
Method Article
Mapeo de las relaciones estructura-función de los factores de transcripción oncogénicos desordenados utilizando el análisis transcriptómico
En este artículo
Resumen
Los dominios intrínsecamente desordenados son importantes para la función del factor de transcripción de fusión oncogénica. Para dirigirse terapéuticamente a estas proteínas, se requiere una comprensión más detallada de los mecanismos reguladores empleados por estos dominios. Aquí, utilizamos transcriptómica para mapear características estructurales importantes del dominio EWS intrínsecamente desordenado en el sarcoma de Ewing.
Resumen
Muchos cánceres se caracterizan por translocaciones cromosómicas que resultan en la expresión de factores de transcripción de fusión oncogénica. Por lo general, estas proteínas contienen un dominio intrínsecamente desordenado (IDD) fusionado con el dominio de unión al ADN (DBD) de otra proteína y orquestan cambios transcripcionales generalizados para promover la malignidad. Estas fusiones son a menudo la única aberración genómica recurrente en los cánceres que causan, lo que las convierte en objetivos terapéuticos atractivos. Sin embargo, apuntar a los factores de transcripción oncogénicos requiere una mejor comprensión del papel mecanicista que desempeñan los IDD de baja complejidad en su función. El dominio N-terminal de EWSR1 es un IDD involucrado en una variedad de factores de transcripción de fusión oncogénica, incluyendo EWS/FLI, EWS/ATF y EWS/WT1. Aquí, utilizamos la secuenciación de ARN para investigar las características estructurales del dominio EWS importante para la función transcripcional de EWS / FLI en el sarcoma de Ewing. Se realiza el primer agotamiento mediado por shRNA de la fusión endógena de las células del sarcoma de Ewing junto con la expresión ectópica de una variedad de construcciones mutantes de EWS. Luego, la secuenciación de ARN se utiliza para analizar los transcriptomas de las células que expresan estas construcciones para caracterizar los déficits funcionales asociados con mutaciones en el dominio EWS. Al integrar los análisis transcriptómicos con información publicada previamente sobre motivos de unión al ADN EWS/FLI y localización genómica, así como ensayos funcionales para la capacidad de transformación, pudimos identificar características estructurales de EWS/FLI importantes para la oncogénesis y definir un nuevo conjunto de genes diana EWS/FLI críticos para el sarcoma de Ewing. Este artículo demuestra el uso de la secuenciación de ARN como método para mapear la relación estructura-función del dominio intrínsecamente desordenado de los factores de transcripción oncogénicos.
Introducción
Un subconjunto de cánceres, incluyendo muchas neoplasias malignas de la infancia y la adolescencia, se caracterizan por translocaciones cromosómicas que generan nuevos oncogenes defusión1,2,3,4,5,6. Las proteínas de fusión resultantes funcionan con frecuencia como factores de transcripción oncogénicos, orquestando cambios generalizados en la regulación transcripcional para promover la tumorigénesis7,8. Los cánceres con estas translocaciones comúnmente poseen un paisaje mutacional por lo demás tranquilo, con pocas aberraciones genómicas recurrentes aparte de la fusión patognomónica4,9. Como tal, dirigirse directamente a la proteína de fusión es una estrategia terapéutica atractiva en estas enfermedades. Sin embargo, estos factores de transcripción oncogénicos comúnmente consisten en un dominio activador transcripcionalmente de baja complejidad, intrínsecamente desordenado, fusionado con un dominio de unión al ADN (DBD)10,11,12,13,14. Tanto los dominios intrínsecamente desordenados (IDD) como los DBD de estas proteínas han demostrado ser difíciles de atacar con los enfoques farmacológicos convencionales. El desarrollo de nuevos enfoques terapéuticos, por lo tanto, requiere una comprensión molecular más detallada de los mecanismos empleados por estas fusiones para regular aberrantemente la expresión génica.
La porción N-terminal idD de EWSR1 se fusiona comúnmente con una DBD en el cáncer, incluyendo EWS/FLI en el sarcoma de Ewing, EWS/WT1 en el tumor difuso de células pequeñas redondas y EWS/ATF1 en el sarcoma de células claras de partes blandas10. El papel mecanicista del EWS IDD en cada una de estas fusiones no se entiende completamente. La familia de fusiones EWS/ETS, específicamente EWS/FLI, es la más funcionalmente caracterizada hasta la fecha. EWS/FLI coordina los cambios epigenéticos y transcripcionales de todo el genoma que conducen a la activación y represión de miles de genes7,11,15,16. Los estudios han demostrado que el IDD es importante para el reclutamiento tanto de coactivadores transcripcionales (como p300, WDR5 y el complejo BAF), como de co-represores (como el complejo NuRD)11,15,17. La fusión del EWS IDD a la porción C-terminal de FLI1 confiere una nueva especificidad de unión al ADN al ETS DBD de FLI1, de modo que la oncoproteína de fusión (EWS/FLI) se une a regiones repetitivas de GGAA-microsatélites del genoma, además del motivo de consenso ETS18,19,20. Combinado con la función de reclutamiento de coactivadores, esta actividad emergente de unión al ADN de EWS/FLI promueve la formación de potenciadores de novo en los sitios de inicio de GGAA-microsatélites distales a la transcripción (TSS) (microsatélites "similares a potenciadores") y recluta ARN polimerasa II para promover la transcripción en GGAA-microsatélites proximales a TSS (microsatélites "tipo promotor")11,15,16,21.
Tomados en conjunto, estos datos nos llevaron a plantear la hipótesis de que los elementos discretos dentro del dominio EWS contribuyen al reclutamiento de distintos co-reguladores para diferentes tipos de sitios de unión EWS / FLI. Sin embargo, el discernimiento de estos elementos dentro de la porción de EWS de EWS / FLI, y cómo funcionan, se ha visto obstaculizado por la naturaleza altamente repetitiva y desordenada del dominio. Aquí utilizamos un sistema de rescate de derribo publicado anteriormente en células de sarcoma de Ewing para mapear funcionalmente estos elementos en el IDD de EWS. En este sistema, EWS / FLI se agota utilizando un shRNA dirigido a los 3'UTR del gen FLI1, y la expresión se rescata con diferentes construcciones de aDNc mutantes EWS / FLI que carecen de los 3'UTR7,17,22. Estos experimentos se centraron en constructos con varias deleciones para mapear la relación estructura-función entre el IDD del EWS y fenotipos oncogénicos importantes, incluida la activación de una construcción de reportero de microsatélites GGAA, ensayos de formación de colonias y validación dirigida de genes activados y reprimidos por EWS / FLI7,17,22 . Sin embargo, estos estudios no lograron encontrar subdominios discretos dentro del IDD de EWS en EWS / FLI que sean de importancia única para la activación o la represión. Todos los constructos probados fueron capaces de activar y reprimir genes diana específicos, lo que llevó a la formación eficiente de colonias, o incapaces de regular cualquiera de los genes diana EWS/FLI, lo que llevó a la pérdida de la formación de colonias7,17,22.
Los análisis transcriptómicos habilitados por la adopción generalizada de la secuenciación de próxima generación se utilizan comúnmente para comparar firmas de expresión génica en dos condiciones, con frecuencia en el contexto de estudios de detección o descriptivos. En su lugar, queríamos aprovechar la capacidad de capturar datos de expresión de todo el genoma utilizando la secuenciación de ARN (RNA-seq) para caracterizar las contribuciones de los IDD a la función del factor de transcripción. En este caso, RNA-seq se empareja con el sistema de derribo-rescate para explorar la relación estructura-función del dominio EWS. Este enfoque es aplicable a otros factores de transcripción de fusión, incluidas otras fusiones de EWS o factores de transcripción de tipo salvaje con una función poco conocida, y tiene múltiples ventajas sobre los otros ensayos utilizados para estudios de mapeo funcional, como ensayos de reportero o qRT-PCR dirigido. Estos incluyen la prueba de determinantes estructurales de la función en el contexto relevante de la cromatina, la capacidad de probar múltiples tipos de elementos de respuesta en un ensayo (es decir, activados y reprimidos, microsatélites GGAA y no microsatélites, etc.), y la capacidad resultante para detectar mejor la función parcial.
La implementación exitosa de este enfoque depende de un sistema basado en células que capture los fenotipos de interés (en este caso células A673 con agotamiento de EWS / FLI mediado por shRNA), y un panel de construcciones mutantes en un vector de expresión apropiado para el sistema basado en células (en este caso, pMSCV-hygro con varios mutantes EWS / FLI marcados con 3x-FLAG que se entregarán por transducción retroviral). Se recomienda la transducción viral de construcciones de agotamiento basadas en CRISPR, construcciones de agotamiento basadas en shRNA y construcciones de expresión de ADNc con la selección adecuada para generar líneas celulares estables sobre transfección transitoria. La interpretación posterior de los resultados se refuerza cuando los datos transcriptómicos se pueden emparejar con otros datos relacionados con la localización del factor de transcripción y otras lecturas fenotípicas cuando estén disponibles.
En este trabajo, aplicamos este enfoque para caracterizar la actividad del mutante DAF de EWS/FLI14. El mutante DAF tiene 17 mutaciones de tirosina a alanina en las regiones repetitivas del EWS IDD de EWS/FLI14. Este mutante EWS en particular había sido reportado previamente y es incapaz de activar la expresión génica del reportero cuando se fusiona con el ATF1 DBD14. Sin embargo, los datos preliminares de qRT-PCR sugirieron que este mutante fue capaz de activar la transcripción del objetivo EWS/FLI NR0B123. El enfoque transcriptómico descrito aquí permitió la detección exitosa de la función parcial del mutante DAF. Al emparejar estos datos transcriptómicos con información sobre motivos de unión y reconocimiento de EWS / FLI, mostramos además que el mutante DAF conserva la función en las repeticiones de microsatélites GGAA. Estos resultados identifican a DAF como el primer mutante EWS/FLI parcialmente funcional y resaltan la función en genes no microsatélites como importante para la oncogénesis (como se informó23). Esto demuestra el poder de este enfoque de mapeo de estructura-función transcriptómica para proporcionar información sobre la función de los factores de transcripción oncogénicos.
Protocolo
1. Configurar un panel de construcciones in vitro
NOTA: Este paso variará dependiendo de la proteína específica a analizar.
- Prepare alícuotas de virus para construcciones de agotamiento y expresión según sea necesario.
- Sembrar una placa de cultivo de tejidos de 10 cm con 3-5 x 106 células HEK293-EBNA o HEK293T para cada construcción necesaria para la transducción viral. Deje que las células se adhieran durante la noche en modified Eagle Media (DMEM) de Dulbecco suplementado con suero bovino fetal (FBS) al 10%, penicilina / estreptomicina / glutamina (P / S / Q) y 0.3 mg / ml G418.
NOTA: Las células HEK293-EBNA y HEK293T se recomiendan para la producción viral porque son fáciles de cultivar, tienen una alta eficiencia de transfección y expresan eficientemente las proteínas recombinantes de los plásmidos episomales. Las células deben estar entre un 50-70% confluentes el día de la transfección. - Prepare una mezcla de transfección para cada construcción de transducción viral. Combine 2 ml de medios séricos reducidos con 90 μL de reactivo de transfección.
NOTA: Se recomienda precalentar los medios séricos reducidos. - Agregue 10 μg cada uno de un plásmido de empaquetamiento viral (por ejemplo, gag-pol), plásmido de envoltura viral (por ejemplo, VSV-G) y uno de agotamiento basado en CRISPR, agotamiento basado en shRNA o construcción de expresión de aDNc (por ejemplo, pMKO o pMSCV) a la mezcla de transfección. Mezclar bien mediante pipeteo suave.
- Deje reposar la mezcla de transfección durante 20 minutos a temperatura ambiente. Retire los medios de crecimiento HEK293-EBNA de las placas de cultivo de tejidos y agregue 3 ml de DMEM suplementado con 10% de FBS, P / S / Q y piruvato de sodio de 10 mM. A cada plato, agregue 2 ml de mezcla de transfección gota a gota. Deje que las células se asienten en medios de transfección durante la noche en una incubadora a 37 ° C y 5% de CO2.
- A la mañana siguiente agregue 20 ml de medios DMEM con 10% de FBS, suplementación con P / S / Q y piruvato de sodio de 10 mM. Incubar las células en él a 37 °C y 5% de CO2 durante la noche.
- A la mañana siguiente, reemplace los medios con medios de recolección viral (VCM) de 5 ml (DMEM suplementado con FBS inactivado al 10% por calor, P / S / Q y HEPES de 20 mM).
- Después de 4 h, recoger VCM de las placas y almacenar en un tubo cónico de 50 ml sobre hielo a 4 °C. Reemplace con 5 ml de VCM fresco.
- Después de 4 h, recoger VCM de las placas en el mismo tubo cónico de 50 ml y almacenar en hielo a 4 °C. Reemplace con 8 ml de VCM fresco para la recolección durante la noche.
- Por la mañana, recoja VCM de las placas y guárdela en el tubo cónico de 50 ml sobre hielo a 4 °C. Reemplace con 5 ml de VCM fresco.
- Después de 4 h, recoger VCM de las placas y almacenar en el tubo cónico de 50 ml sobre hielo a 4 °C. Reemplace con 5 ml de VCM fresco. Después de 4 h, recoja VCM de las placas y agréguelo al tubo cónico de 50 ml.
- Alícuotas recolectan de tubo de 50 ml en criotubos (2 ml por alícuota) después de la filtración a través de un filtro de 0,45 μm. Guarde las alícuotas virales a -80 °C hasta su uso.
NOTA: El protocolo se puede pausar aquí, y las alícuotas virales se pueden almacenar hasta que estén listas para su uso.
- Sembrar una placa de cultivo de tejidos de 10 cm con 3-5 x 106 células HEK293-EBNA o HEK293T para cada construcción necesaria para la transducción viral. Deje que las células se adhieran durante la noche en modified Eagle Media (DMEM) de Dulbecco suplementado con suero bovino fetal (FBS) al 10%, penicilina / estreptomicina / glutamina (P / S / Q) y 0.3 mg / ml G418.
- Células de semillas a la densidad adecuada en una placa de cultivo de tejidos de 10 cm. Objetivo 50% de confluencia. Deje que las células se adhieran durante la noche colocándolas en la incubadora a 37 °C conteniendo un 5% de CO2.
NOTA: Para las células A673, esto es 5 x10 6 células en 10 ml de medios DMEM con 10% de FBS, suplementos de P / S / Q y piruvato de sodio de 10 mM. Estas condiciones pueden variar dependiendo de la tasa de crecimiento de las células utilizadas. - Agotar el factor endógeno de interés. Si las células no necesitan tener la proteína endógena de interés agotada, vaya al paso 1.4.
- Descongelar la alícuota viral para la transducción de shRNA o CRISPR construct dirigido a la proteína de interés. Descongele las alícuotas congeladas rápidamente en un baño de agua a 37 °C.
- Añadir 2,5 μL de polibreno de 8 mg/ml a cada alícuota viral y mezclar mediante pipeteo suave. Retire los medios de las placas de células y agregue suavemente la alícuota viral a la placa de 10 cm pipeteando a lo largo del lado de la placa. Balancea la placa para esparcir los 2 mL de alícuota viral.
- Incubar a 37 °C en la incubadora de cultivo de tejidos durante 2 h. Balancee la placa cada 30 minutos para evitar que las áreas de la placa se sequen.
- Agregue 5 ml de medios DMEM con 10% de FBS, suplementación con P / S / Q y piruvato de sodio de 10 mM, con 5 μL de polibreno de 8 mg / ml. Deje que las células se incuben durante la noche.
- Por la mañana, retire los medios de las células y las células de paso a los medios complementados con un reactivo de selección. Cuando pase las células, sembrarlas de manera que puedan crecer durante 48-72 h y alcanzar el 50% de confluencia.
NOTA: Para las células A673 con pSRP-iEF-2, las células se siembran en una división 1:5 y se seleccionan durante 72 h con 2 μg/ml de puromicina.
- Transducir construcciones de expresión de ADNc.
- Verifique las celdas para confirmar una confluencia del 50-70%.
- Descongelar alícuotas virales para la transducción de constructos de ADNc de interés. Descongele las alícuotas congeladas rápidamente en un baño de agua a 37 °C. Añadir 2,5 μL de polibreno de 8 mg/ml a cada alícuota viral y mezclar mediante pipeteo suave.
- Retire los medios de las células chapadas y agregue suavemente la alícuota viral a la placa de 10 cm pipeteando a lo largo del lado de la placa. Balancea la placa para esparcir los 2 mL de alícuota viral.
- Incubar a 37 °C en la incubadora de cultivo de tejidos durante 2 h. Balancee la placa cada 30 minutos para evitar que las áreas de la placa se sequen.
- Agregue 5 ml de medios DMEM con 10% de FBS, suplementación con P / S / Q y piruvato de sodio de 10 mM, con 5 μL de polibreno de 8 mg / ml. Deje que las células se incuben durante la noche.
- Por la mañana, retire los medios de las células y las células de paso a medios de doble selección. Cultivar y pasar las células según sea necesario durante 7-10 días para permitir la doble selección y expresión de la construcción de ADNc.
NOTA: Esta división de este pasaje puede requerir optimización para diferentes líneas celulares. Para las células A673 con pSRP-iEF-2 y una construcción pMSCV-hygro, las células se pasan sin dividirse en 2 μg/ml de puromicina y 100 μg/mL de higromicina.
2. Recolectar células, validar la expresión de constructos y establecer ensayos fenotípicos correlativos
- Después de 7-10 días de doble selección, recolecte las células en un tubo cónico de 15 ml. Cuente las células recolectadas con un hemocitómetro. Alícuota recolectó células para la secuenciación de ARN y para validar la expresión de construcciones de ADNc.
NOTA: Configure cualquier ensayo fenotípico correlativo requerido por la pregunta de investigación bajo investigación. Los ensayos de formación de colonias son un ejemplo de un ensayo fenotípico correlativo que se utilizan aquí.- Recolectar entre 5 x 105 y 1 x 106 células para la secuenciación de ARN y 2 x 106 células para la extracción de proteínas. Células de pellet por centrifugación a 1.000 x g a 4 °C durante 5 min y retirar el sobrenadante.
- Lave el pellet con 1 ml de PBS frío. Pellet por centrifugación a 1.000 x g a 4 °C durante 5 min y retirar sobrenadante. Congelación instantánea de pellets en nitrógeno líquido y almacenar a -80 °C.
- Configure cualquier ensayo correlativo con las células restantes.
NOTA: El protocolo se puede pausar aquí con muestras recogidas almacenadas en el congelador de -80 °C.
- Validar el derribo de la proteína de interés (si se utiliza) y la expresión del panel de constructos.
- Descongelar pellets celulares para la extracción de proteínas en hielo. Células de resuspend en tampón de extracción nuclear frío de 500 μL (20 mM HEPES pH 7.9, 140 mM NaCl, 10% glicerol, 1.5 mM MgCl2, 1 mM EDTA, 1 mM TDT, 1% IGEPAL) con inhibidor de proteasa. Déjalo reposar durante 5 min sobre hielo.
- Núcleos de pellets por centrifugación a 1.000 x g a 4 °C durante 5 min y eliminar sobrenadante. Lavar núcleos en tampón de extracción nuclear helado de 500 μL (20 mM HEPES pH 7.9, 140 mM NaCl, 10% glicerol, 1.5 mM MgCl2, 1 mM EDTA, 1 mM TDT, 1% IGEPAL) con inhibidor de proteasa.
- Núcleos de pellets por centrifugación a 1.000 x g a 4 °C durante 5 min y retirar el sobrenadante. Núcleos de resuspend en tampón RIPA frío de 200 μL con inhibidor de la proteasa (ajuste el volumen del tampón RIPA según el tamaño del pellet). Déjalo reposar sobre hielo durante 45-60 min con vórtice vigoroso cada 15 min.
- Residuos de células de pellets por centrifugación a 16.000 x g a 4 °C durante 45-60 min. Mantenga el sobrenadante y transfiéralo a un tubo frío fresco
- Prepare muestras para la electroforesis SDS-PAGE hirviendo 5-10 μg de proteína con 1x tampón de carga durante 5 min. Ejecute un gel SDS-PAGE según sea necesario para la proteína de interés.
- Transfiera a una membrana de nitrocelulosa o PVDF según sea necesario para la proteína de interés. Bloquee y seque con los anticuerpos primarios y secundarios apropiados para confirmar el derribo de la proteína endógena (si se usa) y la expresión ectópica de la construcción de ADNc.
NOTA: El protocolo se puede pausar aquí.
- Extraer ARN. Evaluar la calidad y cantidad de ARN.
- Descongele los gránulos de células en hielo. Extraiga el ARN total utilizando un kit de extracción basado en columna de espín de sílice de acuerdo con las instrucciones del fabricante.
- Brevemente, lisa las células usando el tampón de lisis del kit. Aplique el lisado a una columna de espín de sílice con un breve giro a >13000 rpm durante 30-60 segundos o elimine el gDNA aplicando el lisado a una columna de eliminación de gDNA con un breve giro a >13000 rpm durante 30-60 segundos.
- Realice una digestión de ADN en la columna si el lisado se aplicó directamente a una columna de espín de sílice. Si utiliza una columna de eliminación de gDNA, aplique el eluido a una columna de espín de sílice con un breve giro a >13000 rpm durante 30-60 s.
- Lave el ARN en la columna según las instrucciones del fabricante. ARN elutivo en 30 μL de tampón de elución.
- Evalúe la calidad y cantidad de ARN utilizando un fluorómetro o cualquier otro instrumento comparable. Asegúrese de que la relación 260/280 esté cerca de 2 y que haya al menos 2,5 μg de ARN para someter a secuenciación.
NOTA: A medida que se recopilan las réplicas, cada réplica debe procesarse con el mismo protocolo de extracción de ARN. - Utilice una pequeña alícuota de ARN para confirmar el derribo estable de la proteína de interés, si es necesario, mediante qRT-PCR. Almacene la muestra de ARN restante a -80 °C.
- Recolectar réplicas biológicas repitiendo los pasos 1-2 hasta que se hayan recolectado 3-4 conjuntos completos de ARN. Asegúrese de que cada réplica muestre una expresión adecuada de las construcciones de ADNc y un derribo estable de la proteína endógena (si se usa).
3. Secuenciación de próxima generación
- Envíe el ARN extraído para ser secuenciado utilizando una plataforma de secuenciación de próxima generación con un objetivo de 50 millones de lecturas finales paradas de 150 pares de bases (pb). Siga las instrucciones de la instalación que procesa las muestras. Seleccione para ARN poliadenilados y secuenciación específica de hebras.
4. Alineación y canalización de conteo de transcripciones
Nota : este protocolo supone que después del envío y procesamiento de muestras, se devuelve un conjunto de archivos FASTQ emparejados para cada muestra. Estos archivos se comprimen con frecuencia con un sufijo de "fastq.gz". Un análisis más detallado de estos archivos FASTQ requerirá acceso a una instalación de computación de alto rendimiento (HPC) que ejecute un sistema operativo Linux.
- Transferir archivos
- Abra un terminal en el entorno HPC con PuTTY. Haga un directorio para el análisis llamado "proyecto".
- Navegue hasta el directorio "path_to/project" y cree un nuevo directorio para los archivos comprimidos raw fastq.gz llamado "fastq". También haga un directorio llamado "recortado". Esto se muestra en la Figura S1A-C.
- Transfiera los archivos fastq.gz comprimidos sin procesar desde el almacenamiento local al directorio "path_to/project/fastq/" utilizando WinSCP o un programa similar. Compruebe que hay un archivo "R1" y un archivo "R2" para cada ejemplo, como se muestra en la figura S1B.
- Opcional: Si es necesario, instale TrimGalore. Establezca el directorio que contiene el archivo ejecutable trim_galore en la variable de entorno PATH en Linux.
NOTA: Las lecturas y adaptadores de baja calidad se recortan con TrimGalore. TrimGalore está disponible en https://github.com/FelixKrueger/TrimGalore. - Opcional: Navegue al directorio para ver los paquetes de software descargados (es decir, "path_to/software"). Descargue el último paquete trimGalore usando el comando "curl -fsSL https://github.com/FelixKrueger/TrimGalore/archive/[version].tar.gz -o trim_galore-[version].tar.gz".
- Opcional: Descomprima el archivo tar.gz. Utilice el comando "tar -xvzf trim_galore-[version_number].tar.gz".
- Opcional: Haga que TrimGalore sea ejecutable. Utilice el comando "chmod a+x path_to/software/TrimGalore-[version]/trim_galore". Asegúrese de que este nuevo directorio está en la ruta de acceso. Utilice el comando "export PATH=path_to/software/TrimGalore-[version]:$PATH".
- Vaya a path_to/project/fastq/. Utilice TrimGalore para recortar las lecturas de baja calidad de los archivos fastq.gz mediante el comando que se muestra en la figura S1C.
NOTA: Las banderas adicionales para este comando pueden ser relevantes y se pueden encontrar aquí: https://github.com/FelixKrueger/TrimGalore/blob/master/Docs/
Trim_Galore_User_Guide.md - Compruebe si hay archivos fastq.gz recortados en el directorio path_to/project/trimmed. Asegúrese de que se llamen sample1_R1_val_1.fq.gz y sample1_R2_val_2.fq.gz
- Alinee los archivos FASTQ recortados con STAR y genere recuentos de transcripciones.
NOTA: STAR está disponible en https://github.com/alexdobin/STAR)- Opcional: Instale STAR versión 2.6 o posterior. Establezca el ejecutable STAR en la ruta de acceso.
- Opcional: Navegue al directorio para ver los paquetes de software descargados (es decir, "path_to/software").
- Opcional: Descargue el paquete STAR usando el comando "curl -SLO https://github.com/alexdobin/STAR/archive/[version].tar.gz". Descomprima el archivo tar.gz.
- Opcional: Utilice el comando "tar -xzf [version].tar.gz". Haga que STAR sea ejecutable. Utilice el comando "chmod a+x path_to/software/STAR-[version]/bin".
- Opcional: Asegúrese de que este nuevo directorio esté en la ruta de acceso. Utilice el comando "export PATH=path_to/software/STAR-[version_number]/bin/linux_x86_64_static:$PATH".
NOTA: El manual de STAR está disponible en: (https://github.com/alexdobin/STAR/blob/master/doc/STARmanual.pdf). - Asegúrese de que haya un índice de genoma para usar con STAR. Coloque esto en un directorio separado del directorio path_to/project/. Si se generó previamente un índice para experimentos anteriores, úselo. Alternativamente, use un índice pregenerado apropiado si está disponible aquí: http://refgenomes.databio.org/. De lo contrario, construya un nuevo índice utilizando el comando "STAR--runMode genomeGenerate" utilizando las instrucciones del manual STAR.
NOTA: Para el resto de este protocolo, la ruta hacia el índice STAR se denominará "path_to/STAR_index". - Desplácese hasta el directorio path_to/project/. Cree un nuevo directorio llamado "STAR_output" como se muestra en la Figura S1D.
- Desplácese hasta el directorio path_to/project/trimmed/. Utilice el comando que se muestra en la figura S1D para ejecutar STAR y alinear los archivos fastq.gz recortados.
NOTA: Este paso es el más intensivo computacionalmente y se recomienda realizarlo en un clúster hpc con varios subprocesos (es decir, >16) designados para la tarea de alineación. Dependiendo del número de muestras y los recursos computacionales disponibles, este paso puede llevar muchas horas o días. - Encuentre el resultado requerido para los siguientes pasos que contienen los recuentos por transcripción en la siguiente ubicación: path_to/project/STAR_output/sampleN_ReadsPerGene.out.tab.
Nota : en el archivo ReadsPerGene.out.tab, la columna 1 contiene información sobre la característica que se está contando. La columna 2 contiene los recuentos de lectura no descorrida, la columna 3 contiene los recuentos de lectura varada hacia adelante y la columna 4 contiene los recuentos de lectura de trenzado inverso. Las primeras cuatro filas de este archivo tendrán información sobre las lecturas alineadas que no se alinearon con un solo gen. Este protocolo requiere los recuentos de lectura no descorchados. - Utilice RStudio (preferible) o R en el entorno HPC para compilar los datos de la fila 5 y siguientes para las columnas 1 y 2 de cada ejemplo. Establezca el directorio de trabajo en "proyecto" en R.
- Lea en cada archivo ReadsPerGene.out.tab mediante el comando de la figura S2A. Para la primera columna, tome solo los caracteres antes del "." en la columna "Ensembl gene ID" para facilitar el procesamiento posterior.
- Compile recuentos de todos los ejemplos en un marco de datos denominado "totcts" mediante los comandos de la figura S2B. Guarde esta nueva tabla de datos de recuento sin procesar como un archivo de .txt delimitado por tabulaciones, es decir, sample_counts.txt, si lo desea, utilizando el comando "write.table".
NOTA: El orden del ID del gen Ensembl es el mismo para cada archivo ReadsPerGene.out.tab en todas las muestras.
5. Expresión diferencial y análisis descendente
- Normalice los efectos por lotes entre muestras con ComBat.
NOTA: Hay dos variables posibles que explican los cambios en la expresión génica, la primera es el constructo utilizado (es decir, la muestra) y la segunda son los factores externos asociados con el paso de las células a través del tiempo (es decir, el lote). Se recomienda un paso para normalizar las muestras para la variación de lote a lote con el paquete R ComBat.- Instale si es necesario y cargue las bibliotecas para sva, DESeq2, AnnotationDBI, org. Hs.eg.db, pheatmap, RColorBrewer, genefilter, Cairo, ggplot2, ggbiplot, rgl y reshape2 como se muestra en la Figura S2C. Para la instalación, utilice el comando "install.packages" o Bioconductor según la documentación de cada paquete.
- Primero filtre los datos solo a aquellos genes que tengan al menos un recuento por lectura. Guarde esta nueva tabla para denotar el filtrado como se ve en la figura S2D.
NOTA: Con frecuencia, muchos genes tendrán recuentos de lectura muy bajos o nulos. - Prepare una segunda tabla para la normalización de lotes llamada "vars" como se muestra en la Figura S2E. Establezca los nombres de fila en los nombres únicos de cada ejemplo. Establezca los nombres de las columnas en "muestra", "lote" y "construcción".
- Asigne a todas las muestras un número único en la columna "muestra" de 1 a n, siendo n el número de muestras. Asigne números de lote a todas las muestras de la columna "lote" de modo que a_1 y b_1 de condiciones estén asignados 1, y a_2 de condiciones y b_2 de condiciones se asignen 2. Asigne todas las designaciones de condición a todas las muestras de la columna "constructo", de modo que las muestras de condición A sean todas "A" y las muestras de condición b sean todas "B".
- Defina también la variable de lote y una matriz de modelo nulo específica para ComBat como se muestra en la figura S2F. Ejecute ComBat con el comando definido en la figura S2F.
- Mejore aún más los datos redondeando al entero más cercano. También eliminar genes con un valor negativo. Utilice los comandos que se muestran en la figura S3A.
NOTA: La salida de la normalización por lotes tendrá recuentos de lectura no enteros y algunos genes con valores negativos. Este paso es necesario porque el análisis de expresión diferencial descendente no admite recuentos de lectura negativos. - Defina el perfil de expresión diferencial para cada constructo utilizando DESeq2.
- Introduzca el diseño del experimento para DESeq2 como se muestra en la figura S3B. Construya un DESeqDataSet (dds) utilizando la función DESeqDataSetFromMatrix, estime los factores de tamaño y ejecute DESEq2, como se muestra en la figura S3B.
NOTA: Es imperativo que los datos de columna introducidos para "condición" estén en el mismo orden que la columna en la matriz de recuento. - Para evaluar la calidad del análisis, extraiga los recuentos normalizados de rlog utilizados por DESeq2 como se muestra en la Figura S3B.
NOTA: Durante el análisis, deSeq2 transforma los recuentos con un "log regularizado", rlog, transformación para reducir las diferencias de muestra a muestra para genes con recuentos bajos (información baja) con el fin de preservar las diferencias en genes con recuentos más altos entre muestras (información alta). - Al extraer los resultados para cada perfil transcripcional de los resultados de DESeq2, realice comparaciones por pares en referencia a la condición de derribo o al vector vacío basal como se muestra en la Figura S3C. Modifique aún más estos resultados con los símbolos del gen HGNC como se muestra en la Figura S3D.
- Como se ve en la Figura S3E,extraiga los datos de los resultados de DESeq2. Exporte como un solo archivo con el ID de gen de Ensembl, el símbolo HGNC, la expresión media base y los datos de expresión diferencial para todas las construcciones con log2FoldChange y valores p brutos y ajustados.
NOTA: El uso de un valor p ajustado < 0,05 es el punto de corte recomendado para la expresión diferencial. - Evalúe la normalización exitosa del lote y la similitud intramuestrema. Compruebe la agrupación de muestras con PCA y las gráficas de distancia de muestra a muestra utilizando los recuentos normalizados de rlog utilizando el código que se muestra en las figuras S4A-B.
- Introduzca el diseño del experimento para DESeq2 como se muestra en la figura S3B. Construya un DESeqDataSet (dds) utilizando la función DESeqDataSetFromMatrix, estime los factores de tamaño y ejecute DESEq2, como se muestra en la figura S3B.
- Utilice los perfiles de expresión diferencial para generar gráficos de volcanes utilizando el código de la figura S4C. Evaluar los cambios en la expresión génica a través de constructos.
- Utilice los recuentos normalizados de rlog y la agrupación jerárquica para identificar firmas genéticas exclusivas de las diferentes construcciones. Utilice el código que se muestra en la figura S4D.
- Extraiga los 1000 genes más variables en todas las construcciones de una matriz. Utilice pheatmap para realizar la agrupación jerárquica no supervisada de sus muestras en función de estos genes.
- Extraiga los grupos de interés del dendrograma decidiendo en qué nivel del dendrograma aparecen los grupos de interés. Establezca "k" igual al número de clústeres en ese nivel. Vuelva a trazar el mapa de calor ordenado por clúster para determinar qué clústeres son de interés como se muestra en la Figura S5.
- Exporte la lista de genes asociados a cada clúster como se muestra en la Tabla S1. Utilice esta información para determinar los genes en grupos de interés.
- Identificar los roles biológicos para diferentes grupos de genes identificados y comparar entre las clases. Esto se puede realizar utilizando una variedad de herramientas bioinformáticas. ToppGene24 se utiliza aquí y está disponible gratuitamente en línea.
NOTA: Hay muchas herramientas gratuitas que requieren solo una lista de genes para copiar y pegar en un campo en un sitio web. Elija las herramientas analíticas más adecuadas para las preguntas de investigación bajo investigación. - Opcionalmente, si hay datos disponibles sobre la unión genómica que impulsa la salida transcripcional para el factor de transcripción de interés, compare la respuesta transcripcional en genes asociados con diferentes elementos de unión para evaluar aún más la función mutante.
6. Comparación con fenotipos relevantes
- Comparar los fenotipos correlativos con los datos del perfil transcriptómico generados e interpretarlos según corresponda.
Resultados
Los datos preliminares de qRT-PCR sugirieron que un mutante EWS/FLI llamado DAF, con mutaciones específicas de tirosina a alanina en la región repetitiva y desordenada de EWS, mantuvo la capacidad de activar genes diana EWS/FLI, pero no logró reprimir genes diana críticos23. Con el fin de comprender mejor la relación entre estos residuos en el dominio EWS y la función EWS/FLI, se utilizó el protocolo descrito anteriormente y descrito en la Figura 1. Las células del sarcoma de Ewing A673 se transdujeron viralmente con un shRNA dirigido a los 3'UTR de FLI1,lo que resultó en el agotamiento de EWS / FLI endógenos. Después de cuatro días de selección, la función EWS/FLI fue rescatada con la transducción viral de diferentes construcciones mutantes EWS/FLI marcadas con 3XFLAG, con vector vacío como control para ningún rescate. Un mutante no funcional que carecía del dominio EWS, llamado Δ22, se utilizó como control negativo y EWS/FLI de tipo salvaje, llamado wtEF, se utilizó como control positivo(Figura 2A). DAF se utilizó como la construcción de prueba, aunque se puede usar más de una construcción de prueba si se desea. Las células se seleccionaron durante 10 días adicionales para permitir que la expresión de constructo se estabilizara y luego se recolectaron para los ensayos de ARN (con un paso de eliminación de gDNA), proteínas y colonias. Se recolectaron cuatro réplicas y en la Figura 2B-Dse muestran qRT-PCR representativas y western blots que muestran un derribo y rescate efectivos. Cabe señalar que las células rescatadas por DAF no pudieron formar colonias como se muestra en la Figura 2E,lo que sugiere una transformación oncogénica deteriorada.
Después de completar la validación de la réplica y los ensayos fenotípicos, el ARN se envió al Instituto de Medicina Genómica del Nationwide Children's Hospital para la preparación de la biblioteca y la secuenciación de próxima generación con ~ 50 millones de lecturas de extremo pareado de 150 pb recopiladas. Los datos se devolvieron como archivos fastq.gz. Las lecturas de baja calidad se recortaron de estos archivos con TrimGalore y STAR se utilizó para alinear las lecturas con el genoma humano hg19 y contar las lecturas por gen. hg19 se utilizó con fines de compatibilidad con los otros conjuntos de datos seleccionados para EWS/FLI utilizados en el análisis posterior. Estos recuentos de lectura se combinaron en una sola matriz de recuento para todas las muestras, cuyas primeras 6 filas se muestran en la Figura 3.
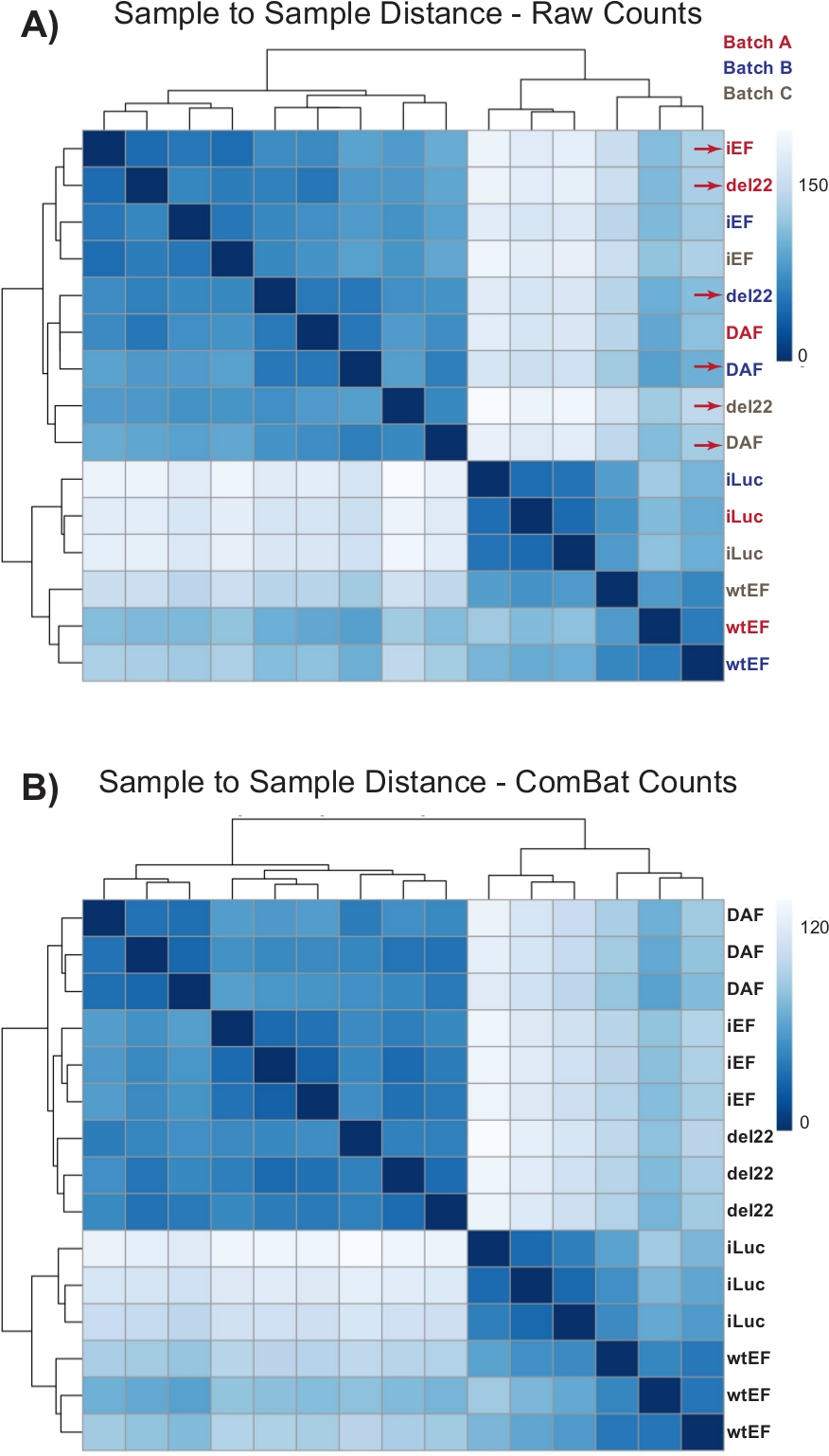
Los recuentos se ejecutaron inicialmente a través de DESeq2 sin normalización de lotes, sin embargo, la inspección visual de la distancia de muestra a muestra mostró posibles efectos de lote de confusión como se muestra resaltado con flechas rojas en la Figura 4A. Esto probablemente surgió debido a la variabilidad biológica introducida por el paso de las células en cultivo y las diferencias en el procesamiento de cada lote. La normalización de los efectos por lotes se realizó con ComBat y generalmente se recomienda. Las distancias de muestra a muestra de los datos normalizados por lotes se muestran en la Figura 4B. Después de la normalización por lotes, se utilizó DESeq2 para generar perfiles transcripcionales para los tres constructos (wtEF, Δ22 y DAF) en relación con la línea de base. Tenga en cuenta que mientras que las células A673 "parentales" (simulacro de derribo y simulacro de rescate, llamado "iLuc" aquí) se incluyeron en el análisis diferencial, la referencia para este experimento son las células con EWS / FLI-agotado, llamadas células iEF. El perfil transcripcional se puede generar para la proteína endógena aquí comparando la muestra de iLuc con iEF, y esto puede ser de interés para comprender cómo funciona el sistema de rescate, pero ese no es el objetivo de este análisis en particular. Los perfiles transcripcionales generados para los mutantes incluyen controles positivos (wtEF) y negativos (Δ22), con respecto a iEF, de modo que estos deberían funcionar como puntos de referencia para otros mutantes. Esto es importante, ya que el control positivo en este ejemplo no recapituló completamente la función de EWS/FLI endógeno como se discutió en otra parte7,23.
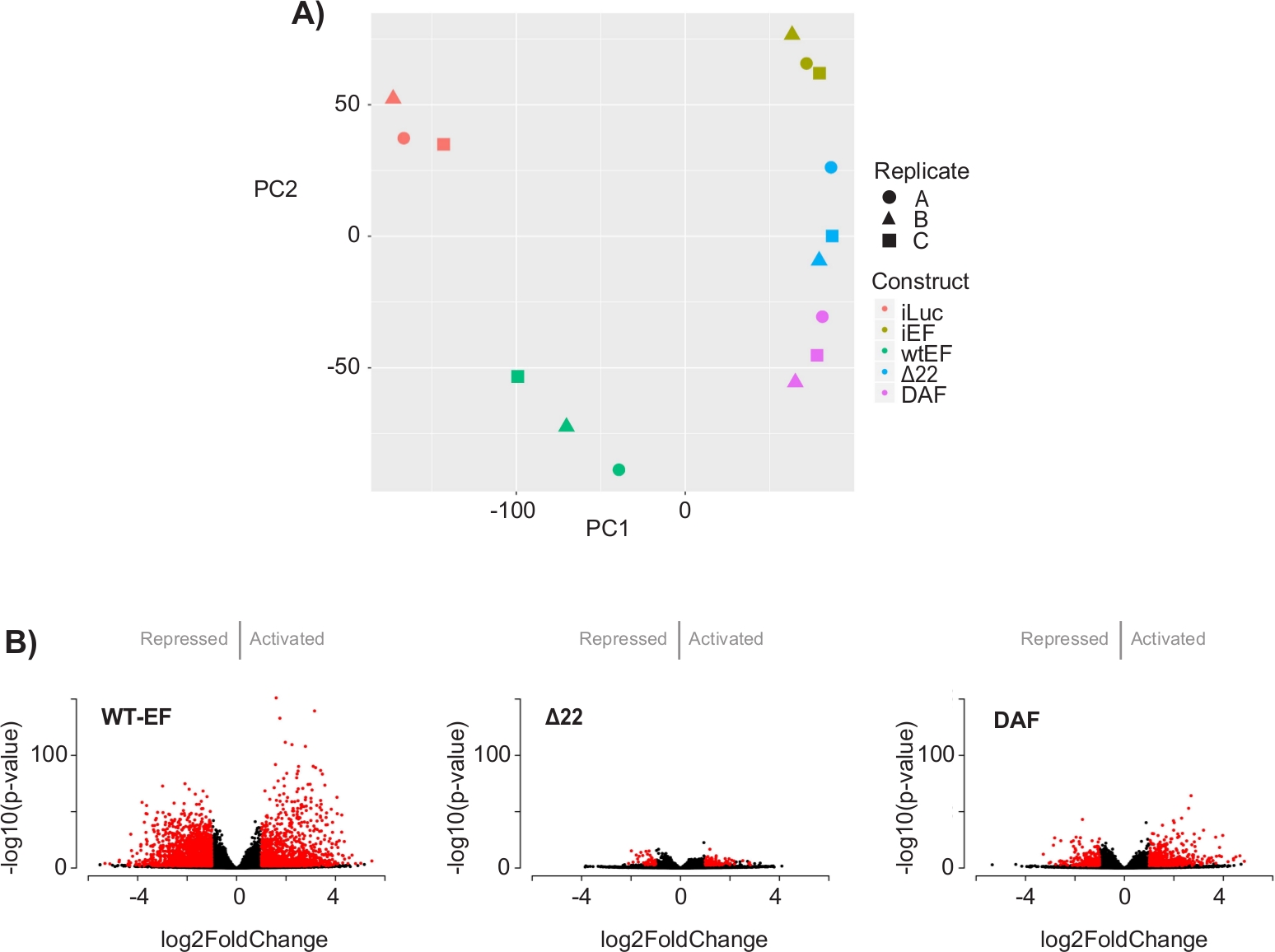
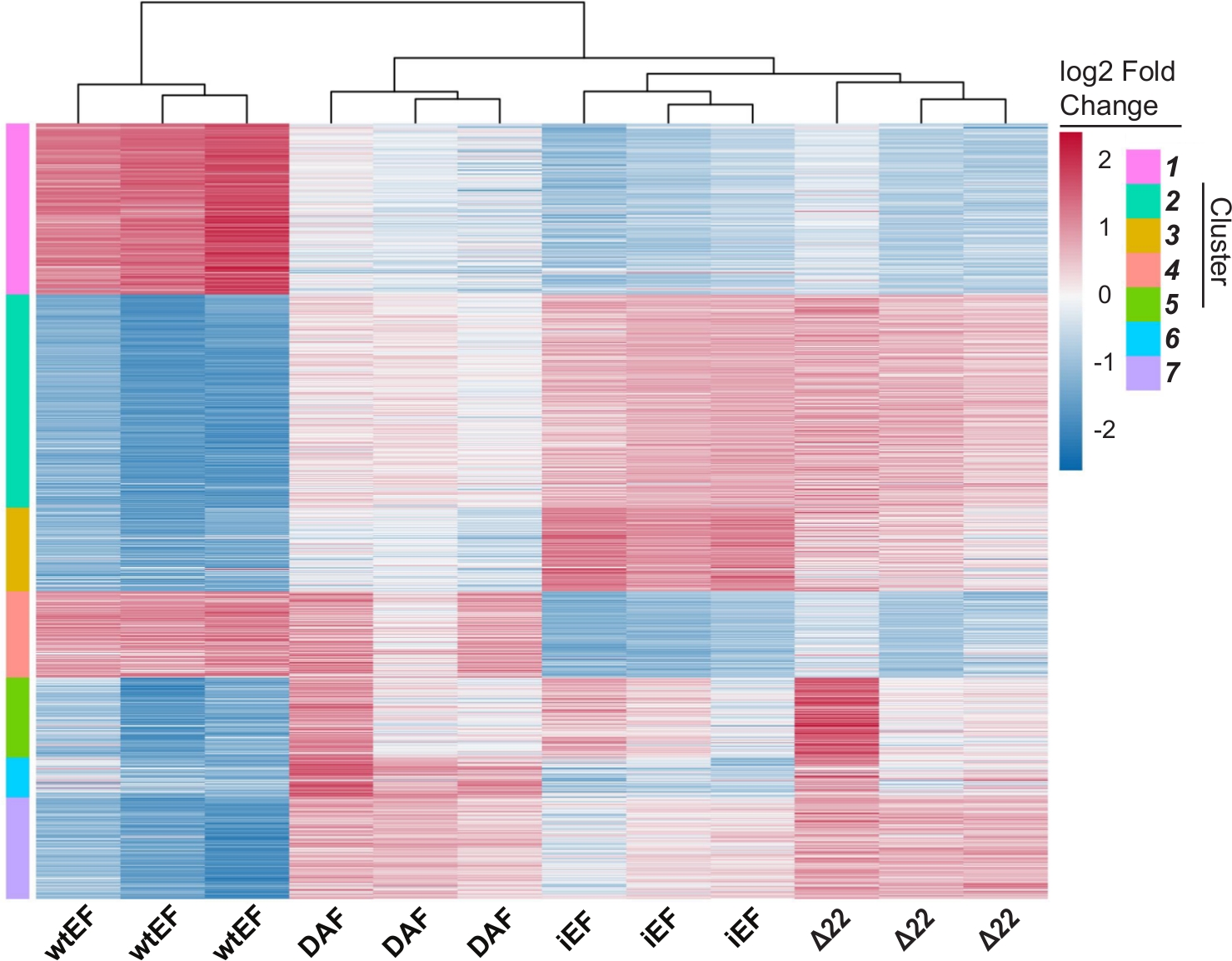
El análisis de componentes principales (ACP) de la Figura 5 sugiere que el perfil transcripcional de DAF es intermedio entre wtEF y Δ22, confirmando la función parcial. Además, la agrupación jerárquica de los 1000 genes más variables en todas las muestras mostró que DAF no pudo reprimir los genes diana EWS / FLI, y solo retuvo parcialmente la actividad de activación de genes como se muestra en la Figura 6A y la Figura S5. El análisis de ToppGene sugirió que las clases de genes que DAF activa son funcionalmente distintas de aquellos objetivos activados por EWS / FLI donde DAF no es funcional (Figura 6B). Curiosamente, la función de los genes activados rescatados por wtEF, pero no por DAF, parece estar relacionada con el control transcripcional y la regulación de la cromatina. Sobre la base de los resultados de los ensayos de formación de colonias, los genes de esta firma genética central deben analizarse más a fondo para determinar su papel en la oncogénesis mediada por EWS / FLI. La importancia de la represión genética mediada por EWS/FLI ha sido descrita previamente17.
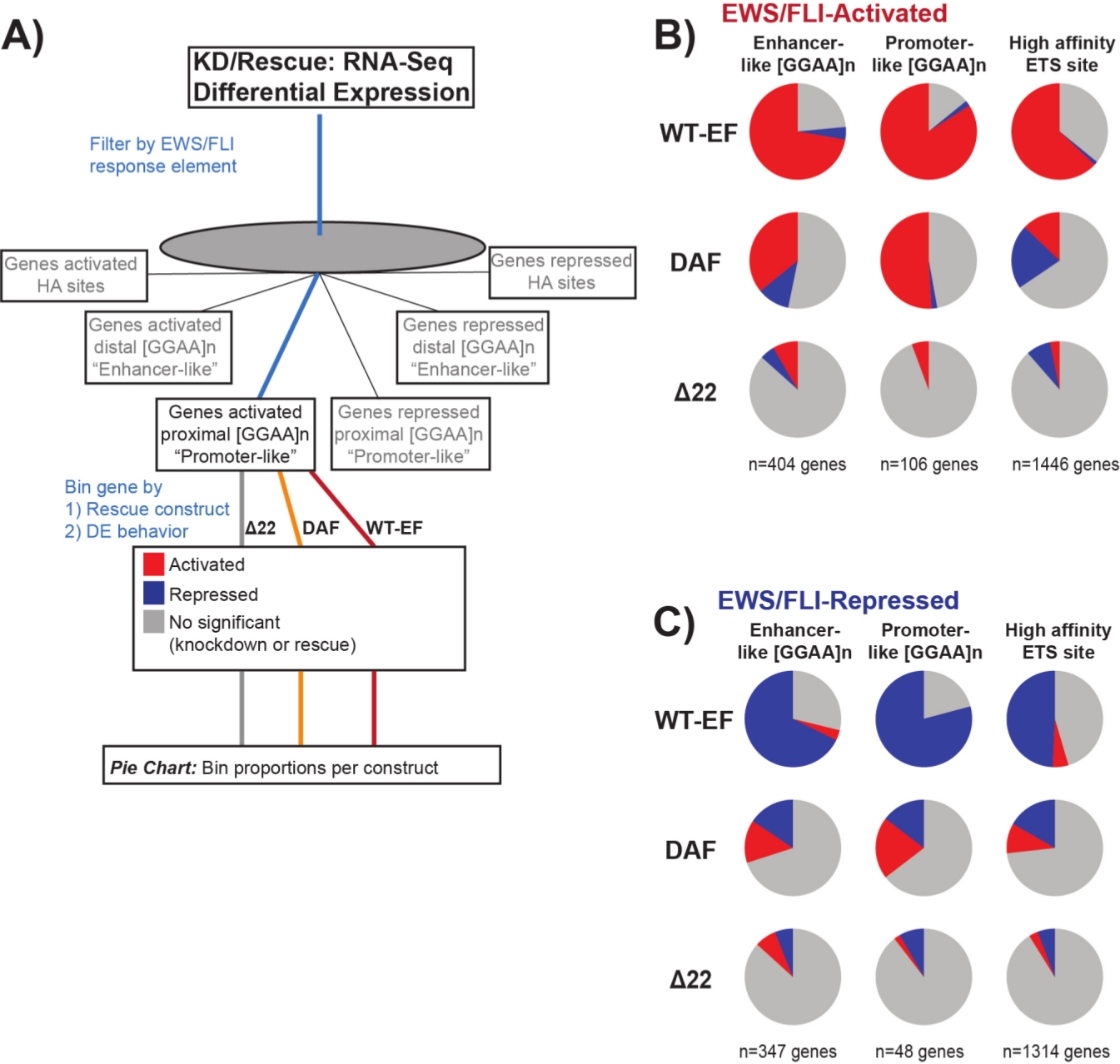
Se sabe que EWS/FLI posee una afinidad de unión única para los elementos de repetición de microsatélites GGAA19,22, y que la unión a estos elementos impulsa la regulación génica aguasabajo 11,15,18,20,22. Estos microsatélites se han caracterizado por estar asociados con la activación o la represión, y ya sea proximal a (< 5 kb) TSS o distal a (> 5 kb) TSS25. Además, existen genes regulados por EWS/FLI con motivos ETS de alta afinidad (HA) proximales a TSS23. Con el fin de analizar más a fondo las características de la función DAF y qué tipos de genes activados por EWS/FLI DAF fue capaz de rescatar, se analizó la expresión diferencial de genes asociados con estas diferentes clases. Curiosamente, DAF fue más capaz de rescatar genes activados por microsatélites GGAA, pero no pudo rescatar genes activados cerca de un sitio ha como se ve en la Figura 7. Como se ve con la agrupación jerárquica, DAF no logra rescatar la represión mediada por EWS / FLI en todas las clases de motivos. Estos datos sugieren que DAF conserva suficientes características estructurales de EWS para unirse y activarse desde microsatélites GGAA, tanto proximales como distales a TSS. Esto probablemente surge del dominio SYGQ intacto que se cree que es importante para la actividad de EWS / FLI en las repeticiones GGAA11. Estos datos también sugieren que las tirosinas específicas mutadas en DAF desempeñan un papel importante, pero poco comprendido, en la regulación génica mediada por EWS / FLI de los sitios de HA, así como en la represión génica, destacando un área importante de investigación adicional.

Figura 1: Flujo de trabajo. Representación del procedimiento paso a paso para realizar el mapeo estructura-función por transcriptómica. Las células se prepararon primero para expresar el conjunto de construcciones requeridas para el mapeo estructura-función. Después de la expresión, las células se cosecharon para ARN y proteínas y se analizaron para fenotipos correlativos. Se validó la expresión de los constructos, y este proceso se repitió 3-4 veces para recolectar réplicas biológicas independientes. El ARN se sometió a la secuenciación de próxima generación (NGS). Cuando se recibieron los datos, los datos se recortaron por calidad, se alinearon y se calcularon los recuentos por transcripción. Se controlaron los efectos por lotes y se determinaron las firmas transcriptómicas y la expresión diferencial mediante DESeq2. Se puede incorporar la agrupación jerárquica y el análisis posterior que integran otros conjuntos de datos -ómicos y diferentes vías o análisis funcionales. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Validación de la expresión de constructo y ensayos correlativos. (A) Esquema que representa las construcciones probadas en este ejemplo. (B) Validación del knockdown de EWS/FLI endógeno y expresión de constructos marcados con 3X-FLAG por immunoblot. (C,D) Validación de la actividad de constructo en un gen diana activado por EWS/FLI(C), NR0B1,y(D)gen diana reprimido, TGFBR2,por qRT-PCR. Los datos se presentan como media +/- desviación estándar. Los valores de P se calcularon con una prueba de significación honesta de Tukey. * p < 0,05, ** p < 0,01, *** p < 0,005 (E) Recuentos de colonias a partir de ensayos de agar blando realizados para evaluar la actividad transformadora de los constructos. Los valores de P se calcularon con una prueba de significación honesta de Tukey. * p < 0,05, ** p < 0,01, *** p < 0,005. Esta figura está adaptada de Theisen, et al.23Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Datos finales de conteo cotejado para el análisis. Captura de pantalla de las primeras 6 filas del archivo de recuento con recuentos de genes para todas las muestras a normalizar y analizar por lotes. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Mapas de calor de distancia de muestra a muestra. (A) Gráfico de distancia de muestra a muestra que muestra la agrupación de muestras de los datos de recuento sin procesar. Las muestras que se agrupan tanto por lote como por muestra se denotan con flechas rojas. (B) Diagrama de distancia de muestra a muestra después de la normalización de lotes con ComBat. Aquí, las muestras de todas las réplicas se agrupan, independientemente del lote. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Resultados del análisis de expresión diferencial. ( A ) El gráfico de análisis de componentes principales (PCA) de las firmas transcriptómicas generadas para todas las muestras muestranunafuerte agrupación intramuestrema y demuestran que DAF está intermediado entre los controles positivos (wtEF) y negativos (Δ22). (B) Gráficos de volcanes que muestran el -log(p-value) trazado contra el log2FoldChange para los genes en cada constructo. Genes con un valor p ajustado < 0,05 y un |log2(FoldChange)| > 1 se consideran significativas y se muestran en rojo. Panel 5B es una adaptación de Theisen, et al.23Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 6: Agrupación jerárquica para identificar clases de genes. (A) La agrupación jerárquica de los 1000 genes más variables en todos los constructos y la línea de base, iEF, muestra que DAF rescata parcialmente la activación de genes mediada por EWS / FLI. (B) La ontología génica (función molecular) resulta de ToppGene que muestra el enriquecimiento funcional de genes activados por EWS / FLI que son rescatados o no rescatados por DAF. Panel 6B está adaptado de Theisen, et al.23Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 7: Análisis detallado de diferentes elementos de respuesta del factor de transcripción a diferentes constructos: (A) Esquema que representa el procesamiento de datos utilizado para generar paneles (B) y (C) mediante la incorporación de otros conjuntos de datos disponibles con los perfiles transcriptómicos aquí. (B,C) Compilación que muestra el rescate de diferentes clases de objetivos directos EWS/FLI- (B) activados y (C) reprimidos. Los genes incluidos fueron solo aquellos genes con expresión diferencial detectable por EWS/FLI endógeno. En cada gráfico circular, el gris representa la porción de genes que no son rescatados por la construcción. El rojo representa la porción de genes que se activan diferencialmente, y el azul representa la porción de genes que se reprimen diferencialmente. Esta figura está adaptada de Theisen, et al.23Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Figura S1: Carga de los archivos fastq.gz en el entorno HPC, recorte y alineación. Haga clic aquí para descargar esta figura.
{kind=link}
Figura S2: Recopilación de recuentos de lectura entre muestras y ejecución de la normalización por lotes con ComBat. Haga clic aquí para descargar esta figura.
{kind=link}
Figura S3: Ejecución de DESeq2 y extracción de resultados del análisis de expresión diferencial. Haga clic aquí para descargar esta figura.
{kind=link}
Figura S4: Análisis de la salida. Haga clic aquí para descargar esta figura.
{kind=link}
Figura S5: Agrupación jerárquica para identificar clases de genes: Agrupación jerárquica de los 1000 genes más variables en todos los constructos y la línea de base, iEF, ordenada en k grupos. En este caso k= 7, pero este parámetro es establecido por el usuario como se muestra en la Figura S4D. Haga clic aquí para descargar esta figura.
{kind=link}
Tabla S1: Lista de genes (Ensembl gene ID) con anotación de clúster. Haga clic aquí para descargar esta tabla.
Discusión
El estudio de los mecanismos bioquímicos de los factores de transcripción oncogénicos es de vital importancia para comprender las enfermedades que causan y diseñar nuevas estrategias terapéuticas. Esto es especialmente cierto en las neoplasias malignas caracterizadas por translocaciones cromosómicas que dan lugar a factores de transcripción de fusión. Los dominios incluidos en estas proteínas quiméricas pueden carecer de interacciones significativas con los dominios reguladores presentes en las proteínas de tipo salvaje, lo que complica la capacidad de interpretar la información estructura-función en el contexto de la fusión26,27,28. Además, muchas de estas fusiones oncogénicas se caracterizan por dominios intrínsecamente desordenados de baja complejidad10,13,29,30.
El dominio EWS es un ejemplo de tal dominio intrínsecamente desordenado que está involucrado en una variedad de fusiones oncogénicas10. La naturaleza intrínsecamente desordenada y repetitiva ha obstaculizado los esfuerzos para comprender los mecanismos moleculares empleados por el dominio EWS. Los esfuerzos previos para investigar la estructura-función han recurrido en gran medida al uso de diferentes mutantes en el contexto de ensayos de genes reporteros o en fondos celulares que no logran recapitular el contexto celular relevante, o carecen de variaciones estructurales que producen una función parcial significativa11,17,25. El método que se presenta aquí aborda estas cuestiones. El mapeo estructura-función se realiza en un contexto celular relevante para la enfermedad y la secuenciación de próxima generación permite el perfil transcriptómico para evaluar la función del factor de transcripción en el entorno de la cromatina nativa. En el caso específico del mutante DAF de EWS/FLI, se informó que DAF mostró poca actividad en ensayos reporteros utilizando elementos de respuesta aislados, pero que mostró actividad en el contexto del promotor genético completo, ya sea en un ensayo reportero o en cromatina nativa, lo que sugiere un fenotipo interesante23. El uso del método descrito aquí resuelve más directamente la cuestión de qué tipo de elementos reguladores en todo el genoma son más sensibles en el entorno de la enfermedad. Al probar todos los genes diana candidatos en su contexto de cromatina nativa simultáneamente, es más probable que un enfoque transcriptómico identifique construcciones con función parcial.
La fuerza inherente de usar un fondo celular relevante para la enfermedad es quizás la mayor limitación de esta técnica. Uno de los factores más importantes es elegir el sistema celular apropiado para estos experimentos. Muchas líneas celulares derivadas de neoplasias malignas con factores de transcripción patognomónicos no toleran fácilmente la eliminación de ese factor de transcripción, y en muchos casos, particularmente para los cánceres pediátricos, la verdadera célula de origen sigue siendo controvertida y la expresión del oncogén en otros fondos celulares es prohibitivamente tóxica31,32 . En estos casos, puede ser útil realizar experimentos en un fondo celular diferente, siempre y cuando el investigador tenga cuidado en la interpretación de los resultados y valide adecuadamente cualquier hallazgo relevante en un tipo de célula más relevante para la enfermedad.
Es de vital importancia validar cuidadosamente la estabilidad y las consecuencias fenotípicas de la expresión del oncogén y enviar solo muestras para secuenciación que cumplan con criterios estrictos. Aquí, esto incluyó western blot para confirmar el derribo y rescate, y qRT-PCR de un pequeño número de genes diana conocidos para validar el control positivo(Figura 2). También es crucial disminuir la mayor variabilidad de lotes posible realizando cuidadosamente las preparaciones de células y ARN de la manera más similar posible a través de cada lote.
El método descrito aquí se vuelve especialmente poderoso cuando se combina con otros tipos de datos genómicos que hablan de la función de todo el genoma del factor de transcripción en estudio. Las direcciones futuras para este tipo de análisis de estructura-función se ampliarían para incluir ChIP-seq y ATAC-seq para determinar la unión del factor de transcripción y cualquier cambio inducido en la accesibilidad a la cromatina. Como suite, este tipo de datos pueden apuntar a dónde los diferentes componentes estructurales de un factor de transcripción oncogénico contribuyen a diferentes aspectos de la función (es decir, la unión al ADN frente a la modificación de la cromatina frente al reclutamiento de co-reguladores). En general, el uso de enfoques basados en NGS para mapear las relaciones estructura-función de los factores de transcripción de fusión puede revelar nuevos conocimientos sobre los determinantes bioquímicos de la función oncogénica de estas proteínas. Esto es importante para mejorar nuestra comprensión de las enfermedades que causan y para permitir el desarrollo de nuevas estrategias terapéuticas.
Divulgaciones
SLL declara un conflicto de intereses como miembro del consejo asesor y accionista de Salarius Pharmaceuticals. SLL también es un inventor incluido en la lista de Patentes de los Estados Unidos No. US 7,393,253 B2, "Métodos y composiciones para el diagnóstico y tratamiento del sarcoma de Ewing", y US 8,557,532, "Diagnóstico y tratamiento del sarcoma de Ewing resistente a los medicamentos". Esto no altera nuestra adhesión a las políticas de JoVE sobre el intercambio de datos y materiales.
Agradecimientos
Esta investigación fue apoyada por el Centro de Computación de Alto Rendimiento en el Instituto de Investigación Abigail Wexner en el Nationwide Children's Hospital. Este trabajo fue apoyado por el Instituto Nacional del Cáncer de los Institutos Nacionales de Salud [U54 CA231641 a SLL, R01 CA183776 a SLL]; Alex's Lemonade Stand Foundation [Premio al Joven Investigador a ERT]; Pelotonia [Beca a ERT]; y el National Health and Medical Research Council CJ Martin Overseas Biomedical Fellowship [APP1111032 a KIP].
Materiales
| Name | Company | Catalog Number | Comments |
| Wet Lab Reagents | |||
| anti-FLI rabbit pAb | Abcam | ab15289 | 1:500 |
| anti-lamin B1 rabbit pAb | Abcam | ab16048 | 1:2000 |
| Cell-based system for introduction of mutant constructs | Determined by cell system used | ||
| Cryotubes | For viral aliquots | ||
| DMEM | Corning Cellgro | 10-013-CV | For viral production |
| Fetal bovine serum | Gibco | 16000-044 | For viral production |
| G418 | ThermoFisher | 10131027 | For viral production |
| HEK293-EBNAs | ATCC | CRL-10852 | For viral production |
| HEPES | Gibco | 15630106 | |
| Hygromycin B | ThermoFisher | 10687010 | |
| M2 anti-FLAG mouse mAb | Sigma | F3165 | 1:2000 |
| Near IR-secondary antibodies | Li-Cor | ||
| Optimem | Gibco | 31985062 | For viral production |
| Penicillin/Streptomycin/Glutamine | Gibco | 10378-016 | For viral production |
| Polybrene | Sigma | TR-1003-G | For viral transduction |
| Puromycin | Sigma | P8833 | Stored at 2 mg/mL stock |
| RNeasy Plus kit | Qiagen | 74136 | Has gDNA removal columns |
| Selection reagents | As dictated by cell system used | ||
| Sodium Pyruvate | Gibco | 11360-070 | For viral production |
| Tissue culture media | Determined by cell system used | ||
| TransIT-LT1 | Mirus | MIR 2304 | For viral production |
| Software | |||
| Access to HPC environment | |||
| AnnotationDbi | 1.38.2 | ||
| Cairo | 1.5-10 | ||
| DESeq2 | 1.16.1 | ||
| genefilter | 1.58.1 | ||
| ggbiplot | 0.55 | ||
| ggplot2 | 3.1.1 | ||
| org.Hs.eg.db | 3.4.1 | ||
| pheatmap | 1.0.12 | ||
| PuTTY | |||
| R | 3.4.0 | ||
| RColorBrewer | 1.1-2 | ||
| reshape2 | 1.4.3 | ||
| rgl | 0.100.19 | ||
| R-studio | |||
| STAR | Version 2.6 or later | ||
| sva | 3.24.4 | ||
| TrimGalore! | |||
| WinSCP |
Referencias
- Miettinen, M., et al. New fusion sarcomas: histopathology and clinical significance of selected entities. Human Pathology. 86, 57-65 (2019).
- Knott, M. M. L., et al. Targeting the undruggable: exploiting neomorphic features of fusion oncoproteins in childhood sarcomas for innovative therapies. Cancer and Metastasis Reviews. 38, 625-642 (2019).
- Yoshihara, K., et al. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene. 34, 4845-4854 (2015).
- Duesberg, P. H. Cancer genes generated by rare chromosomal rearrangements rather than activation of oncogenes. Medical Oncology and Tumor Pharmacotherapy. 4, 163-175 (1987).
- Dupain, C., Harttrampf, A. C., Urbinati, G., Geoerger, B., Massaad-Massade, L. Relevance of Fusion Genes in Pediatric Cancers: Toward Precision Medicine. Molecular Therapy - Nucleic Acids. 6, 315-326 (2017).
- Mitelman, F., Johansson, B., Mertens, F. The impact of translocations and gene fusions on cancer causation. Nature Reviews Cancer. 7, 233-245 (2007).
- Smith, R., et al. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing's sarcoma. Cancer Cell. 9, 405-416 (2006).
- Davicioni, E., et al. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Research. 66, 6936-6946 (2006).
- Gröbner, S. N., et al. The landscape of genomic alterations across childhood cancers. Nature. 555, 321-327 (2018).
- Kim, J., Pelletier, J. Molecular genetics of chromosome translocations involving EWS and related family members. Physiological Genomics. 1, 127-138 (1999).
- Boulay, G., et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell. 171, 163-178 (2017).
- Lessnick, S. L., Braun, B. S., Denny, C. T., May, W. A. Multiple domains mediate transformation by the Ewing’s sarcoma EWS/FLI-1 fusion gene. Oncogene. 10, 423-431 (1995).
- Leach, B. I., et al. Leukemia fusion target AF9 is an intrinsically disordered transcriptional regulator that recruits multiple partners via coupled folding and binding. Structure. 21, 176-183 (2013).
- Ng, K. P., et al. Multiple aromatic side chains within a disordered structure are critical for transcription and transforming activity of EWS family oncoproteins. Proceedings of the National Academy of Sciences U.S.A. 104, 479-484 (2007).
- Riggi, N., et al. EWS-FLI1 Divergent Chromatin Remodeling Mechanisms to Directly Activate or Repress Enhancer Elements in Ewing Sarcoma. Cancer Cell. 26, 668-681 (2014).
- Tomazou, E. M., et al. Epigenome Mapping Reveals Distinct Modes of Gene Regulation and Widespread Enhancer Reprogramming by the Oncogenic Fusion Protein EWS-FLI1. Cell Reports. 10, 1082-1095 (2015).
- Sankar, S., et al. Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene. 32, 5089-5100 (2013).
- Gangwal, K., et al. Microsatellites as EWS/FLI response elements in Ewing's sarcoma. Proceedings of the National Academy of Sciences U.S.A. 105, 10149-10154 (2008).
- Gangwal, K., Close, D., Enriquez, C. A., Hill, C. P., Lessnick, S. L. Emergent Properties of EWS/FLI Regulation via GGAA Microsatellites in Ewing's Sarcoma. Genes & Cancer. 1, 177-187 (2010).
- Guillon, N., et al. The Oncogenic EWS-FLI1 Protein Binds In Vivo GGAA Microsatellite Sequences with Potential Transcriptional Activation Function. PLoS One. 4, 4932(2009).
- Chong, S., et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science. 361, (2018).
- Johnson, K. M., et al. Role for the EWS domain of EWS/FLI in binding GGAA-microsatellites required for Ewing sarcoma anchorage independent growth. Proceedings of the National Academy of Sciences U.S.A. 114, 9870-9875 (2017).
- Theisen, E. R., et al. Transcriptomic analysis functionally maps the intrinsically disordered domain of EWS/FLI and reveals novel transcriptional dependencies for oncogenesis. Genes & Cancer. 10, 21-38 (2019).
- Chen, J., Bardes, E. E., Aronow, B. J., Jegga, A. G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Research. 37, 305-311 (2009).
- Johnson, K. M., Taslim, C., Saund, R. S., Lessnick, S. L. Identification of two types of GGAA-microsatellites and their roles in EWS/FLI binding and gene regulation in Ewing sarcoma. PLOS One. 12, 0186275(2017).
- Kim, P., Ballester, L. Y., Zhao, Z. Domain retention in transcription factor fusion genes and its biological and clinical implications: a pan-cancer study. Oncotarget. 8, 110103-110117 (2017).
- de Mendíbil, I. O., Vizmanos, J. L., Novo, F. J. Signatures of Selection in Fusion Transcripts Resulting from Chromosomal Translocations in Human Cancer. PLOS One. 4, 4805(2009).
- Frenkel-Morgenstern, M., Valencia, A. Novel domain combinations in proteins encoded by chimeric transcripts. Bioinformatics. 28, 67-74 (2012).
- Hegyi, H., Buday, L., Tompa, P. Intrinsic Structural Disorder Confers Cellular Viability on Oncogenic Fusion Proteins. PLoS Computational Biology. 5, 1000552(2009).
- Latysheva, N. S., Babu, M. M. Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Research. 44, 4487-4503 (2016).
- Deneen, B., Denny, C. T. Loss of p16 pathways stabilizes EWS/FLI1 expression and complements EWS/FLI1 mediated transformation. Oncogene. 20, 6731-6741 (2001).
- Kendall, G. C., et al. PAX3-FOXO1 transgenic zebrafish models identify HES3 as a mediator of rhabdomyosarcoma tumorigenesis. eLife. 7, 33800(2018).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados