
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Uso del sistema GAL4-UAS para genética funcional en Anopheles gambiae
En este artículo
Resumen
El sistema bipartito GAL4-UAS es una herramienta versátil para la modificación de la expresión génica de forma espaciotemporal controlada que permite el análisis genético funcional en Anopheles gambiae. Los procedimientos descritos para el uso de este sistema son una estrategia de clonación semi-estandarizada, sexado y cribado de pupas para marcadores de proteínas fluorescentes y fijación embrionaria.
Resumen
El sistema bipartito GAL4-UAS es una herramienta versátil y potente para el análisis genético funcional. La esencia del sistema es cruzar líneas transgénicas "conductoras" que expresan el factor de transcripción de levadura GAL4 de una manera específica del tejido, con líneas transgénicas "respondedoras" que llevan una construcción candidata de interferencia de gen / ARN cuya expresión está controlada por secuencias de activación ascendentes (UAS) que se unen a GAL4. En la progenie subsiguiente, el gen o la construcción silenciadora se expresa de una manera espaciotemporal prescrita, lo que permite ensayar los fenotipos resultantes e inferir la función génica. El sistema binario permite flexibilidad en los enfoques experimentales para detectar fenotipos generados por la expresión transgénica en múltiples patrones específicos de tejidos, incluso si se inducen costos severos de aptitud física. Hemos adaptado este sistema para Anopheles gambiae, el principal vector de malaria en África.
En este artículo, proporcionamos algunos de los procedimientos comunes utilizados durante el análisis GAL4-UAS. Describimos las líneas GAL4-UAS de An. gambiae ya generadas, así como la clonación de nuevas construcciones de respuesta para la regulación ascendente y el derribo de ARNi. Especificamos una guía paso a paso para el sexado de pupas de mosquitos para establecer cruces genéticos, que también incluye la detección de la progenie para seguir la herencia de marcadores genéticos fluorescentes que etiquetan las inserciones del conductor y del respondedor. También presentamos un protocolo para la limpieza de embriones de An. gambiae para estudiar el desarrollo embrionario. Finalmente, introducimos posibles adaptaciones del método para generar líneas conductoras a través de la inserción CRISPR/Cas9 de GAL4 aguas abajo de los genes diana.
Introducción
El sistema bipartito GAL4-UAS es el caballo de batalla de la caracterización funcional de genes en el organismo modelo de insectos Drosophila melanogaster1,2,3. Para utilizar el sistema GAL4-UAS, las líneas conductoras transgénicas, que expresan el factor de transcripción de levadura GAL4 bajo control de una secuencia reguladora, se cruzan con líneas de respuesta que llevan un gen de interés o construcción de interferencia de ARN (RNAi) controlada por una secuencia de activación ascendente (UAS) reconocida por GAL4. La progenie de este cruce expresa el transgén de interés en un patrón espaciotemporal dictado por el promotor que controla la expresión de GAL4 (Figura 1). Los fenotipos mostrados por la progenie de los cruces conductor-respondedor se pueden evaluar para dilucidar la función de los genes candidatos. Aunque D. melanogaster se ha utilizado para examinar genes de otros organismos4,5,6,7, el sistema GAL4-UAS ha sido adaptado para su uso en insectos de importancia médica y agrícola para proporcionar un análisis directo en las especies de interés 8,9,10,11,12,13,14.
En el mosquito africano de la malaria, Anopheles gambiae, el sistema GAL4-UAS se probó por primera vez mediante la coinfección de líneas celulares9. Se ensayaron múltiples construcciones para determinar la eficiencia en diferentes combinaciones por pares y se encontró que 14 UAS repetidos en tándem complementados con un pequeño intrón artificial (UAS-14i) mostraron el rango más amplio de potencial de activación cuando se usaron con un panel de controladores GAL4. Para demostrar la funcionalidad in vivo, estos constructos se utilizaron para crear dos líneas transgénicas separadas de An. gambiae mediante transformación de PiggyBac8: una línea conductora que transporta GAL4 impulsada por un promotor específico del intestino medio, y una línea de respuesta que contiene tanto la luciferasa como los genes de proteína fluorescente amarilla mejorada (eYFP) bajo regulación de secuencias de UAS. La actividad de la luciferasa específica intestinal y la fluorescencia en la progenie indicaron que el sistema era eficiente en Anopheles. Desde entonces, se han creado líneas conductoras que expresan transgenes en otros tejidos importantes para la capacidad vectorial y la resistencia a los insecticidas, incluidos los enocitos15 y hemocitos16, y en un patrón casi ubicuo10. También se han generado numerosas líneas de UAS para evaluar genes que se cree que están involucrados en el metabolismo y la resistencia a insecticidas mediada por el secuestro, la síntesis de hidrocarburos cuticulares y para etiquetar fluorescentemente diferentes tipos de células y tejidos (Tabla 1). Para las líneas de respuesta, la integración dirigida al sitio del transgén ahora se realiza mediante el intercambio de casetes de recombinación catalizados por ΦC311111111 para fijar el contexto genómico de los genes regulados por UAS. De esta manera, la expresión transgénica se normaliza con respecto a la ubicación de la inserción genómica, lo que permite una comparación más precisa de los efectos fenotípicos de diferentes genes candidatos.
Las líneas de respuesta creadas hasta la fecha están diseñadas para expresar el transgén a niveles elevados o para reducir la expresión génica a través de la interferencia de ARN (ARNi). Por lo general, los clones de ADNc se fusionan con la secuencia UAS para generar plásmidos de expresión adecuados, sin embargo, las secuencias genómicas completas también son factibles suponiendo que no sean demasiado grandes para la clonación. Para generar construcciones silenciadoras, hemos utilizado tres métodos diferentes para obtener secuencias invertidas en tándem adecuadas que forman dsRNA de horquilla que estimula el ARNi. Estos han incluido PCR de fusión, PCR asimétrica y síntesis comercial de construcciones de horquilla. Común a cada método es la inclusión de una secuencia de intrón entre las secuencias invertidas para proporcionar estabilidad de clonación. Se han desarrollado plásmidos respondedores en los que se puede insertar un gen de interés/construcción de ARNi15. Estos plásmidos también llevan los sitios attB ΦC31 requeridos para RMCE (descritos en el artículo de Adolfi que acompaña a JoVE que describe la técnica RCME en detalle). En este manuscrito se incluyen protocolos que cubren los pasos importantes requeridos al seleccionar la secuencia para la inserción en uno de estos plásmidos para la sobreexpresión. Además, se describen e ilustran dos protocolos para la creación de construcciones de horquillas de RNAi.
Al crear nuevas líneas, la identificación de individuos transgénicos raros es crucial para reproducirse para establecer y mantener colonias transgénicas. Lo más importante para el sistema GAL4-UAS es la necesidad de distinguir las líneas de respuesta y conductor para establecer cruces e identificar la progenie individual que transporta ambos transgenes. Esto se logra mediante el uso de diferentes genes marcadores seleccionables dominantes vinculados a los casetes del conductor y del respondedor. Más comúnmente, estos son genes marcadores fluorescentes que son claramente distinguibles utilizando filtros ópticos (por ejemplo, eYFP, eCFP, dsRed). Es importante que los marcadores se expresen en un patrón espaciotemporal conocido y confiable, ya que esto facilita la identificación de anomalías y contaminación. La expresión génica del marcador fluorescente es regulada rutinariamente por el promotor sintético 3xP3, que causa la expresión específica de los ganglios oculares y ventrales en todas las etapas del desarrollo de An. gambiae19. Los marcadores fluorescentes controlados por 3xP3 se incluyen en todos los plásmidos de transformación descritos en este artículo. Aquí se incluye un protocolo que detalla los métodos comunes utilizados para detectar líneas fluorescentes an. gambiae pupae GAL4-UAS.
Uno de los elementos clave del sistema GAL4-UAS es la necesidad de cruzar las líneas de conductor y respuesta marcadas diferencialmente. Para hacer esto, los machos y las hembras de cada línea deben separarse antes del apareamiento. Los adultos son fácilmente distinguibles por la vista, sin embargo, para establecer cruces genéticos es sensato separar los sexos antes de la aparición adulta para garantizar que no se haya producido el apareamiento. La diferencia general de tamaño entre las pupas de An. gambiae macho y hembra es demasiado variable para ser un método eficiente y confiable de determinación del sexo20. En cambio, las claras diferencias morfológicas en los genitales externos proporcionan una base confiable para el sexado en An. gambiae. En este artículo, describimos un método confiable para sexar pupas de An. gambiae para establecer cruces apropiados.

Figura 1 - Representación esquemática del proceso para el uso del sistema bipartito GAL4-UAS en Anopheles gambiae. (A) Se representan los componentes principales de un vector de ejemplo (pSL-attB-UAS14-gyp[3xp3-eYFP]), detallando los sitios de restricción disponibles (EcoRI, NheI, XhoI y NcoI) dentro de los múltiples sitios de clonación que son adecuados para su uso para insertar la construcción de horquilla o la secuencia de codificación para el gen de interés. También se representa la estructura de la línea de acoplamiento. (B) El paso de cruce se ilustra indicando el uso de machos de la línea del conductor (que transportan al conductor GAL4 por un promotor de interés y eCFP impulsado por el promotor 3xP3) y hembras de la línea de respuesta (portadores del gen de interés o construcción de horquilla controlada por un promotor de UAS y un marcador eYFP controlado por el promotor 3xP3). (C) Una representación esquemática de GAL4 que impulsa la expresión del gen de interés en la progenie de la cruz en B y una lista de algunos de los fenotipos típicos que se evalúan. Abreviaturas: Sitio de clonación múltiple (MCS), Intercambio de casete mediado por recombinasa (RMCE), Secuencia del activador ascendente (UAS), Proteína fluorescente amarilla mejorada (eYFP), Proteína fluorescente cian mejorada (eCFP). Haga clic aquí para ver una versión más grande de esta figura.
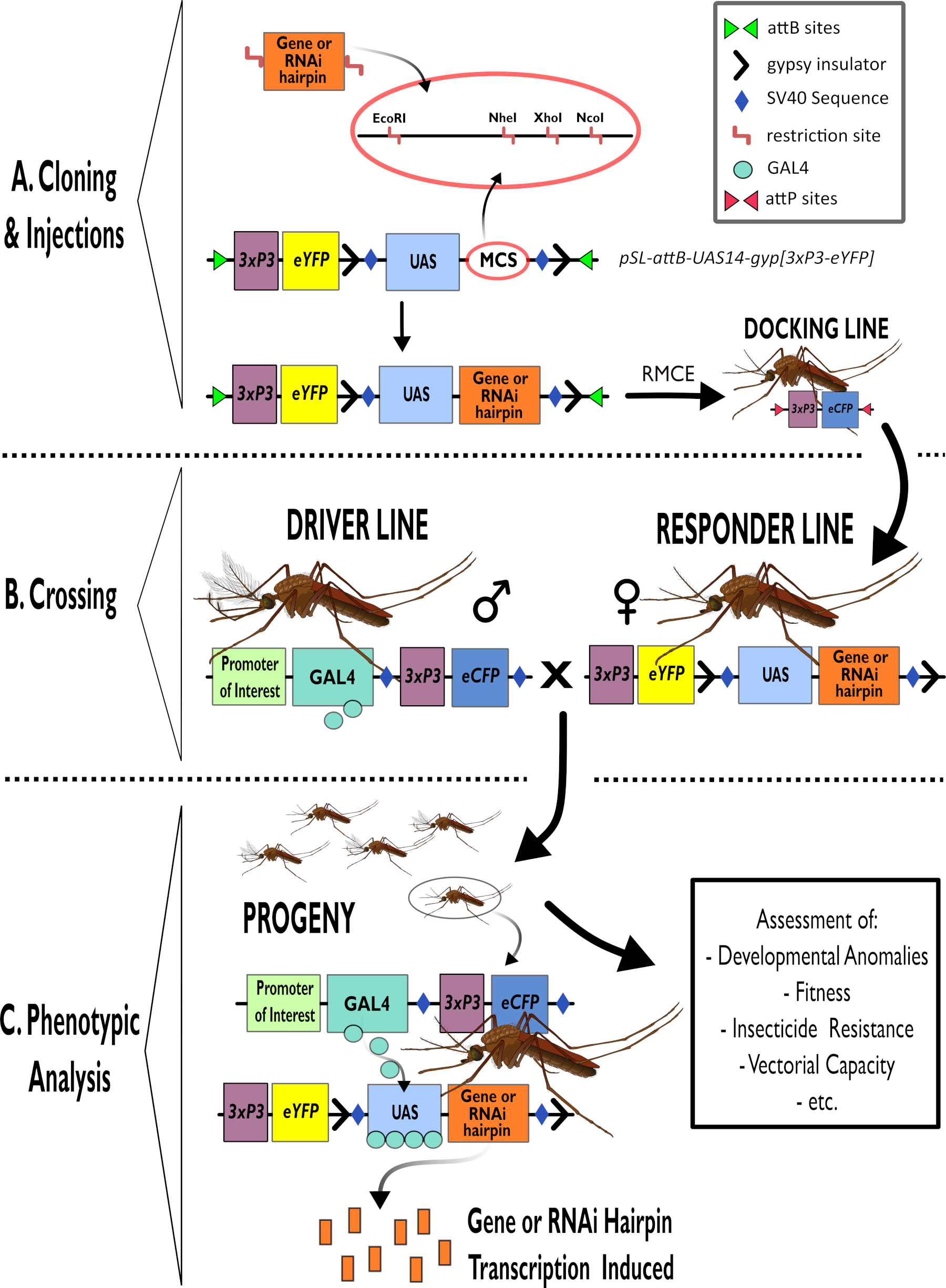{kind=link}
Es el uso de cruces lo que proporciona la naturaleza bipartita del sistema GAL4-UAS, que tiene claras ventajas sobre los enfoques más lineales. Por ejemplo, se pueden evaluar muchas más combinaciones de líneas de conductor y respuesta de lo que sería factible si se tuviera que generar y mantener una nueva línea transgénica para cada combinación promotor/gen. Más importante aún, permite el análisis de genes que producen fenotipos letales o estériles cuando se perturba su expresión que son difíciles de crear/mantener en un sistema lineal. Tales fenotipos letales pueden manifestarse en todas las etapas del desarrollo, dependiendo de la función génica y la expresión espaciotemporal, pero se observan con mayor frecuencia durante el desarrollo embrionario. Visualizar el desarrollo embrionario del mosquito requiere la limpieza del corion opaco que recubre los huevos. Siguiendo los métodos descritos en Trpiš (1970)21 y Kaiser et al. (2014)22, describimos los protocolos que utilizamos para fijar embriones, manteniendo la integridad estructural, y el blanqueamiento para despejar el endodocoro que permite la visualización microscópica y la obtención de imágenes.
Protocolo
1. Diseño y construcción de construcciones de UAS
- Diseño y montaje de vectores para la expresión génica candidata
- Determinar la secuencia que se utilizará para la regulación ascendente de genes candidatos.
- Secuenciar el ADNc/ADNc a partir de la cepa de interés y compararlo con la secuencia publicada para verificar su identidad e identificar posibles SNP y sitios de restricción para la digestión diagnóstica.
- Asegúrese de que el cebador delantero utilizado para la amplificación de genes cubra la secuencia nativa de Kozak y el codón de inicio, cuando corresponda. Una imprimación con ~ 10 pb de unión aguas arriba del codón de inicio abarcará la secuencia de Kozak.
- Incluya el codón de parada en el fragmento amplificado desde el cebador inverso en la mayoría de las circunstancias. Utilice secuencias de terminación de 3' proporcionadas en los vectores plásmidos descritos, o amplifique a partir de secuencias genómicas de genes candidatos.
- Ordene secuencias comerciales con sesgo de codón específico si lo desea.
- Utilice procedimientos de subclonación estándar para insertar casetes de genes en vectores de plásmidos UAS, por ejemplo, pSL-attB-UAS14-gyp[3xP3-eYFP]15 (Figura 1) tanto para la regulación ascendente como para las construcciones de ARNi.
- Producir mosquitos transgénicos creados utilizando el intercambio de casetes mediado por recombinación ΦC31111111111111111111.17,18.23.
- Determinar la secuencia que se utilizará para la regulación ascendente de genes candidatos.
- Creación de construcciones de horquilla de ARNi: amplificación de un solo paso mediante PCR asimétrica15,24
- Extraer ADN genómico (gDNA) de una hembra adulta de An. gambiae portadora del gen candidato deseado utilizando el método Livak25.
- Diseñe la imprimación delantera para unirse al exón objetivo a los 5' del fragmento deseado dirigido hacia el intrón vecino. Diseñe el extremo 3' de una imprimación de puente para que se una al extremo del exón anterior para amplificar el intrón. El extremo 5' es complementario a un pequeño fragmento del exón objetivo inmediatamente después del intrón.
- Ejecutar una reacción de PCR asimétrica como se describe en Xiao (2006)24 (Figura 2).
- Clonar el producto de PCR purificado en un vector adecuado que lleve el promotor UAS (por ejemplo, pSL-attB-UAS14-gyp[3xP3-eYFP]15).
NOTA: Las enzimas dentro del sitio de clonación múltiple que son apropiadas para la clonación de pSL-attB-UAS14-gyp[3xP3-eYFP]15 y los siguientes pasos requeridos se indican en la Figura 1. La digestión de una sola enzima es esencial ya que solo se agrega un sitio de restricción. La desfosforilación del plásmido mejorará la eficiencia de la clonación.
- Extraer ADN genómico (gDNA) de una hembra adulta de An. gambiae portadora del gen candidato deseado utilizando el método Livak25.
- Construcción de construcciones de horquilla rNAi: PCR de fusión de ADNc y gDNA15
- Extraer ADN genómico (gDNA) de una hembra adulta de An. gambiae portadora del gen candidato deseado utilizando el método Livak25.
- Incluir gDNA en una reacción de PCR para amplificar el área objetivo de las secuencias de exones e intrones juntos (Figura 2).
- Diseñe el extremo 3' de la imprimación hacia adelante para unirse a la secuencia de exones objetivo inverso para amplificar hacia la secuencia de intrón objetivo y el extremo 5' para llevar un sitio de restricción para facilitar la clonación.
- Diseñe imprimación inversa (1) para unirse al extremo 5' del intrón y el voladizo final de 5' lleva las primeras bases de la secuencia delantera del exón vecino. Este voladizo se utiliza en la PCR de fusión.
- Purificar el producto de reacción deseado.
- Extraiga el ARN, elimine el ADN utilizando DNasa y prepare el ADNc de la hembra adulta An. gambiae que lleva el gen candidato deseado siguiendo los protocolos del fabricante.
- Utilice el ADNc en una reacción de PCR para amplificar el área objetivo del exón solamente (Figura 2).
- Diseñe la imprimación hacia adelante (2) de modo que el extremo 3' se una en el extremo 3' de la secuencia de exones objetivo complementario y el extremo 5' de la imprimación lleve un sitio de restricción para su uso en la clonación.
NOTA: La imprimación delantera de 1.3.1.2 se puede utilizar de nuevo en esta segunda reacción. Sin embargo, esto significará que una sola digestión enzimática es esencial. El uso de una segunda imprimación hacia adelante con un sitio de restricción diferente permitirá una doble digestión que puede aumentar la eficiencia de la clonación. - Imprimación inversa de diseño (2): el extremo de 3' se une al extremo de 5' del exón vecino amplificando el exón objetivo. El extremo 5' se une al extremo 3' de la hebra delantera de los intrones. Este voladizo se utiliza en la PCR de fusión.
- Purificar el producto de reacción deseado.
- Diseñe la imprimación hacia adelante (2) de modo que el extremo 3' se una en el extremo 3' de la secuencia de exones objetivo complementario y el extremo 5' de la imprimación lleve un sitio de restricción para su uso en la clonación.
- Incluya los productos de los pasos 1.3.1 y 1.3.2 como plantillas para una reacción de PCR de fusión utilizando concentraciones estándar con cebadores Forward 1 y 2. Purificar el producto deseado.
- Digerir el producto purificado para generar los voladizos para la clonación. Clonar en un vector adecuado aguas abajo del promotor UAS. En la Figura 1 se indican las enzimas apropiadas para la clonación de pSL-attB-UAS14-gyp[3xP3-eYFP]15 y los siguientes pasos requeridos.
- Extraer ADN genómico (gDNA) de una hembra adulta de An. gambiae portadora del gen candidato deseado utilizando el método Livak25.

Figura 2 - Representación esquemática de la creación de constructos de ARNi para su inserción en pSL-attB-UAS14-gyp[3xP3-eYFP] mediante dos métodos: (A) PCR asimétrica de un solo paso (adaptado de Xiao. Y H et al (2006) y (B) PCR de fusión de múltiples pasos. Haga clic aquí para ver una versión más grande de esta figura.
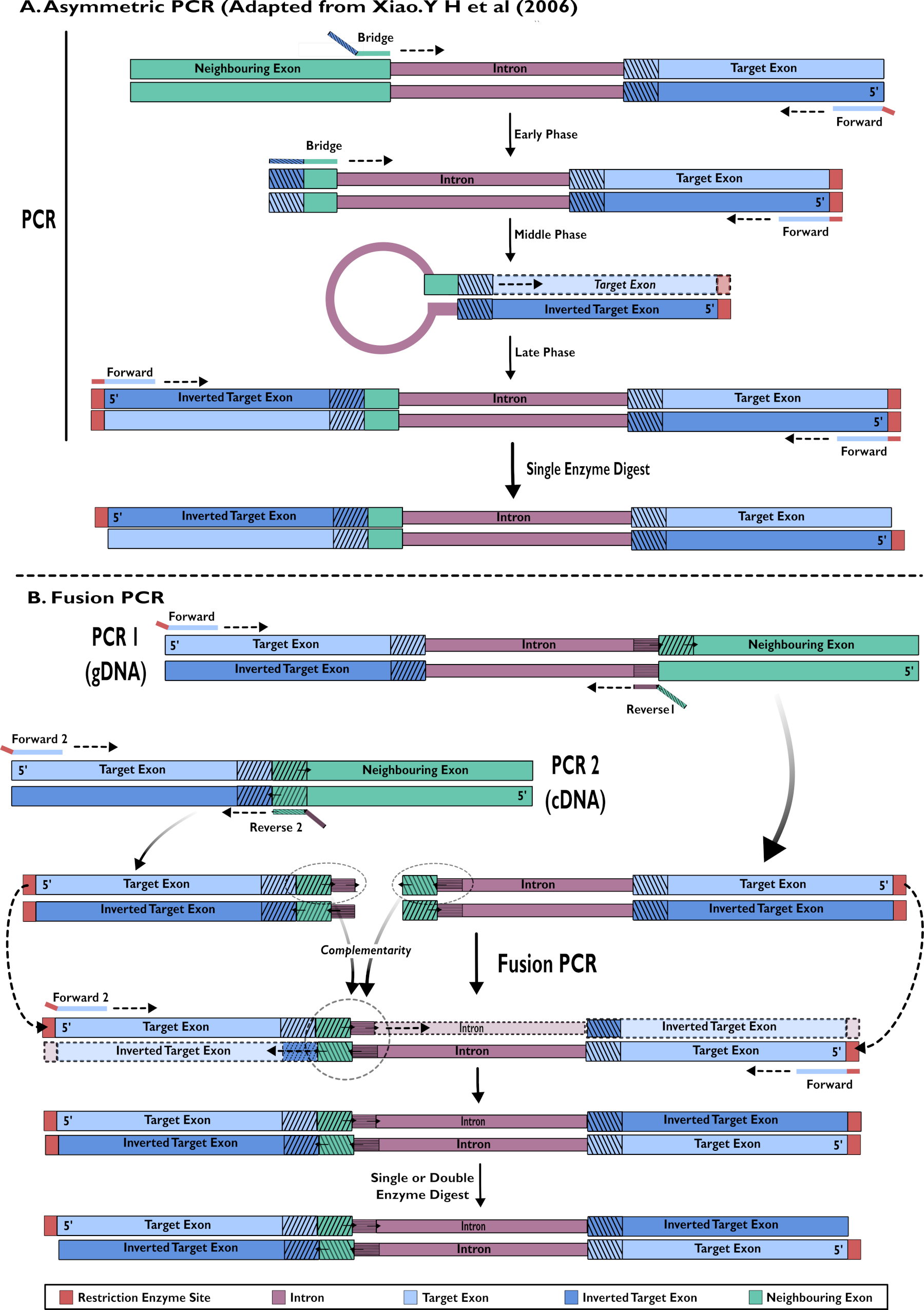{kind=link}
2. Detección de pupas de An. gambiae
- Colección de pupas para caracterización microscópica
NOTA: A lo largo de estos protocolos, el agua se refiere al agua destilada suplementada con 0.01% de sal de estanque.- Mosquitos gambiae de an. trasera utilizando protocolos estándar (por ejemplo, MR426) a la etapa de pupa.
PRECAUCIÓN: Tenga cuidado de no lesionar pupas durante todo este proceso. - Recoja las pupas en un plato plano transparente adecuado para su uso con un estereomicroscopio (por ejemplo, una placa de Petri de plástico de 100 x 15 mm, evitando los bordes).
NOTA: Para recoger pupas utilizamos una pipeta Pasteur de plástico de 3 mL con unos 10 mm cortados desde el extremo para ensanchar el extremo y evitar lesiones a los mosquitos. La detección y el sexado se pueden completar en individuos, sin embargo, esto es muy lento. Se recomienda realizar el cribado y sexado en grupos de 50-200 pupas (el tamaño del grupo posible está limitado por el tamaño del plato utilizado y está sujeto a preferencia personal). Si se está examinando un gran número, la eficiencia se puede aumentar alineando primero las pupas a unas 4 a 5 profundidades en líneas, y moviendo las pupas objetivo fuera de esta línea. - Usando una pipeta Pasteur, retire con cuidado casi toda el agua de alrededor de las pupas. Deje suficiente agua alrededor de las pupas para que sean efectivamente inmóviles, pero se puedan mover fácilmente con un cepillo fino. Si se vuelven difíciles de mover, agregue más agua.
NOTA: Cuando se elimina suficiente agua, las pupas se acostarán de lado, lo que permitirá la visualización de los ojos para la detección de fluorescencia y la identificación de genitales dimórficos (Figura 4DE).
PRECAUCIÓN: Asegúrese de que las pupas no se desecan. Si solo queda un volumen muy pequeño de agua, puede reducirse aún más con el calor de la lámpara del microscopio y cuando se divide entre charcos de pupas. A veces se debe agregar agua adicional durante el proceso utilizando una pipeta Pasteur de 3 ml al grupo (s) deseado (s).
- Mosquitos gambiae de an. trasera utilizando protocolos estándar (por ejemplo, MR426) a la etapa de pupa.
- Identificación de marcadores fluorescentes en pupas
NOTA: El uso de un estereoscopio de bajo aumento permite un cribado de campo amplio, la clasificación se puede hacer en un microscopio compuesto invertido, pero debe hacerse individualmente.- Al detectar un marcador fluorescente, primero es crucial conocer los patrones esperados de expresión y herencia. Considere lo siguiente:
- Color(es): determina qué filtro(s) visualizar la expresión.
- Patrón de expresión espaciotemporal: Comprenda dónde y en qué etapa de la vida espera ver la expresión.
- Ratio de diferentes fenotipos: establecer qué porcentaje de la población debe llevar los marcadores de interés.
- Realice pruebas fluorescentes en la oscuridad, ya que incluso la poca luz puede interferir con la resolución de la fluorescencia. Sin embargo, use una lámpara al lado del estereoscopio cuando se requiera luz para otras manipulaciones.
PRECAUCIÓN: Asegúrese de que el espacio de trabajo alrededor del estereoscopio fluorescente esté despejado antes de apagar las luces. - Encienda la bombilla fluorescente y deje calentar durante el período recomendado por el fabricante (normalmente 10-15 min). Seleccione el filtro requerido en el estereoscopio fluorescente y verifique que haya un haz de luz de color visible que se dirija al centro de la placa del escenario. Si esto no es visible o es muy débil, es posible que la bombilla fluorescente no se haya calentado completamente, que el obturador esté cerrado o que la óptica del microscopio no esté bien alineada.
- Usando luz blanca, centra las pupas en el campo de visión y enfócalas. Es posible que sea necesario cambiar este aumento al cambiar entre diferentes filtros dependiendo de la intensidad de fluorescencia.
- El uso de un pincel de detalle fino asegúrese de que las pupas examinadas no se superpongan.
- Apague la luz blanca del estereoscopio y use el enfoque fino para enfocar el área de las pupas que llevan el fenotipo de interés. El patrón fluorescente debe ser visible. En la Figura 3 se proporcionan ejemplos de fluorescencia controlada por el promotor 3xP3.
- Use el aumento más bajo en el que el fenotipo fluorescente esperado se pueda distinguir de manera confiable de los individuos sin fluorescencia.
- Para las cepas con fluorescencia brillante, use también una luz de campo brillante de baja intensidad durante la detección, si la señal fluorescente aún es claramente identificable.
- Cuando termine la detección primaria, escanee rápidamente las poblaciones bajo otros filtros para detectar una posible contaminación.
PRECAUCIÓN: Asegúrese de que haya una distancia clara entre los grupos de pupas clasificadas para evitar la contaminación por el movimiento de pupas. Tenga en cuenta que el tamaño de los grupos cambiará a medida que las pupas se sexen y que las distancias pueden parecer más grandes cuando se mira bajo aumento. Tenga especial cuidado cuando las piscinas no estén dentro del campo de visión.
- Al detectar un marcador fluorescente, primero es crucial conocer los patrones esperados de expresión y herencia. Considere lo siguiente:

Figura 3 - Anopheles gambiae pupae expresando marcadores fluorescentes impulsados por el promotor 3xP3 (A) eYFP, (B) dsRed y (C) eCFP. Aumento: A = 16X, B, C = 20X.
-
Sexado de pupas
- Recoger pupas. Eliminar el exceso de agua, pero proporcionar lo suficiente para que las paletas anales se separen ligeramente de los genitales para ayudar a la visualización y caracterización morfológica (Figura 4D,E).
- Si alguna pupa / e no está de lado, use un pincel de detalle fino para girar suavemente la pupa y mover las paletas anales para que se puedan identificar los genitales externos.
- Pupas separadas basadas en genitales externos distintivos; los machos tienen un tubo largo que extruye desde el segmento dorsal final aproximadamente la mitad de la longitud de las paletas anales (Figura 4B). Los genitales externos de las pupas femeninas son considerablemente más cortos y bifurcados (Figura 4A).
NOTA: En ocasiones, si el exoesqueleto larval del 4º instar permanece adherido o los genitales externos están dañados (Figura 4C), la identificación segura del sexo es más difícil. Cuando el sexo de una pupa no está claro, es una buena práctica descartarla. Si el individuo debe mantenerse, se debe permitir que la pupa emerja de forma aislada y su sexo se determine utilizando características morfológicas adultas. Es probable que si sus genitales están dañados el individuo no se aparee con éxito. - Haga una piscina para cada sexo en el extremo opuesto del plato a la piscina sin sexo, moviendo las pupas identificadas a través del plato con un pincel de detalle fino. Etiquete la parte inferior del plato donde se reunirán las dos piscinas para identificarlas más tarde.
- Cuando se requiera tanto el sexado como el cribado fluorescente, realice primero el cribado fluorescente, ya que es el proceso más rápido de los dos.

Figura 4 - Sexado de pupas de Anopheles gambiae. Pupas individuales que indican los genitales externos de (A) una hembra (B) un macho y (C) un individuo que no puede ser identificado fácilmente debido al desprendimiento incompleto del exoesqueleto larvario. Imágenes ampliadas a continuación que resaltan los genitales externos. Pupas con ♀ (hembra) y ♂ (macho) indicando los genitales externos de las pupas con (D) ~ 50% de la pupa sumergida en agua y con (E) toda el agua eliminada destacando la diferencia en la facilidad de visualización de los genitales externos. Ampliación: A, B, C = 40x, D, E = 30x. Haga clic aquí para ver una versión más grande de esta figura.
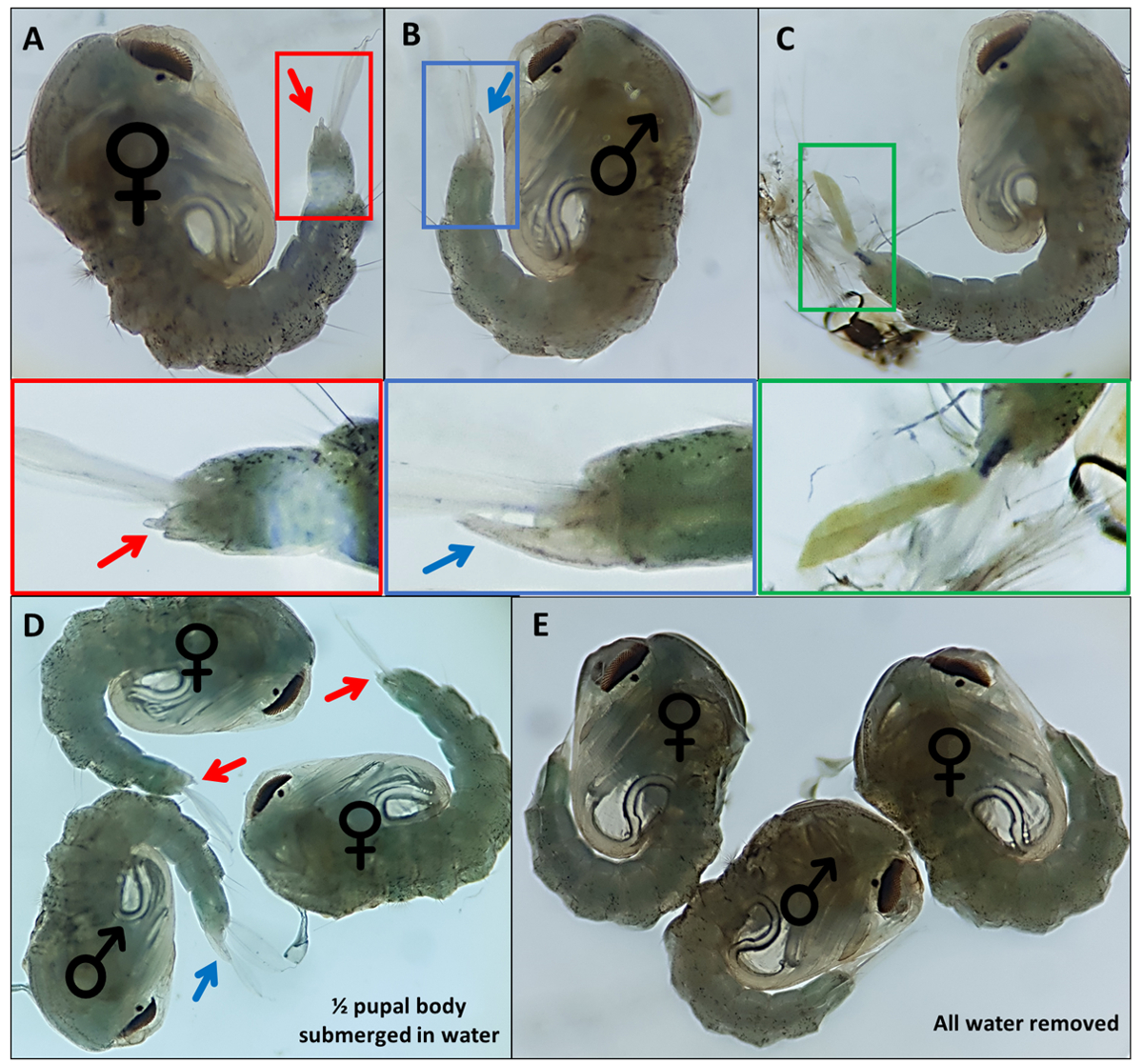{kind=link}
- Confirmación del sexo en adultos
- Hasta que se haya demostrado una tasa de error muy baja, confirme el sexado de pupas por morfología adulta después de la emergencia. Separe las pupas sexadas en grupos de 10 o menos en un tubo transparente de 20 ml con unos pocos ml de agua, sellando con una bola de algodón, etiquetadas con el sexo esperado y dejar emerger durante la noche.
NOTA: Como los adultos son transferidos a la mañana siguiente, no es necesario suministrar alimentos a los adultos emergentes. - Confirme el sexo de los adultos emergidos utilizando características morfológicas al día siguiente.
- Si hay machos presentes en las colecciones femeninas, descarte a las hembras, en caso de que ya se haya producido el apareamiento.
- Si hay hembras presentes en la colección masculina, retire la hembra / s y mantenga a los machos para cruzar.
- Hasta que se haya demostrado una tasa de error muy baja, confirme el sexado de pupas por morfología adulta después de la emergencia. Separe las pupas sexadas en grupos de 10 o menos en un tubo transparente de 20 ml con unos pocos ml de agua, sellando con una bola de algodón, etiquetadas con el sexo esperado y dejar emerger durante la noche.
- Configuración de cruces del sistema GAL4-UAS
- Aspire el número deseado de adultos machos y hembras de los tubos en el paso 2.4 en una jaula o cubo pequeño configurado de la manera estándar para la cría de An. gambiae.
NOTA: Tenga cuidado de no dañar a los adultos durante este traslado. - Use aproximadamente 50 hembras con un número igual de machos, cuando se requieren ~ 2000 adultos de la progenie.
NOTA: Cuando una cruz debe ser alimentada varias veces para generar múltiples lotes de hasta 200 de cada sexo, se puede configurar en jaulas de 30 cm x 30 cm x 30 cm. Cuando solo un pequeño número de hembras (<20) están disponibles para el cruce, agregamos ~ 4 veces el número de machos para aumentar la probabilidad de apareamiento exitoso. - La sangre alimenta a las hembras cruzadas y a la progenie trasera a la etapa apropiada, siguiendo los protocolos estándar26, para realizar una evaluación fenotípica (por ejemplo, resistencia a los insecticidas, capacidad vectorial y ensayos de costos de aptitud física).
- Cuando sea probable el efecto materno de la expresión transgénica, establezca cruces recíprocos de las líneas de conductor y respuesta y evalúe el fenotipo esperado.
NOTA: Los cruces que utilizan poblaciones 'heterocigotas' o mixtas de líneas conductoras y de respuesta, producen progenie con cada uno de los 4 genotipos posibles. Esto proporciona controles de tipo salvaje, solo UAS y GAL4, así como los transheterocigotos GAL4-UAS con los que analizar el fenotipo. Si se cruzan poblaciones homocigotas, establezca cruces adicionales para proporcionar controles apropiados para comparar fenotipos. La progenie debe ser examinada como arriba separando a la progenie portadora de ambos o solo cualquiera de los marcadores, así como negativos, para la evaluación fenotípica.
- Aspire el número deseado de adultos machos y hembras de los tubos en el paso 2.4 en una jaula o cubo pequeño configurado de la manera estándar para la cría de An. gambiae.
- Establecimiento de poblaciones homocigotas a partir de líneas generadas a través de RCME que transportan marcadores fluorescentes alternativos
NOTA: Es esencial que el marcador fluorescente de ambas líneas esté presente en la misma ubicación genómica y sean completamente distinguibles.- Establezca una cruz parental de aproximadamente 200 adultos con el mismo número de machos marcados diferencialmente de una línea y hembras de la otra línea después de la detección para seleccionar individuos que muestren fluorescencia y sexo correctos, como se describió anteriormente. Alrededor de una semana después, la sangre alimenta a la cruz utilizando protocolos establecidos26.
- Remonte la progenie F1 a las pupas utilizando protocolos estándar y recoja pupas como se describió anteriormente.
- Cribado de fluorescencia seleccionando aquellos que llevan ambos marcadores parentales (transheterocigotos). Configura un cruce de F1 con estas pupas.
- Una semana después, la sangre alimenta a las hembras F1 y a la progenie trasera a la etapa de pupa siguiendo los protocolos estándar.
- Revisa las pupas F2 seleccionando aquellas que muestran SOLO uno de los marcadores. Estos serán homocigotos para la inserción. Solo el 25% de la progenie será homocigota para cada inserción, así que asegúrese de que se críe suficiente progenie para proporcionar una jaula de stock (400-500).
NOTA: La selección de la progenie transheterocigota debe ser completamente rigurosa, de lo contrario el proceso se contamina y es posible que no se logre la homocigosidad completa. Verifique toda la progenie seleccionada para el intercross de F1.
- Establezca una cruz parental de aproximadamente 200 adultos con el mismo número de machos marcados diferencialmente de una línea y hembras de la otra línea después de la detección para seleccionar individuos que muestren fluorescencia y sexo correctos, como se describió anteriormente. Alrededor de una semana después, la sangre alimenta a la cruz utilizando protocolos establecidos26.
3. Protocolo de limpieza de embriones de An. gambiae
-
Alimentación y mantenimiento de la sangre
- Mosquitos an. gambiae traseros a adultos siguiendo protocolos estándar (por ejemplo, MR4).
- La sangre alimenta a las mujeres adultas de 5 a 7 días de edad, asegurando que la mayoría estén completamente congestionadas.
PRECAUCIÓN: A lo largo de este protocolo, trabajar rápidamente es esencial para garantizar que no se permita que los huevos se desecan.
-
Puesta de huevos inducida
- 3 días después de la alimentación con sangre, recolecte los huevos a través de la puesta inducida.
- Montar la cámara de oviposición.
- Llene la olla de oviposición con agua a una profundidad de aproximadamente 5 mm. Fije la olla a un extremo de un tubo de polipropileno de 50 ml, previamente cortado con una sierra para que ambos extremos estén abiertos. (Usamos un disco de plástico para una olla (Figura 5); sin embargo, la tapa original del tubo se puede usar en su lugar).
- Cubra el otro extremo del tubo de polipropileno cortado con material (manguera/medias) o secciones de guante de látex aseguradas con una banda elástica, de modo que los adultos puedan ser introducidos pero no puedan escapar (Figura 5). Existen otros diseños alternativos de cámaras de oviposición que pueden utilizarse26.
- Introduzca cuidadosamente 10-15 hembras (sangre alimentada en el paso 3.1.2) en la cámara de oviposición. Cubra la cámara de oviposición para producir oscuridad y deje actuar durante 20 minutos.
PRECAUCIÓN: Evite mover la olla de oviposición una vez que los huevos hayan sido puestos para evitar el varamiento y la desecación de los huevos. - Separe cuidadosamente el tubo de polipropileno de 50 ml de la olla de oviposición, al tiempo que se asegura de no liberar a los mosquitos. Los huevos blancos deben ser visibles. Comprobar que se han establecido suficientes para fines prohibidos. Repetir si es necesario.
- Cubra la maceta (para protegerse contra el polvo) y permita que los huevos maduren hasta la etapa de desarrollo de interés.
- Use un pincel de detalle fino para recoger los huevos de la olla y colóquelos en el agua en un bloque de vidrio excavado de 40 mm2.

Figura 5 - Ejemplo de una cámara de oviposición (A) desmontada para resaltar los componentes y (B) ensamblada. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
-
Fijación de embriones
PRECAUCIÓN: Realice todos los pasos de fijación (paso 3.3) en una campana extractora de humos debido al uso de formaldehído.- Preparar la solución de la FAA como se describe en Kaiser et al. (2014)22. FAA comprende 3,6 M de formaldehído, 0,87 M de ácido acético y 8,5 M de etanol absoluto hecho en volumen con agua destilada (dH2O).
- Para 10 ml de FAA, combine 2,68 ml de 13,42 m de formaldehído, 4,96 ml de etanol de 17,14 m y 0,5 ml de ácido acético de 17,4 m con 1,86 ml de H2O destilado. El fijador se puede mantener durante al menos 3 meses en un recipiente de vidrio herméticamente sellado, guardado en un armario químico designado.
- Retire cuidadosamente el agua del bloque de vidrio con una micropipeta y cubra los huevos en 500 μL de FAA y oscile suavemente (~ 25 RPM) en un agitador orbital a temperatura ambiente durante 30 minutos. Ningún cambio de color es visible en este punto.
- Enjuague bien los huevos con agua destilada. Realice el enjuague 15 veces para eliminar todos los rastros de formaldehído. Usando una micropipeta de 1000 μL, agregue y luego retire 1 ml de dH2O a la vez, asegurándose de no dañar los huevos mientras lo hace.
- Almacene las aguas residuales de los enjuagues en un recipiente designado de descarte de formaldehído para su eliminación de acuerdo con las pautas de seguridad.
- En este punto, los huevos fijos se pueden almacenar a 4 ° C durante la noche en agua para mantenerlos hidratados.
- Preparar la solución de la FAA como se describe en Kaiser et al. (2014)22. FAA comprende 3,6 M de formaldehído, 0,87 M de ácido acético y 8,5 M de etanol absoluto hecho en volumen con agua destilada (dH2O).
-
Blanqueamiento embrionario
PRECAUCIÓN: Realice todos los pasos de blanqueo (paso 4) en una campana de humos debido a la posible liberación de gas cloro cuando se combinan hipoclorito de sodio y ácido acético.- Preparar una solución blanqueadora (solución de Trpiš - descrita en Trpiš (1970)21 y modificada según Kaiser et al. (2014)22). La solución de Trpiš es 0,59 M de hipoclorito de sodio y 0,35 M de ácido acético disuelto en H2O destilado.
- Para un volumen de 10 ml de solución de Trpiš, combine 2,68 ml de hipoclorito de sodio de 2,2 m y 0,2 ml de ácido acético de 17,4 mL con 7,12 ml de H2O destilado.
NOTA: La solución trpiš se puede almacenar durante al menos 3 meses en un recipiente de vidrio herméticamente sellado y mantenerse en un armario químico seguro. La solución puede necesitar ser vórtice después del almacenamiento y siempre debe abrirse en una campana de humos en caso de liberación de gas cloro.
- Para un volumen de 10 ml de solución de Trpiš, combine 2,68 ml de hipoclorito de sodio de 2,2 m y 0,2 ml de ácido acético de 17,4 mL con 7,12 ml de H2O destilado.
- Cubra los huevos fijos con 1 ml de solución de Trpiš e incube a temperatura ambiente durante 30 minutos. Los huevos comenzarán a desarrollar parches pálidos después de unos 5 minutos de incubación, alcanzando finalmente un color blanco lechoso una vez despejados.
- Enjuague los huevos como en el paso 3.3.3 para eliminar la solución de Trpiš.
- Almacene las aguas residuales en un contenedor de desechos designado y deseche con el exceso de agua por el desagüe.
- Preparar una solución blanqueadora (solución de Trpiš - descrita en Trpiš (1970)21 y modificada según Kaiser et al. (2014)22). La solución de Trpiš es 0,59 M de hipoclorito de sodio y 0,35 M de ácido acético disuelto en H2O destilado.
-
Almacenamiento
- Conservar en 500 μL de dH2O y conservar entre 2-8 °C durante unos días. Retire la mayor parte del agua con cuidado antes de ver y tomar imágenes de la masa, pero evite la desecación de los huevos dejando un pequeño volumen de agua en el vidrio del reloj. Esto no interrumpirá la fotografía de los huevos. Los óvulos individuales se pueden colocar en el portaobjetos del microscopio para obtener imágenes de mayor aumento.
Resultados
La expresión 3xP3 de eYFP, dsRed y eCFP proporciona una identificación confiable y fácilmente distinguible de individuos que poseen los genes marcadores que producen expresión en los ojos y los ganglios ventrales de las pupas de An. gambiae (Figura 3). La morfología diferencial observada en los genitales externos masculinos y femeninos utilizados para el sexado y un ejemplo de pupas no identificables se destacan en la Figura 4. La eliminac...
Discusión
Comprender la función genética de los mosquitos es vital para desarrollar nuevos enfoques para controlar Anopheles e impactar la transmisión de la malaria. El sistema GAL4-UAS descrito es un sistema versátil y potente para el análisis funcional de genes candidatos y hasta la fecha hemos utilizado el sistema para examinar las bases genéticas de la resistencia a insecticidas17 y la producción de hidrocarburos cuticulares15,23...
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Agradecemos la financiación de LSTM e IVCC (Adriana Adolfi), BBSRC (New Investigator Award (AL), MRC (PhD studentship to BCP:MR/P016197/1), Wellcome (Sir Henry Wellcome Postdoctoral fellowship to LG: 215894/Z/19/Z) que han incorporado el análisis Gal4UAS en las propuestas.
Materiales
| Name | Company | Catalog Number | Comments |
| 100 x 15 mm plastic Petri dish | SLS | 2175546 | Pack of 10 |
| 1000 µL Gilson Pipette | Gilson | F144059P | |
| 20/25 mL Universal Tubes | Starlab | E1412-3020 | Pack of 400 |
| 3 mL Pasteur Pipettes | SLS | G612398 | Greiner Pasteur pipette 3 mL sterile individually wrapped |
| 50 mL Falcon Tubes | Fisher Scientific | 11512303 | |
| Absolute Ethanol | Fisher Scientific | BP2818-500 | 500 mL |
| Acetic Acid | SLS | 45726-1L-F | 1 L |
| Cages | SLS | E6099 | 30x30x30 with screen port |
| Fine Paint Brushes | Amazon | UKDPB66 | KOLAMOON 9 Pieces Detail Painting Brush Set Miniture Brushes for Watercolor, Acrylic Painting, Oil Painting (Wine Red) |
| Fish food | Amazon | Tetra Min Fish Food, Complete Food for All Tropical Fish for Health, Colour and Vitality, 10 L | |
| Formaldehyde Solution | Sigma Aldrich | F8775 | |
| Mouth Aspirator | John Hock | 612 | |
| Pond Salt | Amazon | Blagdon Guardian Pond Tonic Salt, for Fish Health, Water Quality, General Tonic, pH Buffer, 9.08 kg, treats 9,092 L | |
| Pupae Pots | Cater4you | SP8OZ | 250 pots with lids |
| Small Plastic Buckets | Amazon | 2.5 L White Plastic Pail Complete with White Lid (Pack of 10) | |
| Sodium Hypochlorite | Fisher Scientific | S25552 |
Referencias
- Brand, A. H., Perimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 118 (2), 401-415 (1993).
- Duffy, J. B. GAL4 system in drosophila: A fly geneticist's swiss army knife. Journal of Genetics and Development. 34 (1-2), 1-15 (2002).
- Dow, J. A. . ELS. , (2012).
- Edi, C. V., et al. CYP6 P450 Enzymes and ACE-1 Duplication Produce Extreme and Multiple Insecticide Resistance in the Malaria Mosquito Anopheles gambiae. PLoS Genetics. 10 (3), 1004236 (2014).
- Daborn, P. J., et al. Using Drosophila melanogaster to validate metabolism-based insecticide resistance from insect pests. Insect Biochemistry and Molecular Biology. 42 (12), 918-924 (2012).
- Riveron, J. M., et al. Genome-wide transcription and functional analyses reveal heterogeneous molecular mechanisms driving pyrethroids resistance in the major malaria vector Anopheles funestus across Africa. Genes Genomes Genetics. 7 (6), 1819-1832 (2017).
- Riveron, J. M., et al. A single mutation in the GSTe2 gene allows tracking of metabolically based insecticide resistance in a major malaria vector. Genome Biology. 15 (2), (2014).
- Lynd, A., Lycett, G. J. Development of the Bi-Partite Gal4-UAS System in the African Malaria Mosquito, Anopheles gambiae. PLoS ONE. 7 (2), 31552 (2012).
- Lynd, A., Lycett, G. J. Optimization of the Gal4-UAS system in an Anopheles gambiae cell line. Insect Molecular Biology. 20 (5), 599-608 (2011).
- Adolfi, A., Pondeville, E., Lynd, A., Bourgouin, C., Lycett, G. J. Multi-tissue GAL4-mediated gene expression in all Anopheles gambiae life stages using an endogenous polyubiquitin promoter. Insect Biochemistry and Molecular Biology. 96, 1-9 (2018).
- Kokoza, V. A., Raikhel, A. A. Targeted gene expression in the transgenic Aedes aegypti using the binary Gal4-UAS system. Insect Biochemistry and Molecular Biology. 41, 637-644 (2011).
- O'Brochta, D. A., Pilitt, K. L., Harrell, R. A., Aluvihare, C., Alford, R. T. Gal4-based Enhancer-Trapping in the Malaria Mosquito Anopheles stephensi. Genes Genomes Genetics. 2, 21305-21315 (2012).
- Zhao, B., et al. Regulation of the Gut-Specific Carboxypeptidase: A Study Using the Binary Gal4/UAS System in the Mosquito Aedes Aegypti. Insect Biochemistry and Molecular Biology. 54, 1-10 (2014).
- Imamura, M., et al. Targeted Gene Expression Using the GAL4/UAS System in the Silkworm Bombyx mori. Genetics. 165 (3), 1329-1340 (2003).
- Lynd, A., et al. Development of a functional genetic tool for Anopheles gambiae oenocyte characterisation: application to cuticular hydrocarbon synthesis. bioRxiv. , (2019).
- Pondeville, E., et al. Hemocyte-targeted gene expression in the female malaria mosquito using the hemolectin promoter from Drosophila. Insect Biochemistry and Molecular Biology. 120, 103339 (2020).
- Adolfi, A., et al. Functional genetic validation of key genes conferring insecticide resistance in the major African malaria vector, Anopheles gambiae. Proceedings of the National Academy of Sciences of the United States of America. 116 (51), 25764-25772 (2019).
- Pondeville, E., et al. Efficient integrase-mediated site-specific germline transformation of Anopheles gambiae. Nature Protocols. 9 (7), 1698-1712 (2014).
- Horn, C., Schmid, B. G. M., Pogoda, F. S., Wimmer, E. A. Fluorescent transformation markers for insect transgenesis. Insect Biochemistry and Molecular Biology. 32, 1221-1235 (2002).
- Clements, A. . A. Biology of Mosquitoes, Volume 1: Development, Nutrition and Reproduction. 1, (1992).
- Trpiš, M. A new bleaching and decalcifying method for general use in zoology. Canadian Journal of Zoology. 48, 892-893 (1970).
- Kaiser, M. L., Duncan, F. D., Brooke, B. D. Embryonic Development and Rates of Metabolic Activity in Early and Late Hatching Eggs of the Major Malaria Vector Anopheles gambiae. PLoS ONE. 9 (12), 114381 (2014).
- Grigoraki, L., Grau-Bové, X., Yates, H. C., Lycett, G. J., Ranson, H. Isolation and transcriptomic analysis of Anopheles gambiae oenocytes enables the delineation of hydrocarbon biosynthesis. eLife. 9, 58019 (2020).
- Xiao, Y. -. H., Yin, M. -. H., Hou, L., Pei, Y. Direct amplification of intron-containing hairpin RNA construct from genomic DNA. BioTechniques. 41 (5), 548-552 (2006).
- Livak, K. J. Organization and Mapping of a Sequence on the Drosophila melanogaster X and Y Chromosomes That Is Transcribed during Spermatogenesis. Genetics. 107 (4), 611-634 (1984).
- MR4, CDC, NEI & beiResources. . The MR4 Methods in Anopheles Research Laboratory Manual. 5th Edition. , (2015).
- Sik Lee, Y., Carthew, R. W. Making a better RNAi vector for Drosophila: use of intron spacers. Methods. 30 (4), 322-329 (2003).
- Cha-aim, K., Hoshida, H., Fukunaga, T., Akada, R., Peccoud, J. . Gene Synthesis: Methods and Protocols. , 97-110 (2012).
- Cavener, D. R. Comparison of the consensus sequence flanking translational start sites in Drosophila and vertebrates. Nucleic Acids Research. 15 (4), 1353-1361 (1987).
- Wang, Y., Wang, F., Wang, R., Zhao, P., Xia, Q. 2A self-cleaving peptide-based multi-gene expression system in the silkworm Bombyx mori. Scientific Reports. 5, (2015).
- Galizi, R., et al. A synthetic sex ratio distortion system for the control of the human malaria mosquito. Nature Communications. 5, 3977 (2014).
- Kondo, S., et al. Neurochemical organisation of the Drosophila Brain Visualised by Endogenously Tagged Neurotransmitter Receptors. Cell Reports. 30 (1), 284-297 (2020).
- Lee, P. -. T., et al. A gene-specific T2A-GAL4 library for Drosophila. eLife. 7, 35574 (2018).
- Marois, E., et al. High-throughput sorting of mosquito larvae for laboratory studies and for future vector control interventions. Malaria Journal. 11, 302 (2012).
- Crawford, J. E., et al. Efficient production of male Wolbachia-infected Aedes aegypti mosquitoes enables large-scale suppression of wild populations. Nature Biotechnology. 38 (4), 482-492 (2020).
- Goltsev, Y., et al. Developmental and evolutionary basis for drought tolerance of the Anopheles gambiae embryo. Developmental Biology. 330 (2), 462-470 (2009).
- Rezende, G. L., et al. Embryonic desiccation resistance in Aedes aegypti: presumptive role of the chitinized Serosal Cuticle. BMC Developmental Biology. 8 (1), 82 (2008).
- Vargas, H. C. M., Farnesi, L. C., Martins, A. J., Valle, D., Rezende, G. L. Serosal cuticle formation and distinct degrees of desiccation resistance in embryos of the mosquito vectors Aedes aegypti, Anopheles aquasalis and Culex quinquefasciatus. Journal of Insect Physiology. 62, 54-60 (2014).
- Chang, C. -. H., et al. The non-canonical Notch signaling is essential for the control of fertility in Aedes aegypti. PLOS Neglected Tropical Diseases. 12 (3), 0006307 (2018).
- Clemons, A., Flannery, E., Kast, K., Severson, D., Duman-Scheel, M. Immunohistochemical Analysis of Protein Expression during Aedes aegypti Development. Spring Harbor Protocols. 10, 1-4 (2010).
- Juhn, J., James, A. A. Hybridization in situ of Salivary Glands, Ovaries and Embryos of Vector Mosquitoes. Journal of Visualized Experiments. , e3709 (2012).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados