración de etiquetas de etiquetas y logra datos fiables de forma consistente. Hemos utilizado este protocolo para evaluar perfiles de expresión de proteínas en una variedad de tejidos de ratón (es decir, corazón, músculo esquelético y cerebro), así como células cultivadas in vitro. Además, demostramos cómo evaluar miles de proteínas del conjunto de datos resultante.
Introducción
El término "proteómica" se definió por primera vez como la caracterización a gran escala de todo el complemento proteico de una célula, tejido u organismo1. Los análisis proteómicos permiten la investigación de mecanismos y procesos celulares implicados en el desarrollo de enfermedades, vías terapéuticas y sistemas saludables utilizando técnicas para realizar la cuantificación relativa de los niveles de expresión de proteínas2. Las descripciones iniciales de dichos estudios se publicaron en 1975 y demostraron el uso de electroforesis de gel de poliacrilamida bidimensional (2D-PAGE) para este fin1,3. El método 2D separa las proteínas en función de la carga (enfoque isoeléctrico, IEF) y la masa molecular (electroforesis de gel de sulfato de dodecilo sódico, o SDS-PAGE)4. Durante años, la combinación de 2D-PAGE y la posterior espectrometría de masas en tándem realizada en cada componente de gel fue la técnica de análisis de expresión de proteína no segmentada más común realizada e identificó numerosos perfiles de expresión de proteínas previamente desconocidos5,,6. Las desventajas generales del enfoque 2D-PAGE son que consume mucho tiempo, no funciona bien para las proteínas hidrofóbicas, y hay limitaciones en el número total de proteínas evaluadas debido a la baja sensibilidad7,8.
El etiquetado de isótopos estables por aminoácidos en el cultivo celular (SILAC) método se convirtió en el siguiente enfoque popular para identificar y cuantificar la abundancia de proteínas en las muestras9. Consiste en el etiquetado metabólico de las células que se incuban en medio carente de un aminoácido esencial estándar y complementadas con una versión etiquetada por isótopos de ese aminoácido específico10. La ventaja de esta técnica es su eficiencia y etiquetado preciso9. La principal limitación al enfoque SILAC es principalmente la reducción de la tasa de crecimiento celular causada por la incorporación de etiquetas de isótopos, que puede ser particularmente difícil en líneas celulares relativamente sensibles que modelan enfermedades humanas11.
En 2003, se introdujo en el campo12una técnica novedosa y robusta de proteómica que implica etiquetas isobáricas de etiqueta de masa en tándem (TMT). El etiquetado TMT es un método potente debido a su mayor sensibilidad para detectar los niveles relativos de expresión de proteínas y las modificaciones posttranslacionales13. A partir de esta fecha de publicación, se han desarrollado kits de TMT que pueden etiquetar simultáneamente 6, 10, 11 o 16 muestras. Como resultado, es posible medir la abundancia de péptidos en múltiples condiciones con réplicas biológicas al mismo tiempo14,15,16. Recientemente utilizamos TMT para caracterizar el perfil proteómico cardíaco de un modelo de ratón de Síndrome de Barth (BTHS)17. Al hacerlo, pudimos demostrar una mejora generalizada en los perfiles cardíacos de los ratones BTHS tratados con terapia génica e identificar proteínas novedosas afectadas por BTHS que revelaron nuevas vías terapéuticas implicadas en cardiomiopatías.
Aquí, describimos un método detallado para realizar análisis proteómicos cuantitativos TMT multiplex utilizando muestras de tejido o gránulos celulares. Puede ser beneficioso realizar la preparación y etiquetado de la muestra antes de enviarla a un núcleo porque los péptidos trípticos etiquetados son más estables que las muestras congeladas en bruto, no todos los núcleos tienen experiencia en el manejo de todos los tipos de muestras, y la preparación de muestras en un laboratorio puede ahorrar tiempo para los núcleos, que a menudo tienen largos retrasos. Para obtener descripciones detalladas de la parte de espectroscopia de masas de este proceso, consulte Kirshenbaum et al. y Perumal et al.18,19.
El protocolo de preparación de la muestra consta de los siguientes pasos principales: extracción, cuantificación, precipitación, digestión y etiquetado. Los principales beneficios de este protocolo optimizado son que reduce los costos de etiquetado, mejora la extracción de proteínas y genera constantemente datos de alta calidad. Además, describimos cómo analizar datos TMT para analizar miles de proteínas en un corto período de tiempo. Esperamos que este protocolo anime a otros grupos de investigación a considerar la incorporación de esta poderosa metodología en sus estudios.
Protocolo
El Comité Institucional de Cuidado y Uso de Animales de la Universidad de Florida aprobó todos los estudios con animales.
1. Preparación de reactivos
- Preparar el búfer de lysis CHAPS (150 mM KCl, 50 mM de pH HEPES a 7,4, 0,1% de CHAPS y 1 comprimido de cóctel inhibidor de la proteasa por cada 50 ml de tampón). El tampón sin inhibidores de la proteasa se puede almacenar a 4 oC durante un máximo de 6 meses o tampón con inhibidor de la proteasa almacenado a -20 oC hasta 1 año.
- Preparar bicarbonato de trietimonio de 100 mM (TEAB): Añadir 500 l de 1 M TEAB a 4,5 ml de agua ultrapura.
- Preparar 200 mM de clorhidrato de fosfina (2-carbiretilo) (TCEP): Añadir 70 l de 0,5 M de TCEP, un reactivo desnaturalista, a 70 l de agua ultrapura. A continuación, agregue 35 l del TEAB de 1 M.
- Preparar 5% de hidroxiamina: Añadir 50 l de 50% de hidroxiamina a 450 l de 100 mM TEAB.
2. Extracción de proteínas
- Aísle el músculo del cuádriceps de un ratón eutanizado de acuerdo con un protocolo aprobado por la IACUC. Congele y mantenga a -80 oC o continúe con el protocolo para su uso inmediato.
- Corte para aislar aproximadamente 10 mg de tejido de ratón de cuádriceps fresco o congelado. Separe las fibras usando pinzas cuando trabaje con músculo esquelético. Alternativamente, si se trabaja con cultivos de células, resuspender 3 x 106 celdas en 300 l de búfer de lelisis CHAPS y saltar al paso 2.4.
- Homogeneizar el tejido utilizando un disruptor de perlas utilizando tubos de 2 ml llenos de aproximadamente 200 l de perlas de zirconia/sílice de 1 mm y 500 ml de tampón de lelisis CHAPS. Escalar hacia arriba o hacia abajo según corresponda (p. ej., 5 mg de tejido en 250 ml de tampón de lelisis CHAPS).
- Realizar sonicación (10x para 10 s cada una con 50% de amplitud y 30 s intervalos en hielo) para liberar proteína unida al ADN. Los mismos resultados de degradación del ADN se pueden lograr ya sea con la lisis de jeringa pasando el lisato 10x a través de una aguja de 21 G unida a una jeringa de 1 ml, o mediante incubación de benzonasa (44 U/ml) a 37 oC durante 30 min.
- Centrifugar el izado a 16.000 x g durante 10 min a 4 oC y transfiera el sobrenadante a un nuevo tubo de centrífuga.
3. Medición de proteínas
- Determinar la concentración proteica del sobrenadante utilizando protocolos establecidos (ver Tabla de Materiales).
NOTA: Lo mejor es utilizar muestras a 2 g/l, pero también se pueden utilizar muestras menos concentradas. Si se utiliza una muestra menos concentrada, será necesario ajustar adecuadamente los volúmenes de los reactivos reductores/alquilantes en el paso 5.1. - Prepare una dilución de curva estándar BSA utilizando el búfer de lelisis CHAPS.
- Siga las instrucciones del fabricante, y después de 15 minutos, lea la absorbancia a 750 nm.
4. Reducción/alquilación del tratamiento con reactivos
- Transfiera 200 g de proteína por condición a un nuevo tubo centrífugo y ajútese a un volumen final de 100 l utilizando el tampón de lelisis CHAPS. Es posible escalar hasta 200 l cuando la concentración de proteína es demasiado baja, pero no se olvide de ajustar adecuadamente el volumen de reactivo reductor/alquilante.
- Añadir 5 l del TCEP de 200 mM e incubar muestras a 55oC durante 1 h.
- Inmediatamente antes de su uso, preparar 375 mM de yodoacetamida disolviendo un tubo de iodoacetamida (es decir, 9 mg) en 132 l de 100 mM TEAB. Proteja esta solución de la luz.
- Añadir a la muestra 5 l de iodoacetamida de 375 mM e incubar durante 30 minutos a temperatura ambiente (RT) protegida de la luz.
5. Precipitación de metanol/cloroformo20
- Añadir 400 l de metanol a cada 100 l de proteína y tomar muestras brevemente de vórtice.
- Centrífuga a 9.000 x g durante 10 s en RT. Esto es para incorporar líquidos depositados en los lados del tubo de muestra.
- Añadir 100 l de cloroformo a la mezcla y brevemente vórtice. Utilice 200 l de cloroformo si la muestra tiene una alta concentración de fosfolípidos.
- Centrífuga a 9.000 x g durante 10 s en RT. Esto es para incorporar líquidos depositados en los lados del tubo de muestra.
- Añadir 300 l de agua y vórtice vigorosamente. Es importante obtener una solución homogénea.
- Centrifugar a 9.000 x g durante 1 min en RT. Tenga mucho cuidado de evitar molestar las capas al transferir el tubo a un bastidor.
NOTA: El tubo debe contener ahora tres fases: 1) Capa superior (es decir, sobrenadante), una mezcla de agua y metanol; 2) Capa media (es decir, interfase), proteína blanca precipitada; y 3) Capa inferior (es decir, fase inferior), cloroformo. - Retire con cuidado el sobrenadante.
- Añadir 300 l de metanol a la fase inferior y de interfase restante. Vórtice vigorosamente.
- Centrifugar a 9.000 x g durante 2 minutos en RT. Tenga mucho cuidado de evitar molestar las capas al transferir el tubo a un bastidor.
- Retire con cuidado el sobrenadante.
- Aspirar suavemente tanto líquido como sea posible bajo una corriente de aire (por ejemplo, usando un concentrador de vacío) en RT hasta que el pellet esté un poco húmedo (10 min). Como el tiempo necesario puede ser diferente para cada muestra, compruebe cada 2 minutos para evaluar. Conservar el pellet a -80 oC hasta su posterior procesamiento.
6. Digestión de proteínas
- Resuspender el pellet proteico precipitado en 100 l de tampón de lelisis TEAB.
NOTA: Es opcional medir la concentración de proteínas en este paso. - Inmediatamente antes de su uso, preparar 1 g/l de tripsina añadiendo 100 l de la solución de almacenamiento de tripina (ácido acético de 50 mM) en la parte inferior del vial de vidrio de trippsina de 100 g e incubar durante 5 minutos en RT. Almacene el reactivo restante en dosis de un solo uso a -80 oC.
- Añadir 2,5 ml de trippsina por cada 100 g de proteína. Digerir la muestra durante la noche a 37oC. Este paso es crucial para la solubilización completa de la proteína; no modifique estas condiciones. Después de la digestión, es opcional medir la concentración de proteínas utilizando ensayos proteicos estándar.
7. Etiquetado de péptidos
- Inmediatamente antes de su uso, equilibre los reactivos del kit de etiquetas TMT a RT.
- Disuelva cada uno de los viales de etiqueta TMT de 0,8 mg mediante la adición de 41 ml de acetonitrilo anhidro a cada tubo. Incubar el reactivo durante 5 minutos en RT con vórtices ocasionales. Centrifugar brevemente los tubos.
NOTA: Una concentración de 0,8 mg de etiqueta TMT suele ser suficiente para etiquetar dos conjuntos. Sin embargo, otros investigadores han demostrado que esta concentración puede reducirse aún más y seguir produciendo datos fiables15. - Añada con cuidado 41 l del reactivo de la etiqueta TMT a cada muestra de 100 l.
- Incubar la reacción durante 1 h en RT.
- Añadir 8 l de 5% de hidroxiamina a la muestra e incubar durante 15 minutos para apagar la reacción.
- Divida las muestras en cantidades iguales en un nuevo tubo centrífugo y guárdelos a -80 oC.
NOTA: En este paso las muestras son estables y se pueden enviar para espectroscopia de masas. Es opcional medir la concentración en este punto utilizando ensayos proteicos estándar.
8. Espectroscopia de masas
- Envíe las muestras a una instalación proteómica (este estudio utilizó la instalación UF ICBR Proteomics Core), donde todas las muestras se combinan y purifican utilizando columnas de espín C18.
NOTA: Discuta con la instalación principal cómo enviar las muestras antes de prepararlas para confirmar los pasos exactos que prefieren para los envíos. - Solicite los siguientes procedimientos por muestra multiplexada combinada: extracción de fase sólida, HPLC (SCX, SE), punta zip y LC-MS/MS (gradiente de 2 h para ID de proteína, si >10QE Plus).
- Una vez recopilados los datos, la instalación principal procesará los archivos RAW utilizando el software suministrado por el proveedor para la identificación de proteínas.
9. Análisis de datos
- Normalmente, los datos se entregan desde el núcleo al usuario en el formato 7z, que puede requerir aproximadamente 16 GB de espacio en disco por cada conjunto de datos (en este caso 11 ejemplos). Para el procesamiento de datos, asegúrese de que hay un equipo disponible que tenga al menos 3,4 GHz.
- Extraiga archivos utilizando el Administrador de archivos 7-Zip. Estos archivos extraídos contienen datos RAW, archivo de formato pdStudy y archivo de formato pdResultView. Guarde todos los archivos para análisis posteriores.
- Abra el archivo con El software Proteome Discovery 2.2.
NOTA: El formato de archivo es "Nombre de archivo.pdStudy". Si se abre el "Nombre de archivo.pdResultView" no es posible seleccionar el ejemplo de control. - Seleccione muestras de control en el panel "Muestras".
- Abra Resultado seleccionando ID en el panel " Resultadosdel análisis".
- Exportar a software de hoja de cálculo.
- Guardar datos sin procesar (todas las proteínas identificadas).
- Abra el archivo de software de hoja de cálculo. Esto contendrá todas las proteínas que se han identificado.
- En el archivo de software de hoja de cálculo utilice la función "Filtrar" para examinar "Proteína FDR Confianza: Combinado" en alto (columna B), "#Unique péptidos" superior a 2 (columna K), y cualquiera de "Relación de abundancia" en blanco exclusivamente ( columnaS hasta W).
- Inserte una columna para el cálculo "p-value" con la función
•TTEST (grupo de control, grupo experimental, colas, tipo) - Inserte una columna para "Significancia Estadística" con la función
•IF(p-value<0.05, "Significance","NS") - Utilice la función "Filtrar" para examinar "Significancia estadística" que muestra "Significancia". El resultado muestra las proteínas analizadas con la significancia estadística en el grupo de control y el grupo experimental.
- Determinar significativamente mayor o menor abundancia de expresión proteica en el grupo experimental en comparación con el grupo de control, insertar una columna para "Regulación" con la función
IF(AVERAGE(controlgroup)>AVERAGE(experimentalgroup),"Upregulated","Downregulated")
10. Métodos para evaluar impactos significativos
- Para identificar interacciones proteína-proteína entre los impactos significativos identificados en los estudios de TMT, utilice la herramienta de búsqueda para la recuperación de genes/proteínas que interactúan (STRING) versión 11.021: https://string-db.org/
- Para clasificar por grupos (es decir, función molecular, procesos biológicos y clases de proteínas) utilice el software de clasificación ontológica Análisis de proteínas a través de relaciones evolutivas (PANTHER)22: http://www.pantherdb.org/
- Para identificar interacciones proteicas en una variedad de vías, utilice el software de análisis de vías23.
11. Carga de datos proteómicos en un banco de repositorios
- Para enviar datos proteómicos a la base de datos de Proteomics IDEntificantions (PRIDE) o a Mass Spectrometry Interactive Virtual Environment (MassIVE) se incluyen la siguiente información: los archivos de lista pico (archivos de espectro de masa procesados en un formato estándar como mzXML, mzML, o MGF), archivos de resultados (identificaciones de espectro en un formato estándar como mzIdentML o mzTab) y archivos de espectro sin procesar (archivos de espectro de masas en bruto en un formato no estándar o específico del instrumento, como . RAW o . WIFF).
- Para enviar, cree una cuenta e incluya información como la afiliación y los detalles del proyecto. A continuación, seleccione los archivos enumerados en el paso 11.1 y cárguelos.
- Para crear un conjunto de datos oficial, ejecute un flujo de trabajo de envío en estos archivos cargados.
NOTA: Después del envío, el conjunto de datos será privado en el banco del repositorio. Con la opción privada, los datos solo están disponibles para usuarios autorizados. Hay dos opciones adicionales: 1) Conjunto de datos compartido, que da acceso a los revisores y colaboradores del diario; o 2) Conjunto de datos público, que aparecerá en las búsquedas de conjuntos de datos públicos. Otra característica importante de estos repositorios es la capacidad de actualizar los datos cargados y asociar publicaciones posteriores con el conjunto de datos existente.
Resultados
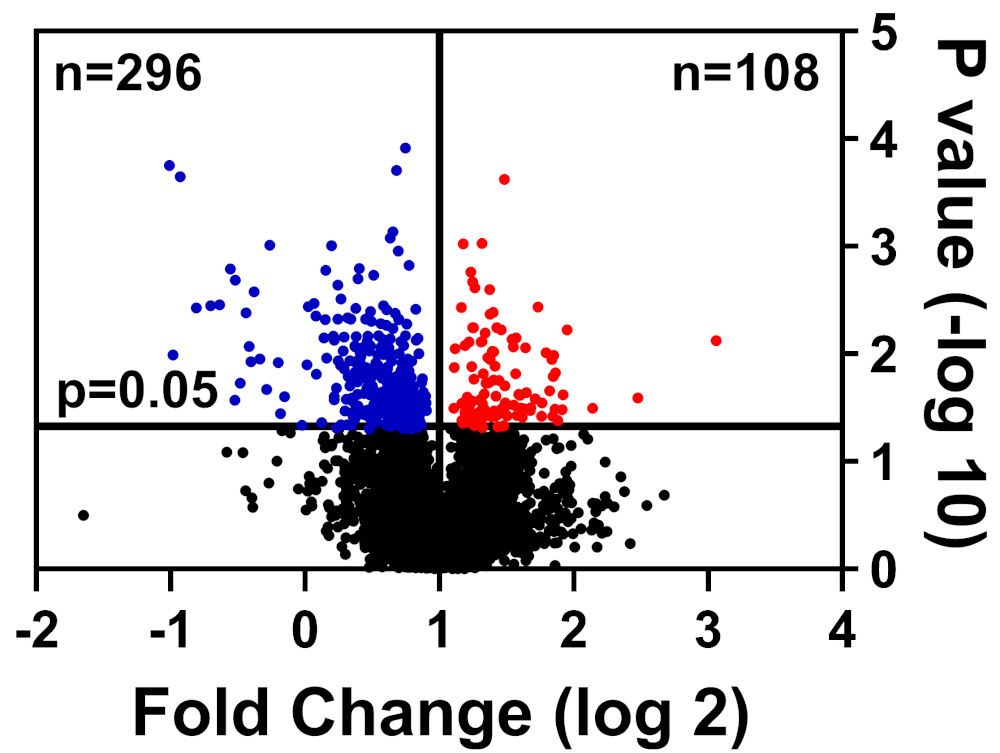
Las células sanas y enfermas fueron coloreadas en búfer CHAPS, preparadas como se detalla en nuestro método de etiquetado TMT, y enviadas al Centro Interdisciplinario de Investigación Biotecnológica de la Universidad de Florida (UF-ICBR) Core para cromatografía líquida con espectrometría de masas en tándem. Después de la adquisición y entrega de datos desde el núcleo, el conjunto de datos se abrió en el software suministrado por el proveedor y se aplicaron los siguientes filtros de corte: 2 péptidos únicos, iones reportero para cada muestra de proteína presente en todos los canales, e incluyen sólo proteínas alteradas significativamente (p a 0,05). La Tabla 1 resume los datos: 39.653 péptidos totales, de los cuales 7.211 tienen igual o mayor que dos péptidos únicos, y 3.829 incluyen iones reportero para todos los canales. Los valores p para estos 3.829 péptidos se calcularon mediante la prueba t del estudiante y p a 0,05 se consideró significativo. Además, se utilizó un corte de cambio plegable para determinar la distribución relativa de proteínas de células enfermas en comparación con las células sanas: reguladas hacia abajo (azul) o reguladas (rojas)(Figura 1).
La lista de expresión de proteínas significativamente desreguladas se evaluó utilizando el sistema de clasificación ontológica PANTHER y los análisis STRING. Los análisis de Pantera mostraron una lista categorizada de proteínas basada en una abundancia significativamente menor(Figura 2A)o mayor en células enfermas basada en la función molecular (Figura 2B). Los análisis de cuerdas de proteínas de abundancia significativamente menor(Figura 2C)y superior(Figura 2D) identificaron múltiples interacciones y fuertes asociaciones entre proteínas.

Figura 1: Gráfica de volcanes que muestra proteínas cuya abundancia no fue alterada significativamente (negro), significativamente reducida (azul), o significativamente aumentada (roja) en células de control enfermas frente a saludables. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Evaluaciones representativas de los impactos significativamente desregulados identificados por PANTHER (A, B) y String (C, D) de proteínas de abundancia significativamente menor o mayor. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Péptidos totales | Total identificado | 2 péptidos únicos | Proteínas cuantificadas | Proteínas significativamente alteradas | |
| Bajo | Alto | ||||
| 39653 | 7211 | 4457 | 3829 | 296 | 108 |
Tabla 1: Tabla representativa de proteínas cuantificadas por análisis de conjunto de datos.
Discusión
Para preparar con éxito las muestras para el análisis proteómico utilizando metodologías de etiquetado de isótopos isópticos isobáricos basados en TMT, es crucial realizar extracciones de proteínas con mucho cuidado a 4 oC y utilizar un tampón de lelisis que contenga un cóctel inhibidor de la proteasa24,,25. El cóctel inhibidor de la proteasa es un reactivo crucial para evitar la degradación inesperada de las proteínas durante la digestión de proteínas. Una diferencia clave entre nuestro protocolo y el actual proporcionado por el proveedor es que recomendamos encarecidamente el uso de tampón de lelisis CHAPS basado en nuestra experiencia con células y tejidos de mamíferos. También sugerimos el uso de un enfoque de precipitación de metanol/proteína cloroforma para pellets celulares y tejidos.
Idealmente, la extracción de proteínas, la medición, los tratamientos de reactivos reductores/alquilantes y las precipitaciones de metanol/cloroformo se realizan el mismo día. Siguiendo esta recomendación dará lugar a concentraciones de proteínas más precisas para su posterior etiquetado. El paso de precipitación de proteínas es importante para la eliminación de reactivos que interferirán con la espectrometría de masas en tándem. Incluyendo el paso de precipitación mejora significativamente la resolución del TMT26. En resumen, las principales ventajas de nuestro protocolo TMT son las altas eficiencias de etiquetado para diferentes tipos de muestras, su reproducibilidad y los datos fiables adquiridos.
A medida que la naturaleza múltiplex de esta estrategia de proteómica no objetivo TMT continúa expandiéndose, mejorará progresivamente la capacidad de los investigadores en una amplia variedad de campos para hacer descubrimientos novedosos. Específicamente en el campo biomédico, nosotros y otros hemos encontrado esta tecnología cada vez más informativa en estudios que exploran nuevos mecanismos de acción en la enfermedad y los impactos relativos de diversas terapias. Por todas estas razones, esta poderosa tecnología complementa el repertorio de otros enfoques OMICS utilizados en estudios de investigación modernos y proporciona información clave que puede guiar un mayor desarrollo terapéutico.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Nos gustaría reconocer la instalación proteómica UF-ICBR para el procesamiento de nuestras muestras. Este trabajo fue apoyado en parte por los Institutos Nacionales de Salud R01 HL136759-01A1 (CAP).
Materiales
| Name | Company | Catalog Number | Comments |
| 1 M Triethylammonium bicarbonate (TEAB), 50 mL | Thermo Fisher | 90114 | Reagent for protein labeling |
| 50% Hydroxylamine, 5 mL | Thermo Fisher | 90115 | Reagent for protein labeling |
| Acetic acid | Sigma | A6283 | Reagent for protein digestion |
| Anhydrous acetonitrile, LC-MS Grade | Thermo Fisher | 51101 | Reagent for protein labeling |
| Benzonaze nuclease | Sigma-Aldrich | E1014 | DNA shearing |
| Bond-Breaker TCEP solution, 5 mL | Thermo Fisher | 77720 | Reagent for protein labeling |
| BSA standard | Thermo | 23209 | Reagent for protein measurement |
| CHAPS | Thermo Fisher | 28300 | Reagent for protein extraction |
| Chloroform | Fisher | BP1145-1 | Reagent for protein precipitation |
| cOmplete, EDTA-free Protease Inhibitor Cocktail Tablet | Roche | 4693132001 | Reagent for protein extraction |
| DC Protein Assay | BioRad | 500-0116 | Reagent for protein measurement |
| Excel | Microsoft Office | Software for data analyses | |
| Heat block | VWR analog | 12621-104 | Equipment for protein digestion incubation |
| HEPES | Sigma | RDD002 | Reagent for protein extraction |
| Methanol | Fisher | A452-4 | Reagent for protein precipitation |
| Pierce Trypsin Protease, MS Grade | Thermo Fisher | 90058 | Reagent for protein digestion |
| Potassium chloride | Sigma | 46436 | Reagent for protein extraction |
| Sigma Plot 14.0 | Sigma Plot 14.0 | Software for data analyses | |
| Sonicator | Fisher Scientific | FB120 | DNA shearing |
| Spectra Max i3x Multi-Mode Detection Platform | Molecular Devices | Plate reader for protein measurement | |
| Thermo Scientific Pierce Quantitative Colorimetric Peptide Assay | Thermo Fisher | 23275 | Reagent for protein measurement |
| Thermo Scientific Pierce Quantitative Fluorescent Peptide Assay | Thermo Fisher | 23290 | Reagent for protein measurement |
| Thermo Scientific Proteome Discoverer Software | Thermo Fisher | OPTON-30945 | Software for data analyses |
| TMT 10plex Isobaric Label Reagent Set 0.8 mg, sufficient reagents for one 10plex isobaric experiment | Thermo Fisher | 90110 | Reagent for protein labeling |
| TMT11-131C Label Reagent 5 mg | Thermo Fisher | A34807 | Reagent for protein labeling |
| Water, LC-MS Grade | Thermo Fisher | 51140 | Reagent for protein extraction |
Referencias
- Graves, P. R., Haystead, T. A. Molecular biologist's guide to proteomics. Microbiology and Molecular Biology Reviews. 66 (1), 39-63 (2002).
- Erdjument-Bromage, H., Huang, F. K., Neubert, T. A. Sample Preparation for Relative Quantitation of Proteins Using Tandem Mass Tags (TMT) and Mass Spectrometry (MS). Methods in Molecular Biology. 1741, 135-149 (2018).
- O'Farrell, P. H. High resolution two-dimensional electrophoresis of proteins. Journal of Biological Chemistry. 250 (10), 4007-4021 (1975).
- Rabilloud, T., Lelong, C. Two-dimensional gel electrophoresis in proteomics: a tutorial. Journal of Proteomics. 74 (10), 1829-1841 (2011).
- Ong, S. E., et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Molecular & Cellular Proteomics. 1 (5), 376-386 (2002).
- Anderson, N. G., Anderson, N. L. Twenty years of two-dimensional electrophoresis: Past, present and future. Electrophoresis. 17 (3), 443-453 (1996).
- Haynes, P. A., Yates, J. R. Proteome profiling-pitfalls and progress. Yeast. 17 (2), 81-87 (2000).
- Bunai, K., Yamane, K. Effectiveness and limitation of two-dimensional gel electrophoresis in bacterial membrane protein proteomics and perspectives. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences. 815 (1-2), 227-236 (2005).
- Sury, M. D., Chen, J. X., Selbach, M. The SILAC fly allows for accurate protein quantification in vivo. Molecular & Cellular Proteomics. 9 (10), 2173-2183 (2010).
- Zhang, G., Neubert, T. A. Use of stable isotope labeling by amino acids in cell culture (SILAC) for phosphotyrosine protein identification and quantitation. Methods in Molecular Biology. 527, 79-92 (2009).
- Wang, X., et al. SILAC-based quantitative MS approach for real-time recording protein-mediated cell-cell interactions. Scientific Reports. 8 (1), 8441 (2018).
- Thompson, A., et al. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Analytical Chemistry. 75, 1895-1904 (2003).
- Cheng, L., Pisitkun, T., Knepper, M. A., Hoffert, J. D. Peptide Labeling Using Isobaric Tagging Reagents for Quantitative Phosphoproteomics. Methods in Molecular Biology. 1355, 53-70 (2016).
- Navarrete-Perea, J., Yu, Q., Gygi, S. P., Paulo, J. A. Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. Journal of Proteome Research. 17 (6), 2226-2236 (2018).
- Zecha, J., et al. TMT Labeling for the Masses: A Robust and Cost-efficient, In-solution Labeling Approach. Molecular & Cellular Proteomics. 18 (7), 1468-1478 (2019).
- Bachor, R., Waliczek, M., Stefanowicz, P., Szewczuk, Z. Trends in the Design of New Isobaric Labeling Reagents for Quantitative Proteomics. Molecules. 24 (4), E701 (2019).
- Suzuki-Hatano, S., et al. AAV9-TAZ Gene Replacement Ameliorates Cardiac TMT Proteomic Profiles in a Mouse Model of Barth Syndrome. Molecular Therapy - Methods & Clinical Development. 13, 167-179 (2019).
- Kirshenbaum, N., Michaelevski, I., Sharon, M. Analyzing large protein complexes by structural mass spectrometry. Journal of Visualized Experiments. (40), e1954 (2010).
- Perumal, N., et al. Sample Preparation for Mass-spectrometry-based Proteomics Analysis of Ocular Microvessels. Journal of Visualized Experiments. (144), e59140 (2019).
- Wessel, D., Flügge, U. I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Analytical Biochemistry. 138, 141-143 (1984).
- Jensen, L. J., et al. STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Research. 37, D412-D416 (2009).
- Mi, H., Muruganujan, A., Casagrande, J. T., Thomas, P. D. Large-scale gene function analysis with the PANTHER classification system. Nature Protocols. 8 (8), 1551-1566 (2013).
- Cirillo, E., Parnell, L. D., Evelo, C. T. A Review of Pathway-Based Analysis Tools That Visualize Genetic Variants. Frontiers in Genetics. 8, 174 (2017).
- Plaxton, W. C. Avoiding Proteolysis during the Extraction and Purification of Active Plant Enzymes. Plant and Cell Physiology. 60 (4), 715-724 (2019).
- Ryan, B. J., Henehan, G. T., Walls, D., Loughran, S. T. . Protein Chromatography: Methods and Protocols. , 53-69 (2017).
- Fic, E., Kedracka-Krok, S., Jankowska, U., Pirog, A., Dziedzicka-Wasylewska, M. Comparison of protein precipitation methods for various rat brain structures prior to proteomic analysis. Electrophoresis. 31 (21), 3573-3579 (2010).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados