
Method Article
Un procedimiento ChIP-Seq semiautomatizado para estudios epigenéticos a gran escala
En este artículo
Erratum Notice
Resumen
Este artículo describe un protocolo ChIP-Seq semiautomatizado y microescalado de regiones de ADN asociadas con una modificación específica de histonas (H3K27ac) utilizando una plataforma de manejo de líquidos ChIP, seguida de la preparación de la biblioteca mediante tagmentación. El procedimiento incluye la evaluación de control con fines de calidad y cantidad y puede adoptarse para otras modificaciones de histonas o factores de transcripción.
Resumen
La inmunoprecipitación de cromatina seguida de secuenciación (ChIP-Seq) es un enfoque poderoso y ampliamente utilizado para perfilar el ADN de cromatina asociado con modificaciones específicas de histonas, como H3K27ac, para ayudar a identificar elementos de ADN cis-reguladores. El proceso manual para completar un ChIP-Seq requiere mucha mano de obra, es técnicamente desafiante y, a menudo, requiere números de células grandes (>100,000 células). El método descrito aquí ayuda a superar esos desafíos. Se describe en detalle un procedimiento completo semiautomatizado y microescalado H3K27ac ChIP-Seq que incluye fijación celular, cizallamiento de cromatina, inmunoprecipitación y preparación de la biblioteca de secuenciación, para lotes de 48 muestras para entradas de número celular inferiores a 100,000 células. La plataforma semiautónoma reduce la variabilidad técnica, mejora las relaciones señal-ruido y reduce drásticamente la mano de obra. De este modo, el sistema puede reducir los costos al permitir volúmenes de reacción reducidos, limitando el número de reactivos costosos como enzimas, perlas magnéticas, anticuerpos y tiempo práctico requerido. Estas mejoras en el método ChIP-Seq se adaptan perfectamente a los estudios epigenéticos a gran escala de muestras clínicas con un número limitado de células de una manera altamente reproducible.
Introducción
El amplio uso de ensayos ChIP-Seq para determinar fragmentos de ADN asociados con modificaciones específicas de histonas se debe en parte a su capacidad para identificar elementos de ADN cis-reguladores, incluidos potenciadores activos, promotores, silenciadores, heterocromatina y otros1,2,3,4. La identificación de regiones reguladoras no codificantes en todo el genoma ha demostrado una valiosa información para comprender mejor la regulación génica en la salud y las enfermedades4. Trabajos previos del laboratorio han utilizado ChIP-Seq para demostrar que los elementos cis-reguladores pueden desempeñar un papel importante en diferentes tipos de células5. Los ensayos chIP del factor de transcripción (TF) se han utilizado para mostrar polimorfismos de un solo nucleótido de riesgo asociado a la enfermedad6.
El uso de ChIP-Seq con muestras clínicas humanas es un desafío, principalmente debido a la limitación del número de células o la muestra de tejido deseada. Como resultado, ha habido un esfuerzo concertado en el campo para mejorar y microescalar estas técnicas y, como resultado, han surgido varios ensayos, como CUT&TAG5,7,8,9,10,11,12. Este ensayo utiliza una transposasa para etiquetar y aislar regiones genómicas unidas por un anticuerpo específico9. Esta técnica ha sido capaz de reducir el número de células a 1.000s y en algunos casos a una sola célula, sin embargo, el uso de esta técnica en la investigación traslacional y la configuración clínica ha mostrado limitaciones debido a los requisitos de uso de células vivas para este método9,12. El requisito de células vivas hace que las muestras clínicas sean logísticamente difíciles de manejar y puede introducir efectos de lote si las muestras no se procesan al mismo tiempo. Otros han optimizado las técnicas a microescala para células fijas de formaldehído, incluido el desarrollo de ChIPmentation11,que se adapta aquí de una manera de alto rendimiento. El uso de celdas fijas permite almacenar muestras hasta la recolección y el posterior procesamiento de todas las muestras juntas para minimizar los efectos por lotes.
Aquí, se describe un ensayo ChIP-Seq microescalado semiautomatizado que reduce el tiempo práctico experimental para perfilar las modificaciones de histonas10. El método semiautomatizado permite ensayos ChIP-Seq de alto rendimiento, lo que permite que hasta 48 muestras se procesen completamente y estén listas para su secuenciación en tan solo 5 días, para tan solo 10,000 células por muestra utilizando un manipulador de líquidos ChIP. El manipulador completa la inmunoprecipitación (IP) y los lavados posteriores de forma autónoma, lo que ayuda a reducir la variabilidad entre muestras. El método semiautomatizado reduce tanto el tiempo práctico en más de 15 h para 48 muestras como la variabilidad técnica, lo que permite realizar estudios epigenéticos a gran escala de manera reproducible y rápida para células primarias o cultivadas. El protocolo explica el proceso de principio a fin para ChIP-Seq de alta calidad. Si las máquinas específicas no están disponibles, el protocolo seguirá siendo un recurso útil para configurar y solucionar problemas de los experimentos ChIP-Seq manualmente.
El ensayo se realizó con tres tipos diferentes de células inmunes humanas primarias y una línea celular cultivada (HUT78 – ATCC: TIB-161). Para mayor claridad, el protocolo se ha dividido en siete secciones: fijación celular, cizallamiento de cromatina a través de sonicación, inmunoprecipitación automatizada de cromatina, preparación de bibliotecas por tagmentación de fragmentos de ADN, amplificación de bibliotecas, purificación de bibliotecas, seguida de cuantificación de ADN. Para obtener recetas de tampón, consulte la Tabla suplementaria 1.
Protocolo
La Junta de Revisión Institucional (IRB) del Instituto la Jolla de Alergia e Inmunología (LJI; Protocolo IRB no. SGE-121-0714) aprobó el estudio. Se reclutaron voluntarios sanos y se les proporcionaron muestras de leucoféresis después del consentimiento informado por escrito.
1. Fijación celular
- Llevar la concentración de suspensión celular a 1 a 2 x10 6 células/ml con medio de cultivo celular completo en un tubo de 15 ml (<10 ml de suspensión) o 50 ml de tubo (10-30 ml de células). Si <1 x 106 celdas, use 0.5 ml del medio en un tubo de 1.5 ml.
- Vórtice suavemente la suspensión celular, agregue 10x búfer de fijación de celdas hacia abajo (1:10; vol:vol) y gire a baja velocidad durante 10 minutos a temperatura ambiente (RT).
- Detenga la reacción vórtice suavemente y agregue 2.5 M de glicina en la proporción de 1:20 (vol: vol). Invierta los tubos varias veces e incube en hielo durante al menos 5 minutos.
- Realice los pasos restantes a 4 °C o sobre hielo. Gire los tubos a 800 x g durante 5 min a 4 °C y deseche el sobrenadante.
- Vuelva a suspender el pellet suavemente con 5 ml de PBS helado 1x e incube durante 2 minutos en hielo.
- Repita 1.4 y 1.5 con 1 ml de PBS 1x helado y transfiera la muestra a un tubo preenfriado de 1.5 ml (etiquetado para almacenamiento a largo plazo). Si procede, se recomienda la preparación de alícuotas.
- Gire los tubos a 1.200 x g a 4 °C y retire la mayor cantidad posible del sobrenadante sin alterar la bolita celular. Congele el pellet en nitrógeno líquido. Conservar a -80 °C.
PRECAUCIÓN: Tome la protección adecuada cuando manipule nitrógeno líquido.
2. Cizallamiento de cromatina
NOTA: Este protocolo está optimizado para el cizallamiento de cromatina de pellets con 0,3 a 3 x 106 celdas en tubos de baja unión de 0,65 ml.
- Retire el tubo de muestras con gránulos de células congeladas de -80 °C y guárdelo sobre hielo seco para evitar cualquier descongelación del gránulo antes de agregar el tampón de lisis. Este paso es crítico.
- Agregue 70 μL de tampón de lisis completa RT fresco al pellet y manténgalo en RT durante 1 min.
- Vuelva a suspender el pellet durante 1 minuto sin la introducción de burbujas y luego incube la suspensión celular en RT durante 1 minuto antes de poner la muestra en hielo.
- Transfiera el pellet resuspendido a un tubo de baja unión de 0,65 ml y manténgalo en hielo.
NOTA: Para obtener una sonicación reproducible, precaliente el sonicador ejecutándolo solo con tubos en blanco durante 3-6 ciclos antes de sonicar las muestras. - Coloque las muestras en el soporte del tubo del sonicador y llene los huecos con tubos de equilibrio llenos de 70 μL de agua. Deje las muestras en el baño de agua durante aproximadamente 1 minuto antes de comenzar la sonicación.
- Realizar sonicación para x ciclos (dependiendo del tipo de célula) con 16 s ON / 32 s OFF por ciclo.
NOTA: Este paso requerirá experimentos de validación para determinar el número óptimo de ciclos para una sonicación eficiente. - Después de cada 3 ciclos, retire las muestras del sonicador, gire suavemente el vórtice y el pulso los tubos antes de volver a colocarlos en el soporte. Asegúrese de que no haya pequeñas gotas en el exterior del tubo, ya que eso puede causar la formación de burbujas.
- Después de completar los ciclos necesarios, girar muestras a >14.000 x g durante 15 min a 4 °C. Transfiera el sobrenadante a un nuevo tubo de baja unión de 0,65 ml preenfriado y de baja unión y manténgalo en hielo.
- Desvincular una fracción de las muestras sonicadas para comprobar la eficiencia de la sonicación.
- Transfiera 1-7 μL del sobrenadante (equivalente a aproximadamente 250 ng de cromatina cortada) a un tubo de PCR de 0,2 ml y haga que el volumen sea de hasta 10 μL con tampón de lisis a corto plazo en RT.
- Añadir 1 μL de RNasa A e incubar durante 30 min a 37 °C a 800 rpm, luego añadir 1 μL de proteinasa K.
- Incubar durante 2 h a 55 °C con agitación a 1.000 rpm.
- Eliminar 2 μL de la muestra desenlazada para la cuantificación del ADN mediante el ensayo de cuantificación fluorescente10 (no se recomienda un espectrofotómetro ya que el jabón y la proteína degradada pueden producir sesgo en los resultados).
- Ejecute la muestra restante en un gel de agarosa al 1,2% durante 1 h a 70 V. Tinción con colorante de ácido nucleico (1:20.000) y lea el gel con un transiluminador UV.
- Preparar alícuotas de material de cromatina para su almacenamiento (diluir la muestra a 25 ng/μL en 20 μL con tampón de lisis completo). Conservar toda la cromatina cortada a -80 °C.
3. ChIP-Seq automatizado para la modificación de histonas
NOTA: Este protocolo está diseñado para ejecutarse en un manipulador de líquidos ChIP. Aunque el sistema puede usar búferes personalizados, todos los búferes se proporcionan con el kit ChIP. Las tiras ChIP con 8 tubos utilizadas en esta sección son específicas para el manipulador de líquidos ChIP.
- Transfiera 16 alícuotas de muestra con 500 ng de cromatina cortada en 20 μL desde -80 °C y colóquelas en hielo para descongelar lentamente la cromatina. Una vez descongelado por completo, vórtice brevemente y pulso-giro.
- Preparación de cromatina
- Pipete 100 μL de tampón tC1 suplementado con 1x inhibidor de la proteasa y 20 mM de butirato de sodio (tampón completo de tC1) en dos tiras de ChIP de 8 tubos.
- Transfiera 20 μL de cada muestra de cromatina a un tubo apropiado de las tiras de 8 tubos ChIP que contienen los 100 μL de tampón tC1 completo. Lave los tubos de cromatina agregando un tampón tC1 completo de 80 μL a los tubos de cromatina y luego transfiéralos de nuevo al tubo apropiado de las tiras de 8 tubos ChIP para un volumen final de 200 μL.
- Preparación del anticuerpo
- Calcule el volumen de anticuerpos de tal manera que haya 0,5 μg de anticuerpos en cada tubo.
Volumen de anticuerpo = ( númerode muestras x anticuerpo por reacción) / concentración de anticuerpos - Agregue la cantidad calculada de anticuerpos en 500 μL de tampón tBW1. Vórtice rápido y pulso-giro.
- Pipetear 70 μL de tBW1 en cada una de las dos tiras de 8 tubos ChIP y agregar 30 μL del anticuerpo + tBW1 a cada uno de los tubos. Esto llevará el volumen total en cada uno de los tubos a 100 μL.
- Calcule el volumen de anticuerpos de tal manera que haya 0,5 μg de anticuerpos en cada tubo.
- Preparación de la cuenta magnética
- Vórtice la proteína A solución de perlas a fondo. Para 0,5 μg de anticuerpos, pipetee 5 μL de perlas en un nuevo conjunto de tiras de 8 tubos ChIP y espín de pulso.
- Llene la última fila del manipulador de líquidos ChIP con tiras de 8 tubos ChIP etiquetadas y vacías.
- Siga las especificaciones del programa ChIP-16-IPure-200D para la colocación de todas las tiras en la máquina manipuladora de líquidos ChIP. Agregue los búferes en la posición correcta, pero use tW4 en lugar del búfer tE1.
NOTA: Organice el día de tal manera que el manipulador de líquidos ChIP realice el ChIP durante la noche. El programa se ejecutará durante aproximadamente 16 h para 16 muestras. Esto marca el final del Día 1.
4. Integración de transposasas de adaptadores de biblioteca para la preparación de bibliotecas
- Ajuste previamente un termomezclador a 37 °C y 500 rpm. Enfríe un imán para tiras de tubo de 0,2 ml sobre hielo.
- Para 16 muestras, prepare 440 μL de tampón de tagmentación en hielo. Pipetear 53 μL en una sola tira nueva de 8 tubos y mantener en hielo.
- En una nueva tira de 8 tubos de 0,2 ml, agregue 220 μL de tampón tC1 frío y manténgalo en hielo. Los tubos de 8 tiras pueden contener este volumen y aún así estar tapados.
- Retire el tubo de tira "MUESTRAS IP" de la máquina manipuladora de líquidos ChIP (fila 12) y tapone los tubos antes de girar por pulsos. Capture las perlas usando el imán para tiras de 8 tubos durante 2 minutos y retire cuidadosamente el sobrenadante.
- Transfiera 25 μL del tampón de tagmentación a las perlas con un multicanal, retire del imán y mezcle suavemente hasta que las perlas sean homogéneas (aproximadamente 5 veces hacia arriba y hacia abajo con la pipeta ajustada a 20 μL).
- Tapa los tubos y colócalos en el termomezclador precalentado e incuba durante 3 min. Aumentar el tiempo disminuirá la eficiencia de la preparación de la biblioteca.
- Transfiera los tubos a un estante de metal refrigerado y agregue un tampón tC1 refrigerado de 100 μL a cada muestra. Ajuste una pipeta multicanal a 80 μL y mezcle la muestra hasta que las perlas sean homogéneas, deteniendo la reacción de etiquetado.
- Vuelva a colocar las muestras en el manipulador de líquidos ChIP y continúe con el procedimiento de lavado Washing_for_IP-reacts_16_Ipure. Asegúrese de que el lavado se realice dos veces con tampón tC1 y dos veces con tW4. La elución debe completarse como lo marca el diseño del programa, con búfer tE1.
- Desenlace del ADN
- Retire las tiras de 8 tubos chIP en la última fila del manipulador de líquidos ChIP y agregue 2 μL RNasa A a cada muestra.
- Tapa los tubos, espín de pulso, mezcla suavemente las perlas con una pipeta multicanal hasta que la mezcla sea homogénea y vuelve a tapar los tubos.
- Incubar las muestras en un termomezclador durante 30 minutos a 37 °C y 900 rpm.
- Retire las muestras del termomezclador, agregue 2 μL de proteinasa K. Siga el mismo procedimiento que 4.9.2 después de la adición.
- Incubar las muestras en un termomezclador durante 4 h a 55 °C y 1.250 rpm, seguido de 65 °C a 1.000 rpm durante la noche.
NOTA: Este es el final del Día 2.
5. Purificación de fragmentos de ADN etiquetados
- Etiquete dieciséis tubos de 1,5 ml con el número de muestra apropiado y agregue 400 μL de tampón de unión al ADN del kit de limpieza de ADN a cada uno.
- Retire las tiras de 8 tubos del termomezclador y haga girar las tiras por pulsos para garantizar que se retenga cualquier producto evaporado. Coloque tiras en un imán de 8 tiras para capturar las cuentas.
- Transfiera 100 μL de ADN desenlazado a cada uno de los tubos de 1,5 ml. Agregue 100 μL del tampón de unión al ADN a las tiras de 8 tubos para lavar las perlas y luego transfiera al tubo apropiado de 1.5 ml.
- Vórtice durante unos 10 s y pulso-giro de los tubos de 1,5 ml.
- Cargue las columnas con los 600 μL que contienen el tampón de unión al ADN y la muestra ChIP.
- Girar muestras durante 20 s a 10.000 x g y volver a cargar la columna con el flow-through. Gira de nuevo con las mismas condiciones y descarta el flujo.
- Lave las columnas dos veces con un tampón de lavado de 200 μL (la misma centrifugación que el paso anterior) y deseche el flujo.
- Secar las columnas centrifugando durante 2 min a 12.000 x g.
- Transfiera la columna a un nuevo tubo de recolección de 1,5 ml y agregue 9 μL de TE Buffer caliente (precalentado a 55 °C) directamente a la matriz de columna. Deje que la columna se incube durante 1 min antes de la centrifugación durante 1 min a 10.000 x g.
- Transfiera los 9 μL del elutio a un nuevo conjunto apropiado de tiras de 8 tubos.
- Complete la elución nuevamente con 8 μL TE Buffer como antes. Transfiera el eluido a las tiras de 8 tubos apropiadas (volumen final 17 μL por muestra) y manténgalo en hielo.
6. Amplificación y selección de tamaño de las muestras purificadas
- Los siguientes pasos utilizan qPCR para determinar el número de ciclos necesarios para una amplificación óptima (determinación CtD – Ct)
- Prepare la mezcla de CtD para todas las muestras multiplicando el contenido del búfer de mezcla de CtD por el número de muestras.
- Dispensar 3,6 μL de mezcla de CtD en una placa qPCR y añadir 1,4 μL de muestras de ADN tagmentado (~10 % del volumen total). Realice la siguiente qPCR: 98 °C durante 3 min, 72 °C durante 5 min, 98 °C durante 30 s, 26 ciclos de 98 °C durante 10 s, 63 °C durante 30 s y 72 °C durante 30 s.
- Prepare la mezcla de amplificadores para todas las muestras multiplicando el contenido del búfer de mezcla de AMP por el número de muestras. Dispensar 14 μL de ADN marcado en pocillos separados de una placa de PCR, luego agregar 2,5 μL de dos cebadores de índice de secuenciación (25 μM) a cada muestra (el volumen de reacción final es de 50 μL).
- Mezclar las muestras mediante pipeta multicanal y realizar el programa de amplificación utilizado en el CtD con el número adecuado de ciclos.
NOTA: Este es un buen punto de parada, ya que las muestras amplificadas se pueden almacenar a -20 ° C durante unas semanas. Sin embargo, la purificación se puede completar el mismo día. Para 48 muestras, los pasos 3 a 6.5 se completaron con otros dos lotes separados y luego se amplificaron en un lote como se describe a continuación. - Realice la post-amplificación, la selección de tamaño y la cuantificación del ADN etiquetado como se describe a continuación. Esto generalmente se puede completar con 48 muestras (se puede completar con menos muestras según se desee).
- Agregue 90 μL de perlas paramagnéticas (proporción 1: 1.8) en cada pozo, mezcle y deje que se incube a RT durante 2 min.
- Captura las cuentas usando un imán de placa y desecha el sobrenadante. Lave las perlas 3 veces con 200 μL de etanol fresco al 80% sin interrumpir el pellet de perlas.
- Retire cualquier exceso de etanol con puntas de 20 μL después del lavado final y deje que las perlas se sequen durante 10 minutos o hasta que aparezcan grietas en los gránulos de perlas.
- Con la placa todavía en el imán, agregue 40 μL de agua precalentada a cada pozo. Selle la placa, vórtice a fondo y haga girar brevemente la placa por pulsos.
- Capture las perlas colocando la placa de nuevo en el imán y transfiera el elutio de 40 μL a una nueva placa de "muestra". Las muestras ahora están purificadas, y los siguientes pasos enriquecen fragmentos que van desde 200-1,000 pb.
- Paso de control de calidad opcional: Retire 4 μL de las muestras y transfiéralo a una placa de control de calidad. Añadir 4 μL de agua a las muestras. Esto determina el porcentaje de fragmentos grandes.
- Agregue 22 μL de perlas paramagnéticas (proporción 1:0.55) a las muestras, mezcle cuidadosamente e incube en RT durante 2 min.
- Coloque en el imán para capturar las cuentas durante 5 minutos y transfiera los sobrenadantes a las columnas 7-12 de la placa de "muestra". Retire la placa del imán y agregue 30 μL de cuentas (relación final de 1: 1.3). Mezcle con cuidado y deje que repose en RT durante 2 minutos.
- Captura las cuentas durante 5 minutos y luego descarta el sobrenadante.
- Lave todas las perlas 3 veces con 200 μL de etanol fresco al 80 % como se describió anteriormente (paso 6.6.2 – 6.6.3).
- Una vez que los gránulos estén secos, eluya el ADN con un tampón TE precalentado de 8 μL a cada pozo, mientras aún está en el imán.
- Retire la placa del imán, el sello y el vórtice a fondo. Deje que la placa se incube durante 2 minutos en RT, espín de pulso y vuelva a colocar la placa en el imán durante 2 minutos. Transfiera el sobrenadante a una placa nueva (Placa 2).
- Para una recuperación máxima, repita la elución con un extra de 8 μL de tampón TE precalentado. Coloque las muestras en los pozos apropiados de tal manera que cada muestra tenga 16 μL de biblioteca final.
NOTA: Al final de este paso debe haber dos placas (una si no se completó ninguna placa de control de calidad). La placa QC tendrá los fragmentos pre-seleccionados y la segunda placa debe tener 48 pocillos de biblioteca final (16 μL en total).
7. Cuantificar las bibliotecas finales y las muestras de control de calidad utilizando un ensayo de fluorescencia
- Cuantificación completa del ADN mediante un ensayo de cuantificación de fluorescencia o un método similar.
- Si se completó la cuantificación del control de calidad, determine el porcentaje de pérdida de muestra que se < 1.000 pb. No debería haber más de alrededor del 20% de pérdida; si hubiera más, podría haber un problema con las proporciones de cuentas aplicadas.
- Determinar el tamaño de los fragmentos de cada muestra, preferiblemente utilizando una máquina de electroforesis capilar. Para calcular la concentración molar utilice la siguiente ecuación: [Concentración de biblioteca (ng/μL) * 106]/[660 * Tamaño medio del fragmento (pb)]).
NOTA: Las bibliotecas están listas para ser agrupadas (cantidades equimolares) y secuenciadas siguiendo los procedimientos estándar de secuenciación de próxima generación.
Resultados
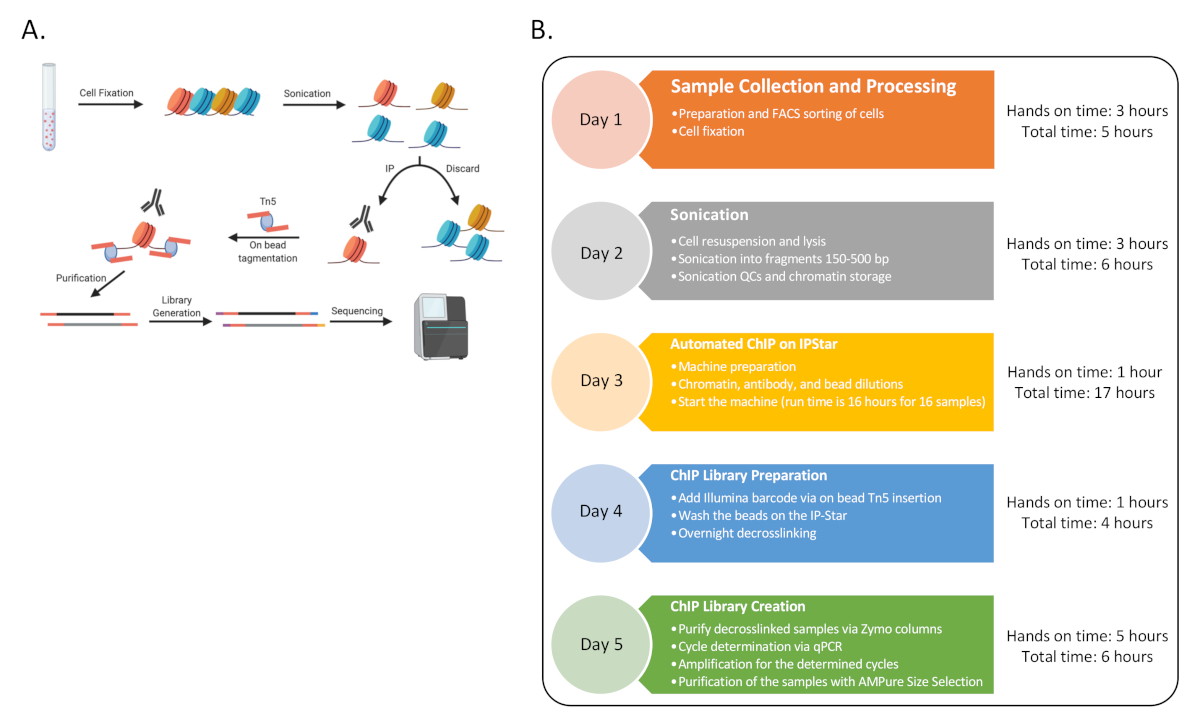
Como prueba de concepto, ChIP-Seq se completó para seis donantes humanos con tres conjuntos de tipos de células inmunes: células T CD4 ingenuas (CD4), monocitos clásicos (MO) y células asesinas naturales (NK), enriquecidas por clasificación FACS como se describió antes13. El procedimiento subrayado consta de nueve procedimientos distintos como se representa en la Figura 1.

Figura 1: Diagrama de flujo general para el procedimiento. (A) Una caricatura del procedimiento general (generado en BioRender). (B) Diagrama de flujo para todos los pasos principales para el protocolo y el tiempo práctico y total estimado asociado con cada día. La secuenciación podría ocurrir al final del Día 5 o más tarde con múltiples rondas. La línea de tiempo también se puede escalonar a lo largo de la semana, donde los días secuenciales 3-4 se pueden completar varias veces en una semana para generar 48 muestras de ChIP. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Después del aislamiento celular por citometría de flujo13,las células clasificadas se centrifugaron y las células se fijaron y almacenaron como se describió anteriormente. Una vez que se recogieron todas las muestras, las muestras se lisaron y prepararon para el cizallamiento cromático en lotes de 12 como se describió anteriormente. Para cada muestra, se completó el número de ciclos para alcanzar la sonicación óptima10. La medición cuantitativa, así como las mediciones del tamaño del fragmento de cromatina cortada mostraron una gran reproducibilidad de nuestro método en los tres conjuntos de células inmunes(Figura 2A). Las diferentes células inmunes humanas se sonicaron en lotes separados y rindieron de manera muy consistente con > el 70% de la muestra entre 100 y 500 pb durante 14 ciclos (16 s ON, 32 s OFF por ciclo). En este punto, las muestras con fragmentos grandes después de la sonicación (< el 70% de la muestra entre 100 - 500 pb) se consideraron fallidas. Estas muestras podrían sonicarse durante 1-2 ciclos adicionales o fueron descartadas y reemplazadas más tarde con células de otro pellet. Nuestro método mostró que ninguna de las muestras requirió más sonicación o fue eliminada, lo que sugiere una robustez absoluta del procedimiento.

Figura 2: Ejemplos de control de calidad de presecuenciación. (A) Los geles de agarosa al 1,2% muestran reproducibilidad de la sonicación. Muestras de sonicación para 6 donantes en tres tipos de células: células T CD4 ingenuas (CD4), monocitos clásicos (MO) y células asesinas naturales (NK). Las muestras fueron sonicadas durante 14 ciclos (16 s ON, 32 s OFF por ciclo). Para cada muestra, se cargaron alrededor de 200 ng de cromatina desenlazada en un gel de agarosa al 1%. Las muestras se consideraron buenas si más del 70 % de los fragmentos están dentro de 100-500 pb. (B) Top - Análisis de curvas de amplificación qPCR para determinar el número óptimo de ciclos para la amplificación (Ct donde hay 1/2 de la intensidad máxima). Las muestras ideales tienen un Ct de aproximadamente 15 y la amplificación se puede completar hasta 2 ciclos más del Ct medido. La flecha es un ejemplo de un mal ejemplo donde el Ct es mayor que 18. Abajo - Se muestra un ejemplo de un conjunto pobre de muestras que tienen un Ct mayor que 18. Estas muestras también mostraron una menor intensidad de fluorescencia. (C) Izquierda - Los rastros de electroforesis del analizador de fragmentos mostraron la distribución de las bibliotecas etiquetadas finales después de la amplificación y la selección de tamaño. Las muestras con más del 85 % de la biblioteca de fragmentos dentro de 200-1.000 pb se consideraron como buenas muestras. La medición de la intensidad máxima de la fluorescencia también se considera un parámetro importante de control de calidad, de hecho, si la señal es baja, es poco probable que la muestra se secuencia bien. Derecha: se muestran ejemplos de muestras positivas en CD4, MO y NK. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Después de la cuantificación, las muestras se ejecutaron en un manipulador de líquidos ChIP con anticuerpos H3K27ac, seguido de la etiquetación con la enzima transposasa Tn5. Para determinar el número apropiado de ciclos de amplificación por qPCR, se utilizó el 10% de las muestras etiquetadas. Para la determinación del número de ciclos para la amplificación de las muestras, encontramos el ciclo en el que la intensidad de la muestra es la mitad del máximo promedio para la determinación del ciclo (Figura 2B). Las muestras con valores de Ct de más de 18 no tuvieron un buen desempeño después de la secuenciación y, por lo tanto, su valor de Ct fue indicativo de una muestra de ChIP fallida. Estas muestras generalmente también produjeron una menor cantidad de ADN después de la amplificación. Las muestras (entrada de 100.000 células) con un valor de Ct igual o inferior a 15 fueron ideales y las muestras entre 15 y 18 fueron aceptables pero menos consistentes después de la secuenciación. Para muestras con menos de 100,000 celdas de entrada, los valores de Ct generalmente se encontraron entre 15 y 18, pero no necesitaron más de 18 ciclos para producir suficiente producto para la secuenciación.
Después de la amplificación del ADN etiquetado, las bibliotecas se purificaron y seleccionaron el tamaño para obtener una distribución de tamaño ideal, que oscilaba entre 200 y 1.000 pb, para la secuenciación NextGen. La evaluación de la distribución del tamaño en cada una de las bibliotecas se completó porque se obtuvieron los mejores datos de secuenciación cuando más del 85% de los fragmentos de ADN oscilaban entre 200 y 1.000 pb(Figura 2C). En particular, a medida que se cargaba la misma cantidad de ADN (medida por cuantificación de fluorescencia), se notó que las muestras con menor intensidad de fluorescencia generalmente se secuenciaban mal(Figura 2C).
Post secuenciación, se aplicaron controles de calidad estándar basados en las directrices ENCODE ChIP-Seq5,14,15.

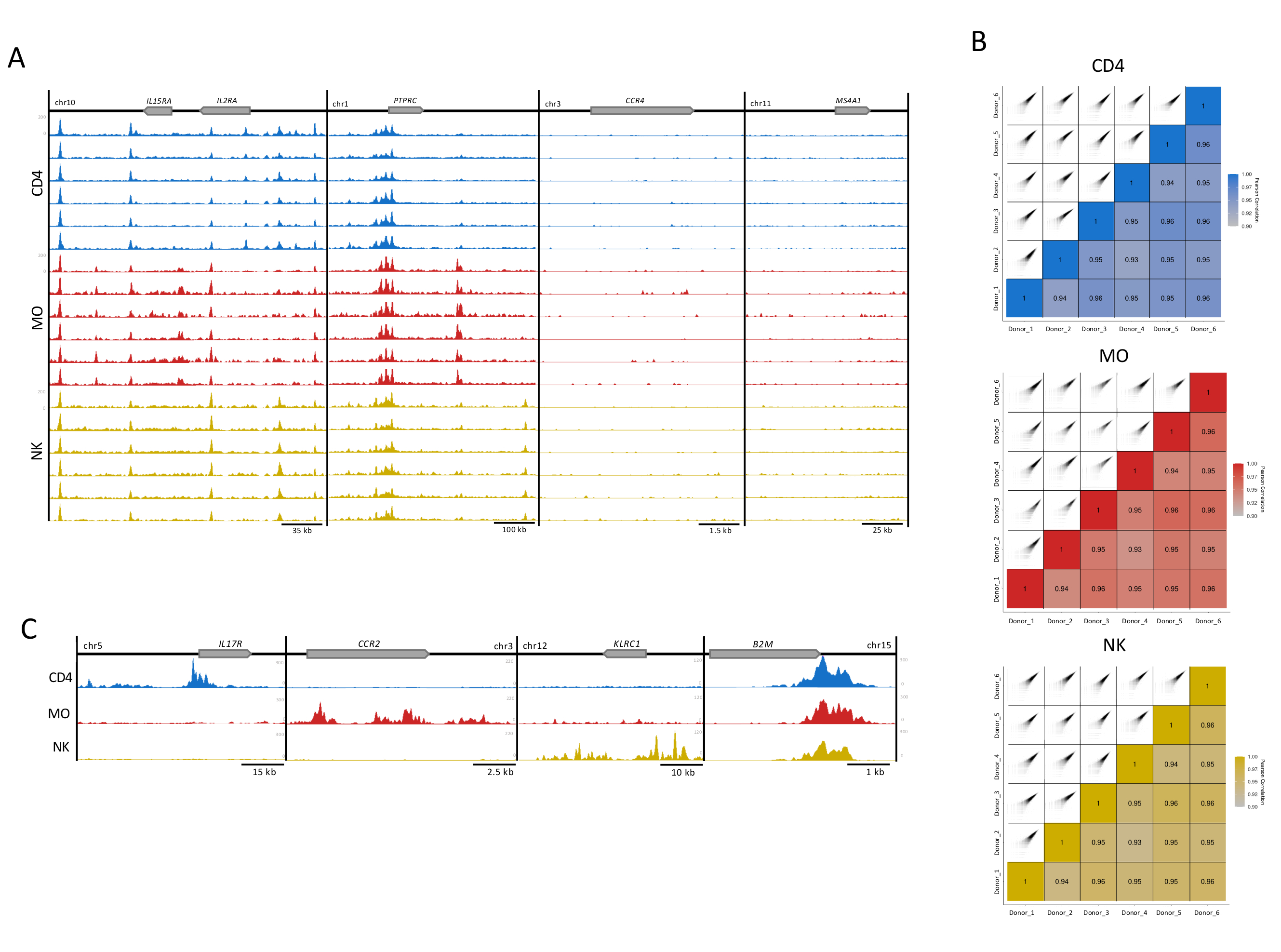
Figura 3: Reproducibilidad de las muestras de células inmunes. (A) H3K27ac rastrea (UCSC Genome Browser, intensidad máxima, función de suavizado de 4, todos con eje Y igualmente escalado) para 6 donantes (100,000 células por réplica) en cada tipo de célula (CD4, MO y NK). Se muestran cuatro loci ejemplares, dos con (locus IL2RA y PTPRC) y dos sin enriquecimiento para H3K27ac (CCR4 y MS4A1). (B) La correlación de Pearson entre los donantes y las gráficas de correlación correspondientes generadas utilizando una extensión de 300 pb y una ventana de 500 pb dentro del paquete MEDIPS para cada uno de los tipos de célula replica16. (C) Archivos de donantes fusionados para cada tipo de célula que muestran pistas H3K27ac (intensidad máxima del UCSC Genome Browser, función de suavizado de 4) en regiones específicas del tipo de célula (IL17R para CD4, CCR2 para MO y KLRC1 para NK) y el gen de mantenimiento doméstico B2M, presente en todos los tipos de células. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Para el control de calidad visual, se prepararon pistas de enriquecimiento H3K27ac para su visualización en el navegador del genoma UCSC. Para cuatro loci genéticos, las pistas individuales para cada muestra mostraron una alta calidad de mapeo y una relación señal-ruido que refleja la alta consistencia y robustez de nuestro ensayo(Figura 3A). Los dos loci a la izquierda albergan genes bien expresados en estos tipos de células, mientras que los genes en los dos loci a la derecha no se expresan y sirven como controles de fondo13 (Figura 3A). Además, se utilizó el paquete de análisis MEDIPS como variable post-secuenciación para evaluar el índice de correlación entre réplicas técnicas(Figura 3B)5,16,17,estableciendo el grado de correlación para lecturas de nivel de enriquecimiento para contenedores de 500 pb16. Para la mayoría de las comparaciones por pares, los índices de correlaciones de Pearson mostraron una correlación de más del 90%, lo que sugiere un alto nivel de consistencia entre las réplicas biológicas(Figura 3B). Las réplicas con correlación aceptable se fusionaron para aumentar la relación señal-ruido. Mientras que los loci específicos del tipo celular mostraron un alto enriquecimiento en las células apropiadas, un gen de mantenimiento doméstico (B2M) mostró una modificación de histonas muy consistente(Figura 3C). Para el análisis, la fusión de pistas de réplicas aumentará el enriquecimiento, reforzará la señal específica, incluso para importantes potenciadores específicos del tipo de célula, y reducirá la variabilidad interindividual inherente a las muestras humanas5.
Aunque se utilizaron 100,000 células para este estudio, hubo una alta reproducibilidad para tan solo 10,000 células en una línea de células T cultivadas en humanos (HUT78). El análisis de correlación entre el conjunto de datos ChIP-Seq realizado a partir de muestras con menos de 100.000 células mostró una alta reproducibilidad y correlación hasta 10.000 células(Figura 4A).

Figura 4: Reproducibilidad de muestras de baja entrada. (A) Ejemplos de la consistencia de H3K27ac ChIP-Seq para células 100,000 hasta 10,000 en células HUT-78 (una línea celular de linfoma de células T). Las pistas (UCSC Genome Browser, intensidad máxima, función de suavizado de 4, todas con eje Y igualmente escalado) muestran el locus IL4. (B) Correlaciones de Pearson de las réplicas utilizando una extensión de 300 pb y una ventana de 500 pb dentro del paquete MEDIPS16. (C) Correlaciones de Pearson entre los diferentes grupos de números celulares (100.000, 50.000 y 10.000 células) utilizando los mismos parámetros MEDIPS que en (B)16. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
El análisis de correlación de Pearson mostró un alto índice de correlación (83% a 92%), lo que sugiere el mantenimiento de la señal en muestras de bajo número de células. Sin embargo, hubo un aumento de fondo a medida que se redujeron los números de células, así como una disminución de los coeficientes de correlación(Figura 4B). Para mantener señales de fondo bajas, se fusionaron duplicados técnicos y se probó la correlación entre grupos(Figura 4C).
| Búfer de fijación de celdas 10X | |
| Compuesto | Concentración final |
| Solución de formaldehído | 11% |
| NaCl | 100 metros |
| EDTA, pH 8.0 | 1 mM |
| EGTA, pH 8.0 | 0,5 mM |
| HEPES, pH 7.5 | 50 metros |
| Búfer de lisis completo | |
| Compuesto | Concentración final |
| Tris-HCI, pH 8.0 | 50 metros |
| EDTA, pH 8.0 | 10 mM |
| SDS | 0.25% |
| Butirato de sodio | 20 metros |
| Cóctel inhibidor de la proteasa | 1X |
| Tampón de lisis a corto plazo | |
| Compuesto | Concentración final |
| Tris-HCI, pH 8.0 | 50 metros |
| EDTA, pH 8.0 | 10 mM |
| SDS | 0.25% |
| Mezcla de tagmentación | |
| Compuesto | Concentración final |
| Tris-HCI, pH 8.0 | 10 mM |
| MgCl2 | 5 mM |
| N,N-dimetilformamida | 10% |
| Enzima de tagmentación Illumina | 1:24 vol:vol |
| Mezcla de CtD | |
| Compuesto | Por muestra (μL) |
| NextEra Índice Primer A (25 μM) | 0.275 |
| NextEra Índice Primer B (25 μM) | 0.275 |
| 2X KAPA HiFi HotStart Ready Mix | 2.75 |
| 1:1000 SYBR Tinte verde | 0.11 |
| Tinte pasivo ROX | 0.11 |
| Agua | Relleno a 4 μL |
| Mezcla amp | |
| Compuesto | Por muestra (μL) |
| 2X KAPA HiFi HotStart Ready Mix | 27.5 |
| Agua | Llenar a 31 μL |
Tabla suplementaria 1: Recetas tampón.
Tabla suplementaria 2: Correlaciones de muestras de Spearman y Pearson para los 6 donantes y cada tipo de célula. Haga clic aquí para descargar esta tabla.
Discusión
El método descrito aquí amplía el procedimiento de ChIPmentation11, que implementa un protocolo de preparación de la biblioteca de tagmentación antes de la purificación del ADN, mediante la automatización y microescala del protocolo. Desde el inicio de ChIP-Seq, el número de células requeridas se ha reducido drásticamente, de aproximadamente 20 millones de células para histonas a cientos e incluso células individuales1,7,10,12,18,19,20,21. Estos métodos recientemente desarrollados han permitido una comprensión más profunda de cómo los mecanismos cis-reguladores están funcionando en las células al aumentar la sensibilidad y permitir que se prueben poblaciones de células clínicas raras5,6,12,17. Por ejemplo, uno de los procedimientos más recientes y populares, llamado CUT&TAG, como alternativa ChIP-Seq robusta y sensible9. Produce una excelente relación señal-ruido ya que la enzima Tn5 se une covalentemente a la proteína A y reconoce la cadena Fc del anticuerpo ChIP con alta especificidad9. La actividad de fondo de la enzima Tn5 se reduce ya que la enzima no es funcional antes de unirse al anticuerpo objetivo9. Sin embargo, la implementación de este método en un contexto clínico es limitada, ya que requiere células vivas no fijas. Además, la eliminación de fragmentos de ADN del núcleo hipotónico podría tener efectos negativos en la cromatina a medida que se elimina durante el ensayo. El requisito necesario para trabajar con células frescas y vivas es una fuente de problemas para muestras clínicas raras y para grandes cohortes de muestras, ya que las cohortes grandes pueden tardar numerosos años en recolectar5. Otro tipo de método, drop-ChIP, utiliza elegantemente un dispositivo de microfluídica para generar etiquetas basadas en gotas antes de procesar el ChIP19. Sin embargo, utiliza un dispositivo microfluídico altamente especializado y, si bien es posible completar ChIP-Seq de una sola célula, también se limita al uso de células vivas7,8,9,18,19. Los métodos más nuevos que se basan en ChIP-Seq, como PLAC-Seq o HiChIP, intentan comprender las interacciones de 3 dimensiones (3D) entre los picos ChIP-Seq22,23. Estos métodos 3D son emocionantes, ya que están identificando interacciones cis-reguladoras o mediadas por TF en todo el genoma y una mejor comprensión de la regulación de la expresión génica en tipos celulares de interés, en tejidos sanos y en el contexto de la enfermedad.
Hay algunos pasos críticos a considerar para que el protocolo tenga éxito, como la calidad de la cromatina sonicada y la calidad del anticuerpo. La eficiencia de cizallamiento es crítica, si la cromatina no se sonica bien, la eficiencia del ensayo disminuye drásticamente24. La sonicación es un aspecto desafiante de ChIP-Seq debido a los números de celda requeridos. En el sonicador utilizado en el protocolo, la eficiencia se redujo drásticamente en menos de 300.000 células. Este es un aspecto desafiante en ChIP-Seq, ya que sonicar por debajo de ese nivel a menudo requeriría fragmentación enzimática, que es menos imparcial. Como resultado, la sonicación es un factor limitado importante para el verdadero ChIP-Seq a microescala. Otras plataformas de sonicación y kits disponibles comercialmente se probaron para sonicar la cromatina, pero el sonicador utilizado aquí tuvo los resultados más robustos y reproducibles. Otra ventaja del sonicador es no tener que comprar tubos especializados para ejecutar la sonicación, lo que reduce los costos cuando se trata de una gran cantidad de muestras. Para una sonicación óptima, en primer lugar, es importante precalentar el sonicador como se describió anteriormente. En segundo lugar, para lisar el pellet, se recomienda que la punta de la pipeta toque la parte inferior del tubo mientras se lisa para romper las células con más restricciones físicas. En tercer lugar, cualquier formación de burbujas antes de la sonicación dificulta la capacidad de la muestra para ser sonicada de manera uniforme. Si hay burbujas formadas durante la lisis, es importante eliminarlas con una pipeta. Esto puede ser un desafío sin eliminar una gran cantidad de muestra, pero si la punta se presiona ligeramente contra la burbuja, se puede extraer lentamente sin pérdida de mucha muestra. Por último, al determinar el número de ciclos, complete un curso de tiempo donde cada tres ciclos, la muestra se elimina, se purifica y se ejecuta en un gel de agarosa. Evite la sonicación excesiva / insuficiente de las muestras, ya que esto disminuye la eficiencia de ChIP. Si la muestra está subsónica, los fragmentos grandes pueden tener un efecto negativo en la calidad ChIP-Seq24. Por otro lado, si la muestra está sobresonada, existe el riesgo de que el epítopo objetivo se pierda en el proceso.
Otra parte esencial de ChIP-Seq es la calidad del anticuerpo. Antes de realizar cualquier estudio a gran escala, es necesario optimizar el anticuerpo que se utilizará. El objetivo es obtener una relación señal/ruido significativamente alta de las regiones conocidas del genoma y otra es la reproducibilidad. Si el anticuerpo está tirando de una gran cantidad de señal de fondo, se puede recomendar usar una entrada más grande o probar un lote / proveedor diferente. Esto agregará tiempo antes de comenzar un experimento a gran escala, pero es un paso esencial. Para detectar la señal a ruido, se recomienda usar qPCR con regiones que se sabe que son un objetivo de su anticuerpo y otra región que se sabe que está ausente. Se ha notado que las modificaciones de histonas son más robustas y fáciles de optimizar que las TF.
El protocolo descrito anteriormente proporciona un método robusto para la modificación de histonas de alto rendimiento ChIP-Seq de una manera semiautónoma y microescalada. El método limita la cantidad de tiempo práctico y aumenta la reproducibilidad sobre chIP-Seq manual. Estudios previos realizados en el laboratorio utilizaron ChIP manual sobre réplicas técnicas y obtuvieron una correlación de Spearman promedio de 0,505,sin embargo, con el sistema semiautomatizado, la correlación de Spearman entre diferentes donantes con un promedio de células NK de 0,66(Tabla suplementaria 2). Esto también se completó con aproximadamente un 40% menos de tiempo práctico. El método descrito aquí ha sido optimizado para modificaciones de histonas (H3K27ac se muestra aquí, pero el protocolo no debería necesitar ninguna modificación para otros) y solo requeriría modificaciones menores para ser implementado para TF ChIP-Seq. A pesar de la calidad del anticuerpo, la principal modificación sería para el tiempo de sonicación y potencialmente los buffers utilizados durante la IP. Por lo general, para los ensayos de TF ChIP, el método puede funcionar mejor con fragmentos ligeramente más largos de cromatina (con un rango de alrededor de 350-800 pb) ya que los complejos TF: DNA probablemente sean menos capaces de mantenerse a través de una sonicación rigurosa6. Es posible que los búferes también deban cambiar a una mezcla personalizada u otros kits disponibles en la industria, ya que los TF pueden comportarse de manera diferente a las modificaciones de histonas.
Aunque el manipulador de líquidos ChIP automatizado se probó para tan solo 10,000 células, hubo una disminución notable en la reproducibilidad a concentraciones más bajas de cromatina. Debido a esto, el protocolo no se recomendó a menos de 10,000 células, siendo 100,000 células las condiciones óptimas. El protocolo también se completó utilizando búferes ChIP de la industria, lo que fue un gasto adicional pero proporcionó datos de mayor calidad. El protocolo podría modificarse con respecto a las condiciones de sonicación (siempre que la cromatina cortada se mantenga dentro del mismo rango), los tampones podrían personalizarse para la inmunoprecipitación (IP; se puede requerir optimización) o no se puede usar el manipulador de líquidos ChIP. Una limitación del protocolo es el uso del manipulador de líquidos ChIP, que puede ser una inversión costosa y solo puede ejecutar 16 muestras a la vez. El manipulador de líquidos ChIP se limita a reacciones a pequeña escala y no se recomienda un número de células superior a un millón. Sin embargo, el protocolo podría completarse sin él, completando los pasos de IP y lavado manualmente. Si la IP y los lavados se completaron a mano, el tiempo para completar el ensayo aumentará y la reproducibilidad puede disminuir, pero esta guía seguirá siendo útil para ejecutar un experimento ChIP-Seq de alta calidad. Cabe destacar que otros manipuladores de líquidos podrían adaptarse para ejecutar reacciones ChIP semiautomatizadas.
En resumen, los principales beneficios de este sistema es la naturaleza de alto rendimiento, ya que los pasos de IP y lavado se completan de forma autónoma. Como tal, se pueden completar rondas secuenciales de experimentos ChIP, lo que permite que hasta 48 muestras se procesen completamente y estén listas para la secuenciación en 5 días, con un tiempo práctico limitado en comparación con los experimentos manuales ChIP-Seq. Otro beneficio es el aumento de la reproducibilidad ya que ChIP-Seq puede ser difícil de obtener resultados altamente reproducibles. Otros métodos requieren células vivas, sistemas complejos de microcantilización o que el trabajo se complete a mano. Este sistema tendrá que ser optimizado para muestras de baja entrada (<10.000 células), permitiendo en última instancia reacciones ChIP de una sola célula. El sistema también es capaz de ser adaptado para los nuevos métodos ChIP, como PLAC-Seq y HiChIP22,23.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Agradecemos a los miembros del laboratorio de Vijayanand por la ayuda técnica y las discusiones constructivas y al Dr. Sharron Squazzo y al Sr. Geoffrey Berguet de Diagenode por la asistencia técnica con el sonicador y la máquina y los protocolos de manejo de líquidos ChIP. Este trabajo fue apoyado por las subvenciones de los NIH AI108564, R01HL114093, S10RR027366 (BD FACSAria II) y S10OD016262 (Illumina HiSeq 2500).
Materiales
| Name | Company | Catalog Number | Comments |
| 200 µl tube strips (8 tubes/strip) + cap strips | Diagenode | C30020002 | Strip tubes for use on the IP Star; ChIP 8-tube strip |
| AMPure XP for PCR Purification | Beckman Coulter | A63880 | SPRI beads |
| Axygen 0.6 mL MaxyClear Snaplock Microcentrifuge Tube | Corning | MCT-060-C | 0.65 mL low binding tube |
| Bioruptor Pico Sonicator | Diagenode | B01060010 | Sonicator used in the lab but others can be used |
| ChIP DNA Clean & Concentrator (Capped Columns) | Zymo Research | D5205 | DNA clean-up kit |
| Dynabeads Protein A for Immunoprecipitation | ThermoFisher | 10001D | |
| EDTA (0.5 M), pH 8.0, RNase-free | ThermoFisher | AM9260G | |
| EGTA pH 8.0 | Millipore Sigma | E3889-25G | |
| Eppendorf ThermoMixer C | Eppendorf | 2231000667 | |
| Formaldehyde solution | Millipore Sigma | 252549-1L | |
| Glycine | Millipore Sigma | 50046-250G | |
| H3K27ac polyclonal antibody - Premium | Diagenode | C15410196 | |
| HEPES (1 M) pH 7.5 | ThermoFisher | 15630080 | |
| IDT for Illumina Nextera DNA Unique Dual Indexes | Illumina | 20027213 | |
| Illumina Tagment DNA Enzyme and Buffer Small Kit | Illumina | 20034197 | |
| IP-Star Compact Automated System | Diagenode | B03000002 | Automated system for ChIP-Seq studies; ChIP liquid handler |
| KAPA HiFi HotStart ReadyMix | Roche | KK2601 | PCR mix |
| Medium reagent container for SX-8G IP-Star Compact | Diagenode | C30020003 | |
| MgCl2 (magnesium chloride) (25 mM) | ThermoFisher | R0971 | |
| N,N-Dimethylformamide | Millipore Sigma | D4551-250ML | CAUTION - low flash point |
| NaCl (5 M), RNase-free | ThermoFisher | AM9760G | |
| PBS (10X), pH 7.4 | ThermoFisher | 70011044 | |
| PCR Flex-free 8-tube stripes, attached individual optical caps | USA Scientific | 1402-4700 | 8 strip tubes, 0.2 mL 8-tube strip |
| Proteinase Inhibitor Cocktail | Millipore Sigma | P8340 | |
| Proteinase K Solution (20 mg/mL), RNA grade | ThermoFisher | 25530049 | |
| PureLink RNase A (20 mg/mL) | ThermoFisher | 12091021 | |
| Quant-iT PicoGreen dsDNA Reagent | ThermoFisher | P7581 | Used in the flourescence quantification |
| QuantStudio 6 Flex Real-Time PCR System | ThermoFisher | 4485699 | qPCR |
| ROX Reference Dye | ThermoFisher | 12223012 | |
| Sodium butyrate | Millipore Sigma | 303410-100G | |
| SYBR Gold Nucleic Acid Gel Stain (10,000X Concentrate in DMSO) | ThermoFisher | S11494 | nucleic acid dye |
| SYBR Green I Nucleic Acid Gel Stain - 10,000X concentrate in DMSO | ThermoFisher | S7563 | |
| TE Buffer | ThermoFisher | 12090015 | |
| Tips (bulk) | Diagenode | C30040020 | Tips for the IP Star |
| True MicroChIP Kit | Diagenode | C01010130 | Contains all the buffers for the IP; ChIP kit |
| UltraPure 1M Tris-HCI, pH 8.0 | ThermoFisher | 15568025 | |
| UltraPure SDS Solution, 10% | ThermoFisher | 24730020 |
Referencias
- Barski, A., et al. High-resolution profiling of histone methylations in the human genome. Cell. 129 (4), 823-837 (2007).
- Johnson, D., Mortazavi, A., Myers, R., Wold, B. Genome-Wide Mapping of in Vivo Protein-DNA Interactions. Science. 316, 1497-1502 (2007).
- Mikkelsen, T. S., et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 448 (7153), 553-560 (2007).
- Furey, T. S. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nature Reviews Genetics. 13 (12), 840-852 (2012).
- Seumois, G., et al. Epigenomic analysis of primary human T cells reveals enhancers associated with TH2 memory cell differentiation and asthma susceptibility. Nature Immunology. 15 (8), 777-788 (2014).
- Schmiedel, B. J., et al. 17q21 asthma-risk variants switch CTCF binding and regulate IL-2 production by T cells. Nature Communication. 7, 13426 (2016).
- Ai, S., et al. Profiling chromatin states using single-cell itChIP-seq. Nature Cell Biology. 21 (9), 1164-1172 (2019).
- Brind'Amour, J., et al. An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nature Communication. 6, 6033 (2015).
- Kaya-Okur, H. S., et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nature Communication. 10 (1), 1930 (1930).
- Youhanna Jankeel, D., Cayford, J., Schmiedel, B. J., Vijayanand, P., Seumois, G. An Integrated and Semiautomated Microscaled Approach to Profile Cis-Regulatory Elements by Histone Modification ChIP-Seq for Large-Scale Epigenetic Studies. Methods Molecular Biology. 1799, 303-326 (2018).
- Schmidl, C., Rendeiro, A. F., Sheffield, N. C., Bock, C. ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nature Methods. 12 (10), 963-965 (2015).
- Skene, P. J., Henikoff, S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife. 6, 21856 (2017).
- Schmiedel, B. J., et al. Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell. 175 (6), 1701-1715 (2018).
- Landt, S. G., et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Research. 22 (9), 1813-1831 (2012).
- Chen, L., et al. Genetic Drivers of Epigenetic and Transcriptional Variation in Human Immune Cells. Cell. 167 (5), 1398-1414 (2016).
- Lienhard, M., Grimm, C., Morkel, M., Herwig, R., Chavez, L. MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics. 30 (2), 284-286 (2014).
- Engel, I., et al. Innate-like functions of natural killer T cell subsets result from highly divergent gene programs. Nature Immunology. 17 (6), 728-739 (2016).
- Grosselin, K., et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nature Genetics. 51 (6), 1060-1066 (2019).
- Rotem, A., et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nature Biotechnology. 33 (11), 1165-1172 (2015).
- Wang, Q., et al. CoBATCH for High-Throughput Single-Cell Epigenomic Profiling. Molecular Cell. 76 (1), 206-216 (2019).
- Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Fang, R., et al. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Research. 26 (12), 1345-1348 (2016).
- Mumbach, M. R., et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods. 13 (11), 919-922 (2016).
- Diagenode. The Ultimate Guide for Chromatin Shearing Optimization with Bioruptor Standard and Plus. Diagenode. , (2012).
Erratum
Formal Correction: Erratum: A Semiautomated ChIP-Seq Procedure for Large-scale Epigenetic Studies
Posted by JoVE Editors on 9/14/2020. Citeable Link.
An erratum was issued for: A Semiautomated ChIP-Seq Procedure for Large-scale Epigenetic Studies. An author's name was updated.
The name was corrected from:
Pandurangan Vijayanad
to:
Pandurangan Vijayanand
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados