
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Un protocolo integral de alta eficiencia para el aislamiento, cultivo, polarización y caracterización glucolítica de macrófagos derivados de la médula ósea
En este artículo
Resumen
Este protocolo proporciona métodos detallados y completos para el aislamiento, cultivo, polarización y medición del estado metabólico glucolítico de macrófagos vivos derivados de la médula ósea (BMDM). Este documento proporciona instrucciones paso a paso con ilustraciones visuales realistas para el flujo de trabajo y la evaluación glucolítica de BMDM en tiempo real.
Resumen
Los macrófagos se encuentran entre las células presentadoras de antígenos más importantes. Se han identificado muchos subconjuntos de macrófagos con firmas metabólicas únicas. Los macrófagos se clasifican comúnmente como subtipos M1 (inflamatorios) y M2 (antiinflamatorios). Los macrófagos tipo M1 son macrófagos proinflamatorios que son activados por LPS y/o citocinas proinflamatorias como INF-γ, IL-12 e IL-2. Los macrófagos polarizados tipo M1 están involucrados en varias enfermedades al mediar la defensa del huésped contra una variedad de bacterias y virus. Esto es muy importante para estudiar los macrófagos M1 inducidos por LPS y sus estados metabólicos en enfermedades inflamatorias. Los macrófagos similares a M2 se consideran macrófagos antiinflamatorios, activados por citocinas y estimuladores antiinflamatorios. Bajo el estado proinflamatorio, los macrófagos muestran un aumento de la glucólisis en la función glucolítica. La función glucolítica se ha investigado activamente en el contexto de la glucólisis, la capacidad glucolítica, la reserva glucolítica, la glucólisis compensatoria o la acidificación no glucolítica utilizando analizadores de flujo extracelular (XF).
Este artículo demuestra cómo evaluar los estados glucolíticos en tiempo real con pasos fáciles de seguir cuando los macrófagos derivados de la médula ósea (BMDM) están respirando, consumiendo y produciendo energía. Utilizando inhibidores y activadores específicos de la glucólisis en este protocolo, mostramos cómo obtener una visión sistémica y completa de los procesos metabólicos glucolíticos en las células y proporcionar resultados más precisos y realistas. Para poder medir múltiples fenotipos glucolíticos, proporcionamos un método de normalización fácil, sensible y basado en el ADN para la evaluación de la polarización de los BMDM. El cultivo, la activación/polarización y la identificación del fenotipo y el estado metabólico de los BMDM son técnicas cruciales que pueden ayudar a investigar muchos tipos diferentes de enfermedades.
En este trabajo, polarizamos los macrófagos M0 naïve a macrófagos similares a M1 y M2 con LPS e IL4, respectivamente, y medimos un conjunto completo de parámetros glucolíticos en BMDM en tiempo real y longitudinalmente a lo largo del tiempo, utilizando análisis de flujo extracelular y activadores e inhibidores glucolíticos.
Introducción
Los macrófagos son una de las células más críticas del sistema inmunitario innato tipo M1. Están implicados en la eliminación de enfermedades infecciosas, fagocitosis, presentación de antígenos y regulación de la inflamación2. Además, se requiere que los macrófagos regulen otras células inmunitarias a través de varias citocinas que liberan3. Existe un amplio espectro en los fenotipos de macrófagos4. Dependiendo de las señales a las que están expuestos los macrófagos, se polarizan hacia diferentes estados inflamatorios ymetabólicos 5. Los macrófagos manifiestan alteraciones metabólicas en diversas enfermedades, dependiendo del tejido en el que residan los macrófagos6. Los macrófagos polarizados tienen la capacidad de reprogramar o cambiar su metabolismo glucolítico, metabolismo lipídico, metabolismo de aminoácidos y fosforilación oxidativa mitocondrial (OXPHOS)7,8. Los macrófagos tipo M1 activados clásicamente y los macrófagos similares a M2 alternativamente activados son los dos fenotipos de macrófagos más estudiados3. Los macrófagos inactivos no activados se denominan macrófagos M0. La polarización de los macrófagos M0 hacia un fenotipo similar a M1 puede inducirse mediante la estimulación de BMDMs ingenuos con lipopolisacárido bacteriano (LPS)9. La vía de señalización PI3K-AKT-mTOR-HIF1a puede activarse en macrófagos en presencia de citocinas inflamatorias, interferón-gamma (IFN γ) o factor de necrosis tumoral (TNF)10. Los macrófagos similares a M1 tienen niveles aumentados de metabolismo de glucólisis, niveles disminuidos de fosforilación oxidativa (OXPHOS), produciendo citocinas inflamatorias involucradas en enfermedades infecciosas e inflamatorias8. Por otro lado, la polarización hacia el fenotipo tipo M2 puede ser inducida por la interleucina (IL)-4, a través de las vías JAK-STAT, PPAR y AMPK, o por (IL)-13 y TGFβ pathays11,12.
A diferencia de los macrófagos M1, los macrófagos M2 tienen una glucólisis disminuida y un aumento de OXPHOS y están involucrados en actividades antiparasitarias y de reparación de tejidos 8,13. Los BMDM son un sistema ampliamente utilizado para el estudio de los macrófagos que se derivan de las células madre de la médula ósea. La glucólisis y el OXPHOS son las dos principales vías de producción de energía enlas células. En función de su microentorno, los BMDM pueden optar por utilizar cualquiera de estas vías; En algunos casos, cambie de uno a otro, o utilice ambas vías14. En este estudio, nos centramos en el metabolismo de la glucólisis en macrófagos proinflamatorios activados. Cuando la glucosa en el citoplasma se convierte en piruvato y luego en lactato, las células producen protones en el medio que causan una elevación en la tasa de acidificación en el medio rodeado de células similares a M15. Se utilizó un analizador de flujo extracelular para medir la tasa de acidificación de los medios celulares. Los resultados se informan como Tasa de Acidificación Extracelular (ECAR) o como Tasa de Eflujo de Protones.
Un método optimizado, rápido y fácil para acceder a los niveles de glucólisis en macrófagos polarizados es esencial para determinar el fenotipo glucolítico, los cambios en los metabolitos y los efectos de los inhibidores/activadores y fármacos en los macrófagos polarizados. El método descrito en este manuscrito ha sido optimizado para proporcionar información sobre factores específicos de glucólisis (glucólisis, capacidad glucolítica, reserva glucolítica y acidificación no glucolítica), así como la reprogramación metabólica del metabolismo glucolítico. El inhibidor (2DG) que se ha utilizado en este estudio se dirige explícitamente a la vía de la glucólisis.
Este protocolo optimizado ha sido modificado y mejorado en base a la combinación de un protocolo publicado16, el análisis de flujo extracelular de ensayos glucolíticos de las guías de usuario del fabricante y la comunicación directa con los científicos de investigación y desarrollo del fabricante.
Access restricted. Please log in or start a trial to view this content.
Protocolo
Los ratones fueron sacrificados humanamente de acuerdo con las pautas de la Evaluación y Acreditación del Cuidado de Animales de Laboratorio (AAALAC) y la Asociación Americana para la Ciencia de los Animales de Laboratorio (AALAS) y utilizando protocolos aprobados por el Comité Institucional de Cuidado y Uso de Animales de la Universidad de Texas A&M (IACUC).
1. Recolección de médula ósea de ratones y cultivo de BMDM
- Sacrificar al ratón (6-10 semanas de edad C57Bl/6 ratones estaban en este protocolo) y colocarlo sobre su lado ventral, cortar la piel y la capa peritoneal y despegar suavemente las patas.
NOTA: Utilice la exposición al gas CO2 para aplicar la eutanasia al ratón. - Separe ambas patas traseras de la cadera hacia abajo, teniendo cuidado de no cortar el hueso.
- Coloque toda la pierna en un tubo cónico vacío de 50 ml (con los pies hacia arriba para tener un agarre fácil de sacar más tarde) sobre hielo y proceda a recolectar ambas patas del ratón.
2. Exposición del fémur
NOTA: Realice los siguientes pasos en una cabina de bioseguridad.
- Extraiga el fémur cortando la tibia de cada pierna y extraiga la mayor cantidad posible de tejido que rodea el fémur con tijeras y papel de laboratorio.
- Coloque los fémures recolectados y "limpios" en una placa de 10 cm que contenga un trozo de papel de laboratorio saturado con medio de cultivo de tejidos (TC) o PBS. Colócalos sobre hielo.
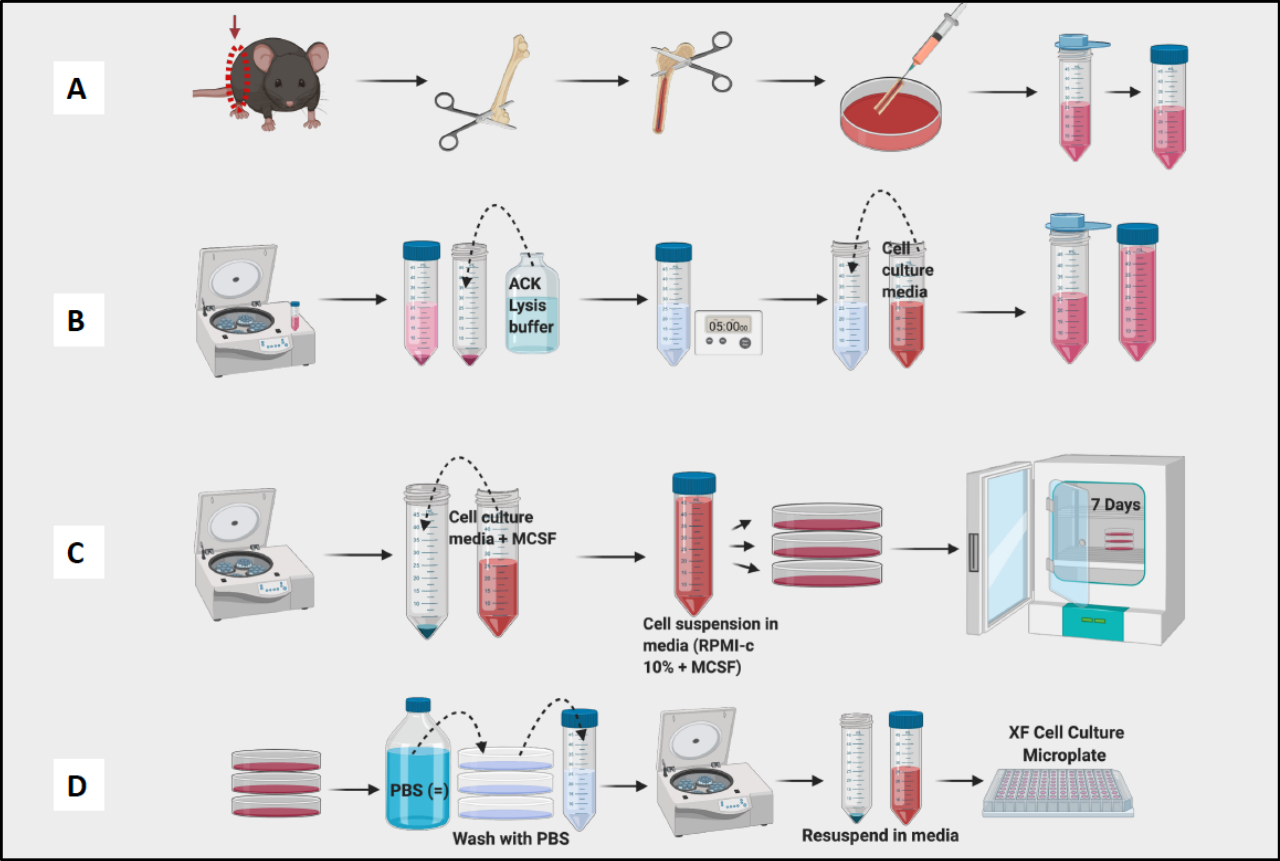
- Continúe recolectando fémures y retirando tejido de todos los fémures antes de proceder a la etapa de lavado (Figura 1A).
3. Enjuague de tuétano
- Para lavar la médula de los fémures, use una jeringa de 3 ml llena de medio TC o PBS con una aguja de 23 G. Llene la jeringa antes de exponer la médula.
- Usa tijeras para exponer la médula cortando el extremo del fémur en ambas epífisis.
- Inserte la punta de la aguja en el fémur y enjuague suavemente la médula en un plato de 10 cm.
- Pasa la aguja por toda la longitud del fémur y enjuaga hasta que el color del hueso se vuelva blanco. Por lo general, la mayoría de la médula se puede lavar con 2-3 mL de medio.
- Enjuague todos los fémures y la médula ósea del pool en el plato. Use una aguja para romper los grumos visibles. Filtre la médula en un tubo cónico de 50 mL (Figura 1A).
4. Lisis de glóbulos rojos
- Centrifugar el tuétano a 190 x g durante 10 min. Aspirar el sobrenadante.
- Vuelva a suspender el pellet en 4 mL de tampón de lisis ACK con una pipeta. Deje que el tampón de lisis RBC actúe durante 5 minutos a temperatura ambiente.
- Añadir 4 mL de medio TC RPMI-C 10% (RPMI 1640 -GlutaMAX) suplementado con 2-mercaptoetanol, gentamicina, estreptomicina y 10% FCS a la suspensión de médula y centrifugar a 1300 x g durante 10 min.
- Cuele nuevamente para eliminar los desechos de glóbulos rojos y vuelva a suspender en un pequeño volumen de RPMI-C 10% para contar.
- Cuente las celdas con un contador de celdas (Figura 1B). Se utilizó un contador Vi-Cell para determinar el recuento y la viabilidad de las células en la suspensión.
5. Enchapado y cultivo
- Agregue 10 mL de RPMI-C 10% + 10 ng/mL M-CSF (Factor Estimulante de Colonias de Macrófagos, un regulador esencial de la proliferación, diferenciación y supervivencia de monocitos/macrófagos) a tantas placas de 10 cm como desee.
- Agregue un volumen apropiado de celdas contadas de modo que cada placa de 10 cm contenga 1 x 106 celdas. Coloque las placas en una incubadora a 37 °C (definida como día 0).
- El día 3, agregue suavemente 5 mL de RPMI-C fresco 10% + 10 ng/mL M-CSF a cada placa.
NOTA: El día 7, los BMDM deben estar listos para la prueba (Figura 1C).
6. Cosecha de platos
- Use un microscopio óptico para confirmar que la mayoría de las células se han adherido a las placas.
- Aspire suavemente los medios. A continuación, añada 3 ml de PBS y agite suavemente la placa. Aspire este pozo para eliminar cualquier célula no adherente restante.
- Agregue de 7 a 10 ml de PBS frío a la placa, use una pipeta P1000 para lavar la parte inferior de las placas y recoja todas las células restantes en un tubo de recolección.
NOTA: Mantenga los tubos en hielo, ya que los macrófagos se adhieren muy firmemente y se adherirán al interior del tubo. Si las células se mantuvieran frías, estarían menos adheridas. - Celdas de centrífuga, recuento y placa para experimentos (Figura 1D). Mediante citometría de flujo, las células resultantes deben tener una positividad del >95% para CD11b y F4/80. (La polarización de los macrófagos se determinó mediante tinción con marcadores similares a M1 de CD38, TNF-a y MCP-1 y marcador similar a M2 de CD206.
NOTA: Realice los pasos 6.1-6.3 en la cabina de bioseguridad y realice el paso 6.4 en la mesa de trabajo. Mantener técnicas asépticas durante todo el procedimiento.

Figura 1: Flujo de trabajo gráfico del cultivo de médula ósea de ratón de macrófagos derivados de BM. (A) Extracción de piernas, exposición del fémur y lavado de la médula; b) lisis de glóbulos rojos; C) Enchapado y cultivo; (D) Recolección de células de las placas. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
7. El día antes del ensayo del analizador de flujo metabólico: siembra y polarización de las células para la prueba glucolítica
- Caliente el analizador de flujo metabólico a 37 °C encendiendo el instrumento.
- Hidratar un cartucho añadiendo 200 μL de una solución calibrante e incubar el cartucho en una incubadora sin CO2 durante la noche (Figura 2A). La humedad de la incubadora sin CO2 no es importante para la hidratación del cartucho.
- Una hora antes del experimento, sumerja la placa unas cuantas veces hacia arriba y hacia abajo, lo que ayudará a eliminar las burbujas de aire.
- Diseñe el mapa de placas en el software en la prueba de esfuerzo de glucólisis predeterminada-inyección aguda, siguiendo las instrucciones de la prueba.
- Haga clic en el icono del software y luego haga clic en Prueba de inyección aguda de estrés de glucólisis. En el icono de definición de grupo, genere nombres de grupo.
- Hay cinco ciclos de medición con una duración de 18 minutos y cuatro inyecciones. Cambie la inyección del puerto A a glucosa, el puerto B a oligomicina, el puerto C a rotenona y antimicina A (Rot/AA) y el puerto D a 2DG.
- Vuelva a suspender las celdas en medio RPMI-C 10% y siembre 50k celdas por pocillo, excepto los cuatro bordes de la placa (A1, A12, H1 y H12; Agregue solo medios, no células) en una microplaca del analizador de flujo metabólico hasta un volumen final de 100 μL. Normalmente se requiere un mínimo de 40k células para realizar este ensayo.
- Deje que las celdas se asienten a temperatura ambiente durante 45 minutos para evitar el efecto de borde de las celdas. El efecto de borde es cuando el medio de alrededor del perímetro de la placa se evapora parcialmente, lo que provoca cambios de volumen y concentración y reduce la viabilidad de la célula.
- Agregue 10 ng/mL de LPS para polarizar los macrófagos naïve hacia células similares a M1 y agregue 20 ng/ml de IL-4 para polarizarlos hacia células similares a M2. Utilice al menos de 3 a 6 pocillos por condición (Figura 2B).
- Revise las células bajo el microscopio y coloque la placa en una incubadora a 37 °C y 5% de CO2 durante 24 horas.

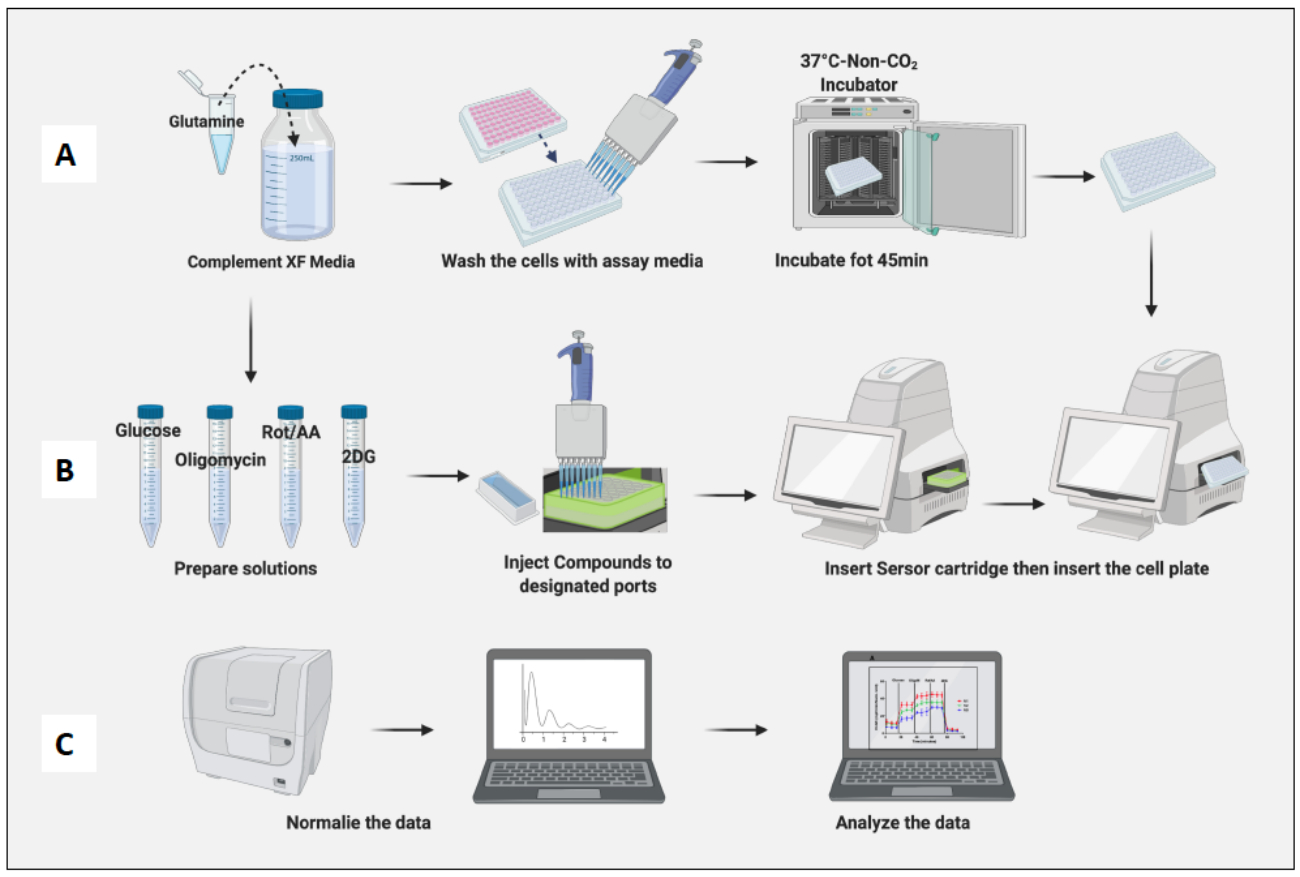
Figura 2: Demostración gráfica de la siembra y polarización de las células. (A) Configuración del analizador de flujo extracelular e hidratación del cartucho; (B) Polarización de las células e incubación durante la noche. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
8. Día del ensayo: XF Preparación del medio y del compuesto
- Complemente 100 mL de medio de ensayo XF RPMI (pH 7.4) con 2 mM de glutamina.
- Filtre-esterilice los medios utilizando un sistema de filtro de vacío de 0,2 μm.
- Coloque el medio de ensayo en un baño de agua a 37 °C durante 20 minutos.
- Retire las celdas plateadas de la incubadora a 37 °C con 5% de CO2 . Lave las células con medios de ensayo dos veces y reemplace el medio anterior con medio de ensayo hasta el volumen final de 180 μL.
- Use un microscopio para asegurarse de que todos los pocillos tengan células confluentes y marque los pocillos que tengan rasguños por el pipeteo. Si hay algún rasguño, retire esa placa antes de analizar.
- Coloque la placa que contiene células en una incubadora sin CO2 durante 45 minutos (Figura 3A).
- Uso de los compuestos y los medios de ensayo para hacer soluciones madre de glucosa (100 mM), oligomicina (100 μM), Rot/AA (50 μM) y 2DG (500 mM) (Tabla 1).
- Realice una mezcla de inyección 10x de cada compuesto utilizando medios de ensayo (Tabla 2).
| Culatas de inyección (proporcionadas en los kits) | Añadir Medios de ensayo completos (mL) | Concentración de stock final (μM) |
| Glucosa | 3 | 100K |
| Oligomicina | 0.72 | 100 |
| 2-DG | 3 | 100 mil |
Tabla 1. Culatas de inyección
| Puertos en el cartucho | Soluciones de stock | Añadir volumen de stock | Adición de medios de ensayo | Concentración final de inyecciones (10x) | Agregue este volumen al puerto designado (μL) | Concentración final después de la inyección en cada pocillo |
| Un | Glucosa (100 mM) | 3000 μL + 0 μL | 100 mM | 20 | 10 mM | |
| B | Oligomicina (100 μM) | 300 μL + 2700 μL | 10 μM | 22 | 1,0 μM | |
| C | Rotenona/Antimicina A (50 μM) | 300 μL + 2700 μL | 5 μM | 25 | 0,5 μM | |
| D | 2-DG (500 mM) | 300 μL + 0 μL | 500 mM | 28 | 50 mM | |
Tabla 2. Concentraciones finales de inyección
9. Día del ensayo: Realización de la prueba glucolítica aguda en macrófagos polarizados
- Abra la plantilla de ensayo de esfuerzo de glucólisis (inyección aguda) guardada desde el software. La prueba de glucoestrés agudo predeterminada tiene 3 minutos de mezcla y medición antes de cada inyección.
- Verifique la plantilla y los detalles del ensayo y, cuando esté listo, haga clic en Ejecutar y siga las instrucciones del ensayo predeterminado. Sin embargo, todos los parámetros se pueden personalizar.
- Retire el cartucho sensor de la incubadora que no sea de CO2 , retire la tapa e insértelo en el instrumento de manera que el pocillo A1 de la placa del cartucho caiga en la esquina superior izquierda del panel de inserción de la máquina. Por lo general, la calibración tarda entre 20 y 45 minutos.
- Una vez finalizada la calibración, el dispositivo expulsará la placa que contiene la solución calibrante y contendrá el cartucho del sensor. Retire el calibrante que contiene la placa.
- Retire la placa de celdas de la incubadora sin CO2 , retire la tapa de la placa e insértela en la máquina. Haga clic en Ejecutar (Figura 3B).
- Una vez finalizado el ensayo, la máquina expulsará la placa de celda y el cartucho del sensor.
- Retire el medio de la placa y congélelo a -20 °C para una mayor normalización.
- Utilice el kit comercial de ensayo de proliferación celular (por ejemplo, CyQUANT) para normalizar las células.
- Agregue 1 mL de compuesto B o tampón de lisis a 19 mL de agua destilada sin nucleasa.
- Añadir 100 μL de solución de trabajo compuesta A o GR a la solución mencionada anteriormente.
- Asegúrese de que las células de la placa estén descongeladas y, a continuación, añada 200 μL de la solución a cada pocillo.
- Incubar durante 5 min a temperatura ambiente (RT).
- Mida la fluorescencia en longitudes de onda de excitación de 480 nm y de emisión de 520 nm utilizando un lector de placas.
- Normalice las celdas en el panel de normalización del software.
- Normalice las células en función del recuento de células de macrófagos ingenuos (Figura 3C). Considere el promedio de los macrófagos ingenuos como 1 (dividiendo el número de células de cada pocillo por el número promedio de celdas de los macrófagos naïf) y aplíquelos a todos los macrófagos.

Figura 3: Día del ensayo: preparación del medio y del compuesto y ejecución del ensayo. (A) Preparación de las células para el ensayo; (B) Preparación de compuestos, calibración y ejecución del ensayo; C) Normalización y análisis de datos. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
Resultados
La glucólisis y la fosforilación oxidativa mitocondrial son las dos principales vías de producción de ATP en las células (Figura 4A). Algunas células tienen la capacidad de cambiar entre estas dos vías para satisfacer sus demandas de energía. La conversión de glucosa en piruvato en el citoplasma se denomina glucólisis. El piruvato tiene dos destinos; se convertirá en lactato o se metabolizará aún más a través d...
Access restricted. Please log in or start a trial to view this content.
Discusión
Como se mencionó anteriormente, la máquina analizadora de flujo extracelular puede proporcionar información en tiempo real sobre dos de las principales vías de producción de energía de las células mediante la medición de OCR (tasa de consumo de oxígeno), un indicador de la actividad mitocondrial de OXFOS, y ECAR (tasa de acidificación extracelular), que es un indicador de glucólisis. Los macrófagos pueden utilizar ambas vías, dependiendo de su microentorno. También pueden c...
Access restricted. Please log in or start a trial to view this content.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Agradecemos a la Sra. Joanna Rocha por su asistencia editorial. El trabajo fue parcialmente apoyado por los Institutos Nacionales de Salud (NIH, por sus siglas en inglés) R01DK118334 (a los Dres. Sun y Alaniz) y (NIH) R01A11064Z (a los Dres. Jayaraman y Alaniz).
Access restricted. Please log in or start a trial to view this content.
Materiales
| Name | Company | Catalog Number | Comments |
| 23G needles | VWR | BD305145 | |
| 2-mercaptoethanol | Life Technologies | 21985023 | |
| 50ml Conical Tube | VWR | 21008-951 | |
| ACK lysis buffer | Thermo Fisher Scientific | A1049201 | It can be lab-made |
| Agilent Seahorse XF glycolysis stress test kit | Agilent Technologies | 103020-100 | |
| Agilent Seahorse XF Glycolysis Stress Test Kit User Guide | Agilent Technologies | 103020-400 | |
| Agilent Seahorse XF Glycolytic Rate Assay Kit | Agilent Technologies | 103344-100 | |
| Agilent Seahorse XF Glycolytic Rate Assay Kit User Guide | Agilent Technologies | 103344-100 | |
| Alexa Fluor 488 anti-mouse CD206 (MMR) Antibody | BioLegend | 141710 | |
| anti-mouse CD11b eFluor450 100ug | eBioscience | 48-0112-82 | |
| BD 3ML - SYRINGE | VWR | BD309657 | Other syringes are acceptable too |
| Cell counter-Vi-CELL- XR Complete System | BECKMAN COULTER Life Sciences | 731050 | Cells can be manually counted too |
| Cell Strainer-70µm | VWR | 10199-656 | |
| CyQUANT Cell Proliferation Assay Kit | Thermo Fisher Scientific | C7026 | |
| F4/80 monoclonal antibody (BM8) pe-Cyanine7 | eBioscience | 25-4801-82 | |
| Fetal Bovine Serum | Life Technologies | 16000-044 | |
| Flow cytometer: BD LSFRFortessa X-20 | BD | 656385 | |
| Kim Wipes | VWR | 82003-822 | |
| LPS-SM ultrapure (tlrl-smpls) 5 mg | Invivogen | tlrl-smlps | |
| MCSF | Peprotech | 315-02 | |
| Murine IL-4 | Peprotech | 214-14 | |
| PE Rat Anti-Mouse CD38 | BD Biosciences | 553764 | |
| Penicillin-Streptomycin (10,000 U/mL) | Life Technologies | 15140122 | |
| Petri Dish 100mm x 15 mm | Fisher Scientific | F80875712 | |
| RPMI, Glutamax, HEPES | Invitrogen | 72400-120 | |
| Seahorse Calibrant Solution | Agilent Technologies | 103059-000 | |
| Seahorse XF 200mM Glutamine Solution | Agilent Technologies | 103579-100 | |
| Seahorse XF Glycolytic Rate Assay Kit | Agilent Technologies | 103344-100 | |
| Seahorse XFe96 FluxPaks | Agilent Technologies | 102416-100 | |
| XF Glycolysis Stress Test Kit | Agilent Technologies | 103020-100 | |
| XF RPMI Medium, pH 7.4 without phenol Red | Agilent Technologies | 103336-100 |
Referencias
- Rosowski, E. E. Determining macrophage versus neutrophil contributions to innate immunity using larval zebrafish. Disease Models & Mechanisms. 13 (1), (2020).
- Martinez-Pomares, L., Gordon, S. The Autoimmune Diseases. , Elsevier. 191-212 (2020).
- Orecchioni, M., Ghosheh, Y., Pramod, A. B., Ley, K. Macrophage polarization: different gene signatures in M1 (LPS+) vs. classically and M2- (LPS–) vs. alternatively activated macrophages. Frontiers in immunology. 10, 1084(2019).
- Barrett, T. J. Macrophages in atherosclerosis regression. Arteriosclerosis, Thrombosis, and Vascular Biology. 40 (1), 20-33 (2020).
- Ni, Y., et al. Adipose tissue macrophage phenotypes and characteristics: the key to insulin resistance in obesity and metabolic disorders. Obesity. 28 (2), 225-234 (2020).
- Chu, C., et al. Modulation of foreign body reaction and macrophage phenotypes concerning microenvironment. Journal of Biomedical Materials Research Part A. 108 (1), 127-135 (2020).
- Batista-Gonzalez, A., Vidal, R., Criollo, A., Carreño, L. J. New insights on the role of lipid metabolism in the metabolic reprogramming of macrophages. Frontiers in Immunology. 10, 2993(2020).
- Kang, S., Kumanogoh, A. The spectrum of macrophage activation by immunometabolism. International Immunology. , (2020).
- Shi, Y., et al. M1 But Not M0 Extracellular Vesicles Induce Polarization of RAW264. 7 Macrophages Via the TLR4-NFκB Pathway In Vitro. Inflammation. , 1-9 (2020).
- Zhang, L., Li, S. Lactic acid promotes macrophage polarization through MCT-HIF1α signaling in gastric cancer. Experimental Cell Research. 388 (2), 111846(2020).
- Geng, T., et al. CD137 signaling induces macrophage M2 polarization in atherosclerosis through STAT6/PPARδ pathway. Cellular Signalling. , 109628(2020).
- Liu, Q., et al. Combined blockade of TGf-β1 and GM-CSF improves chemotherapeutic effects for pancreatic cancer by modulating tumor microenvironment. Cancer Immunology & Immunotherapy. , 1-16 (2020).
- Rigoni, T. S., et al. RANK Ligand Helps Immunity to Leishmania major by Skewing M2 Into M1 Macrophages. Frontiers in Immunology. 11, 886(2020).
- Viola, A., Munari, F., Sánchez-Rodríguez, R., Scolaro, T., Castegna, A. The metabolic signature of macrophage responses. Frontiers in Immunology. 10, (2019).
- Ivashkiv, L. B. The hypoxia–lactate axis tempers inflammation. Nature Reviews Immunology. 20 (2), 85-86 (2020).
- Van den Bossche, J., Baardman, J., de Winther, M. P. Metabolic characterization of polarized M1 and M2 bone marrow-derived macrophages using real-time extracellular flux analysis. Journal of Visualized Experiments. (105), e53424(2015).
- Van den Bossche, J., et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell reports. 17 (3), 684-696 (2016).
- Liu, P. S., Ho, P. C. Metabolic Signaling. , Springer. 173-186 (2019).
- Wang, F., et al. Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell metabolism. 28 (3), 463-475 (2018).
- Romero, N., Rogers, G., Neilson, A., Dranka, B. Quantifying Cellular ATP Production Rate Using Agilent Seahorse XF Technology. , Agilent Technologies, Inc. (2018).
- Mookerjee, S. A., Brand, M. D. Measurement and analysis of extracellular acid production to determine glycolytic rate. Journal of Visualized Experiments. (106), e53464(2015).
- Mookerjee, S. A., Goncalves, R. L., Gerencser, A. A., Nicholls, D. G., Brand, M. D. The contributions of respiration and glycolysis to extracellular acid production. Biochimica Et Biophysica Acta-Bioenergetics. 1847 (2), 171-181 (2015).
- Yang, S., et al. Macrophage polarization in atherosclerosis. Clinica Chimica Acta. 501, 142-146 (2020).
- Hörhold, F., et al. Reprogramming of macrophages employing gene regulatory and metabolic network models. PLoS Computational Biology. 16 (2), e1007657(2020).
- Han, X., Ma, W., Zhu, Y., Sun, X., Liu, N. Advanced glycation end products enhance macrophage polarization to the M1 phenotype via the HIF-1α/PDK4 pathway. Molecular and Cellular Endocrinology. , 110878(2020).
- Müller, E., et al. Toll-like receptor ligands and interferon-γ synergize for induction of antitumor M1 macrophages. Frontiers in Immunology. 8, 1383(2017).
- Soto-Heredero, G., Gómez de las Heras, M. M., Gabandé-Rodríguez, E., Oller, J., Mittelbrunn, M. Glycolysis–a key player in the inflammatory response. The FEBS Journal. , (2020).
Access restricted. Please log in or start a trial to view this content.
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados