
Method Article
Visualización en 3D de poblaciones de células inmunitarias en tejidos infectados por el VIH mediante microscopía de aclarado, inmunotinción, confocal y fluorescencia de lámina de luz
En este artículo
Resumen
El aclaramiento de tejidos, combinado con la microscopía de inmunofluorescencia, permite la visualización espacial y la cuantificación de las poblaciones de células inmunitarias y las proteínas del virus dentro de los tejidos intactos. El corte óptico de tejidos aclarados con microscopía de fluorescencia confocal y de lámina de luz puede generar modelos 3D de entornos de tejidos complejos y revelar la heterogeneidad espacial exhibida durante la infección por VIH.
Resumen
El Virus de Inmunodeficiencia Humana (VIH), el agente causante del Síndrome de Inmunodeficiencia Adquirida (SIDA), es un importante problema de salud mundial, con casi 40 millones de personas infectadas en todo el mundo y sin una cura ampliamente accesible. A pesar de los intensos esfuerzos, la comprensión detallada de las interacciones entre el virus y la célula huésped en los tejidos durante la infección y en respuesta a la terapia sigue siendo incompleta. Para abordar estas limitaciones, se aplican las técnicas de limpieza de tejidos a base de agua CUBIC (Clear, Unobstructed Brain/Body Imaging Cocktails and Computational analysis) y CLARITY (Clear Lipid-traded Acrylamide-hybridized Rigid Imaging/Immunostaining/in situ-hybridization-Tissue hYdrogel) para visualizar interacciones complejas entre el virus y la célula huésped en tejidos infectados por el VIH de modelos animales y humanos utilizando microscopía de fluorescencia confocal y de lámina de luz. El corte óptico de tejidos intactos y el análisis de imágenes permiten la reconstrucción rápida de la información espacial contenida en tejidos completos y la cuantificación de las poblaciones de células inmunitarias durante la infección. Estos métodos son aplicables a la mayoría de las fuentes de tejidos y a diversas cuestiones biológicas, incluidas las enfermedades infecciosas y el cáncer.
Introducción
La creciente necesidad de imágenes cuantitativas de tejidos espaciales en la investigación biológica condujo recientemente a la aparición de técnicas de limpieza de tejidos para generar imágenes de mayor volumen (mm3-cm 3) de tejidos intactos con resolución de una sola célula. Los tejidos incluyen organizaciones complejas de biomoléculas con estructuras, composiciones y funciones definidas de manera única. Desafortunadamente, muchas biomoléculas presentes en los tejidos (por ejemplo, lípidos y cromóforos) dispersan, absorben o emiten luz cuando se obtienen imágenes mediante microscopía óptica, lo que dificulta la obtención de imágenes de gran volumen. Además, los tejidos a menudo exhiben un índice de refracción que no coincide con las soluciones de imagen estándar y las lentes ópticas, lo que resulta en distorsiones ópticas durante la obtención de imágenes. Un enfoque óptimo para obtener imágenes de grandes volúmenes de tejido con un microscopio óptico debe implicar hacer coincidir el índice de refracción de los tejidos, las soluciones de imagen y los objetivos, al tiempo que permite la penetración de la luz profundamente en el tejido sin interrumpir las características biológicas de los tejidos intactos durante el procesamiento. Los primeros intentos de reducir las diferencias en el índice de refracción entre los tejidos y las soluciones de imagen mediante la limpieza de muestras de tejido opaco fueron llevados a cabo por el anatomista alemán Werner Spalteholz afinales del siglo XIX. Esta técnica de limpieza de tejidos involucró solventes químicos agresivos, que pueden dañar las muestras de tejido, pero sin embargo representó la primera imagen de mayor volumen reportada de tejidos intactos. Los métodos modernos de microscopía óptica, combinados con la potencia de cálculo para la captura y el análisis de imágenes, han vuelto a poner de moda recientemente la limpieza de tejidos como método para obtener imágenes de muestras de tejido grandes e intactas con resolución de una sola célula. Durante las últimas dos décadas, surgieron docenas de técnicas avanzadas de limpieza de tejidos, incluidas las orgánicas y las basadas en agua, cada una con fortalezas y debilidades para aplicaciones específicas.
Las imágenes de tejidos en 3D pueden sondear interacciones biológicas más complejas que no se pueden reproducir en cultivos celulares. Por ejemplo, los patrones de señalización celular2, las distribuciones espaciales de distintos tipos de células3 y la conectividad cerebral4 se mapearon previamente de manera cuantitativa utilizando métodos de imágenes de tejidos/órganos completos. Aquí se describe una aplicación de protocolos de limpieza de tejidos a base de agua para limpiar, inmunotinción y visualizar distintas poblaciones de células diana del VIH dentro de tejidos linfoides intactos infectados por el VIH durante la infección activa. Dentro del cuerpo, el VIH infecta predominantemente a las células T CD4+ e integra una copia de su genoma en los genomas de las células huésped infectadas. Posteriormente, el virus secuestra la maquinaria de la célula huésped infectada para replicarse, lo que resulta en la diseminación del virus, la muerte de la célula huésped, la disfunción inmune y la progresión a largo plazo hacia el SIDA. Es importante tener en cuenta que los comportamientos de las células T infectadas en tejidos y cultivos celulares son notablemente discrepantes. Las células T CD4+ cultivadas con el VIH pueden producir sincitios masivos inducidos por el VIH que pueden incluir docenas de núcleos5, mientras que experimentos similares con células T CD4+ primarias cultivadas en hidrogeles de matriz extracelular (MEC) en 3D o muestras de tejido de ratones humanizados infectados por el VIH (ratones hu) generalmente producen sincitios con 2-5 núcleos6. Comprender la transmisión local de célula a célula y la diseminación sistémica del virus dentro de las personas infectadas por el VIH es probablemente aún más complicado, ya que implica el transporte del virus por múltiples tipos de células infectadas desde los tejidos hasta los vasos sanguíneos y los nuevos tejidos, donde los viriones libres y las células productoras de virus pueden acceder a un gran número de linfocitos susceptibles7. Estos escenarios no son actualmente posibles de recapitular en sistemas de cultivo celular, y los tejidos de modelos animales y humanos siguen siendo un recurso importante para comprender la patogénesis del virus en el contexto de un organismo complejo con un sistema inmunitario en funcionamiento.
Las terapias antirretrovirales (TAR) actuales aumentan en gran medida la esperanza y la calidad de vida de las personas con VIH (PCH) al inhibir la replicación del VIH y detener la progresión de la enfermedad hacia el SIDA. Desafortunadamente, el TAR no elimina las células inmunitarias infectadas latentemente que contienen una inserción del genoma retroviral que están inactivas y no producen virus activamente. Aunque el virus no es detectable en la sangre de la mayoría de las personas que reciben tratamiento antirretroviral, las cargas virales se recuperan rápidamente después de que se interrumpe el tratamiento antirretroviral y la progresión de la enfermedad continúa8. La naturaleza persistente de la infección por el VIH causada por el reservorio latente de células infectadas representa un gran impedimento para establecer una cura del VIH. Los reservorios tisulares del VIH siguen siendo poco conocidos, y es crucial establecer una comprensión más profunda de estos reservorios en los tejidos linfoides antes, durante y después de la TAR, para caracterizar completamente la patogénesis del virus y evaluar nuevos tratamientos que eliminen eficazmente las células infectadas latentemente que no producen activamente el virus.
Aquí, CUBIC3 y CLARITY9, dos protocolos de limpieza de tejidos a base de agua previamente adaptados, se aplicaron para obtener imágenes de poblaciones de células inmunitarias dentro de numerosos tejidos linfoides intactos de ratones infectados por el VIH con sistemas inmunitarios humanizados (ratones humanos), primates no humanos (NHP) infectados con SIV/SHIV y humanos infectados por el VIH. Estos protocolos son adaptables tanto a la microscopía confocal como a la microscopía de fluorescencia de lámina ligera en función de los objetivos de la imagen (mayor resolución frente a mayor volumen) y de la instrumentación disponible. Aunque la microscopía óptica no puede resolver viriones individuales, el uso de la inmunofluorescencia puede identificar regiones de tejido que contienen virus y células productoras de virus que se pueden analizar más a fondo con métodos de mayor resolución. Los métodos presentados aquí se pueden adaptar para visualizar casi cualquier tejido del cuerpo con resolución de una sola célula con el fin de cuantificar las relaciones espaciales entre tipos específicos de células en diferentes condiciones durante la infección y son fácilmente traducibles a muestras de pacientes humanos altamente relevantes para el estudio de enfermedades infecciosas o cáncer.
Protocolo
Todos los experimentos con animales se llevaron a cabo de acuerdo con los protocolos institucionales de cuidado animal aprobados. Todos los tejidos humanos se adquirieron de acuerdo con las directrices institucionales aprobadas por la ética de la investigación en seres humanos.
1. Recolección y fijación de tejidos (lo mismo para CUBIC y CLARITY)
- Identificar y diseccionar los tejidos linfoides como se ha descrito anteriormente10.
- Extraiga los tejidos linfoides con tijeras de disección y pinzas en cuestión de minutos después de la autopsia, cuando sea posible de manera segura.
- Coloque las muestras de tejido en un tampón fijador recién hecho y helado que contenga un 8% de paraformaldehído (PFA), 5% de sacarosa en trihidrato de cacodilato de sodio 0,1 M para preservar adecuadamente las muestras de tejido para microscopía óptica (LM), microscopía electrónica (EM) o inmuno-EM. Alternativamente, fije las muestras para LM con 4% de PFA en 0,1 M PBS. Fije las muestras durante la noche antes de comenzar el proceso de eliminación para garantizar la desactivación completa del virus.
PRECAUCIÓN: El paraformaldehído es tóxico por contacto con la piel y por inhalación y también es un sólido inflamable; Manéjelo con cuidado y guárdelo en un gabinete de almacenamiento inflamable. El cacodilato de sodio trihidrato es tóxico si se ingiere o inhala. - Tome una imagen de referencia del tejido antes de comenzar el proceso de limpieza.
NOTA: Las muestras de LM se pueden almacenar durante al menos 1 año en estas condiciones. Para trabajar con muestras que expresan proteínas fluorescentes endógenas, mantenga siempre las muestras en la oscuridad en los pasos siguientes.
2. Limpieza de tejido cúbico
- Enjuague las muestras de tejido linfoide en PBS estéril de 0,1 M tres veces agitando a temperatura ambiente durante 15 minutos para asegurar la eliminación de PFA durante cada cambio de tampón.
NOTA: Deseche los líquidos que contengan PFA de acuerdo con las pautas institucionales. - Sumerja la muestra de tejido linfoide en el reactivo CUBIC-1 (ver tabla de materiales) a 37 °C durante 3 días con una agitación suave. Tome imágenes de referencia periódicas para controlar el proceso de decoloración a lo largo del tiempo.
- Cambie el reactivo-1 por 3-4 días adicionales de inmersión, o hasta que se complete la decoloración del tejido. El tiempo necesario para el aclarado depende tanto del volumen como del tipo de tejido. Para acelerar el proceso de decoloración de los tejidos, actualice el reactivo CUBIC-1 diariamente y utilice volúmenes más grandes.
- Lave las muestras de tejido linfoide tres veces con 0,1 M PBS durante 30 min a temperatura ambiente con una agitación suave.
- Sumerja las muestras de tejido linfoide en CUBIC Reagent-2 (consulte la tabla de materiales) a 37 °C con una agitación suave durante 2-7 días o hasta que se logre una transparencia completa. Si las muestras no alcanzan una transparencia completa, repita los pasos 2.2-2.5 hasta que el borrado ya no progrese. Tome imágenes de referencia periódicas para monitorear el proceso de limpieza a lo largo del tiempo.
- Lave las muestras de tejido linfoide tres veces con 0,1 M PBS durante 30 min a temperatura ambiente con una agitación suave.
- Almacene las muestras en el reactivo CUBIC-2 con azida sódica al 0,01 % volúmen/volumen (V/V) en la oscuridad (consulte la tabla de materiales).
NOTA: Las muestras se pueden almacenar durante al menos 6 meses utilizando este método.
PRECAUCIÓN: La azida de sodio es altamente tóxica y representa un grave peligro de inhalación. Se recomienda comprar soluciones diluidas de azida de sodio al 5% o menos.
3. Bloqueo e inmunotinción de muestras cúbicas
- Lave las muestras de tejido linfoide tres veces con 0,1 M PBS durante 30 minutos cada una a temperatura ambiente con una agitación suave.
- Para obtener imágenes con un microscopio confocal, corte el tejido en rodajas de ~0,5-1 mm de grosor utilizando una matriz de corte de tejido. Para realizar la microscopía de fluorescencia de lámina de luz (LSFM), bloquee toda la región del tejido.
- Bloquear las muestras con 5 mL de solución bloqueante CUBIC durante la noche a 4 °C con agitación (ver Tabla de Materiales). Cuando trabaje con NHP o muestras humanas, utilice FcR antihumano. Al trabajar con muestras de ratón, utilice FcR anti-ratón en la solución de bloqueo.
- Teñir las muestras con 5 mL de anticuerpos primarios (ver Tabla de Materiales) en solución de bloqueo (sin FcR específico de la especie) durante 3 días a temperatura ambiente con agitación (Opcional: centrifugar el stock de anticuerpos concentrados a 2.300 x g durante 5 min antes de su uso, para reducir la adición de anticuerpo agregado).
- Lave la muestra teñida a temperatura ambiente agitándola durante un período de tiempo mínimo de 5 h en total con al menos cinco intercambios de tampón de solución de lavado (consulte la tabla de materiales).
- Teñir las muestras con anticuerpos secundarios (ver Tabla de Materiales) en solución bloqueante (sin FcR específico de la especie) durante 3 días a temperatura ambiente con agitación (Opcional: centrifugar los anticuerpos a 2.300 x g durante 5 min antes de su uso para minimizar la agregación de anticuerpos).
- Lave la muestra manchada cinco veces con solución de lavado a temperatura ambiente agitando durante al menos 5 h en total.
- Tiñir las muestras con 5 mL de solución de tinción DAPI (ver Tabla de Materiales) a cada muestra de tejido e incubar durante 10 min a temperatura ambiente. Deje que las muestras permanezcan en la solución de tinción DAPI en la oscuridad a 4 °C para obtener imágenes más tarde.
- Lave las muestras de tejido linfoide con solución de lavado tres veces a temperatura ambiente agitándolas durante 30 minutos cada una.
- Sumerja la muestra teñida en CUBIC Reagent-2 durante la noche a temperatura ambiente en la oscuridad antes de montar la muestra.
4. Limpieza de tejidos CLARITY
- Enjuague las muestras de tejido linfoide en PBS estéril de 0,1 M tres veces agitando a temperatura ambiente durante 15 minutos cada una para eliminar el PFA.
- Colocar las muestras de tejido en 15 mL de solución de acrilamida recién hecha e incubar a 4 °C durante la noche con una agitación suave (ver Tabla de Materiales).
PRECAUCIÓN: La acrilamida no polimerizada es una potente neurotoxina y se absorbe fácilmente a través de la piel. Evite cualquier contacto con la piel y enjuague inmediatamente si se produce contacto. - Deje que las muestras de tejido se calienten a temperatura ambiente.
- OPCIONAL: Desgasifique las muestras de tejido burbujeando nitrógeno en la solución de acrilamida durante 1 min. Tenga cuidado de utilizar un caudal bajo que evite salpicaduras de acrilamida no polimerizada tóxica (~1-2 burbujas/s).
- Colocar las muestras de tejido en un baño de agua a 37 °C durante 1-3 h para polimerizar, invirtiendo cada 15 min. Retire las muestras tan pronto como se detecte una polimerización notable, como lo indica un líquido viscoso, la aparición de líneas de Schleren durante la mezcla o la formación de una cápsula transparente alrededor del tejido.
NOTA: Si se produce la polimerización completa de la solución de acrilamida, recorte el exceso de hidrogel de la muestra y continúe con el protocolo. - Lave las muestras de tejido con PBS estéril de 0,1 M tres veces durante 30 minutos cada una a temperatura ambiente agitando suavemente para eliminar la solución de acrilamida.
- Coloque las muestras de tejido en 15 mL de SDS al 8% en PBS de 0.1 M a 37 °C con un balanceo suave durante 2-5+ días para permitir el aclarado. Actualice periódicamente la solución SDS al 8% y utilice hasta 50 ml de la solución para acelerar el aclarado, si es necesario. Detenga el proceso de limpieza cuando las muestras sean visualmente transparentes o ya no progresen. Tome imágenes de referencia periódicas para monitorear el proceso de limpieza a lo largo del tiempo.
- Lave las muestras de tejido con PBS estéril 0,1 M cinco veces durante 1 día a temperatura ambiente con una agitación suave.
- Mantenga las muestras temporalmente en 0,1 M PBS (más 0,01% de volumen/volumen (v/v) NaN3 para almacenamiento a largo plazo) en la oscuridad hasta que estén listas para obtener imágenes de fluorescencia endógena.
- Coloque el tejido en 5 mL de medio de imagen RI-2 (consulte la tabla de materiales). Incubar durante la noche a temperatura ambiente en la oscuridad para verificar que el proceso de limpieza esté completo antes de la inmunotinción. Tome imágenes de referencia para controlar la transparencia del tejido.
5. Bloqueo e inmunotinción de muestras CLARITY
NOTA: Estos pasos son similares al bloqueo y la inmunotinción de los tejidos aclarados CUBIC, pero utilizan diferentes formulaciones para soluciones de bloqueo, lavado y tinción.
- Lave las muestras de tejido linfoide tres veces con 0,1 M PBS durante 30 min cada vez a temperatura ambiente con una agitación suave.
- Para obtener imágenes con un microscopio confocal, corte el tejido en rodajas de ~0,5-1 mm de grosor con un cortador de tejido de 0,5 mm y una matriz. Para realizar LSFM, bloquee toda la muestra de tejido.
- Bloquear las muestras con 5 mL de solución bloqueadora CLARITY (ver Tabla de Materiales) durante la noche a 4 °C con agitación.
- Teñir las muestras con 5 mL de anticuerpos primarios (ver Tabla de Materiales) en solución bloqueante (sin FcR específico de la especie) durante 3 días a temperatura ambiente con agitación (Opcional: centrifugar los anticuerpos a 2.300 x g durante 5 min antes de su uso para minimizar la agregación de anticuerpos).
- Lave la muestra manchada cinco veces con la solución de lavado a temperatura ambiente agitando durante al menos 5 h en total (consulte la tabla de materiales).
- Teñir las muestras con 5 mL de anticuerpos secundarios (ver Tabla de Materiales) en solución de bloqueo (sin FcR específico de la especie) durante 3 días a temperatura ambiente con agitación (Opcional: centrifugar los anticuerpos a 2.300 x g durante 5 min antes de su uso para minimizar la agregación de anticuerpos). Para acortar la duración total del protocolo, utilice anticuerpos primarios conjugados con fluoróforos para eliminar la necesidad de incubación con anticuerpos secundarios.
- Lave la muestra manchada cinco veces con la solución de lavado a temperatura ambiente agitando durante al menos 5 h en total.
- Tiñir las muestras con 5 mL de solución de tinción DAPI (ver Tabla de Materiales) a cada muestra de tejido e incubar durante 10 min a temperatura ambiente. Deje que las muestras permanezcan a 4 °C en la oscuridad en la solución de tinción DAPI para obtener imágenes más tarde.
- Lave las muestras de tejido linfoide con la solución de lavado tres veces a temperatura ambiente agitándolas durante 30 minutos cada vez.
- Coloque el tejido en 5 mL de medio de imagen RI-2 (R.I. = 1,46) e incube durante la noche a temperatura ambiente en la oscuridad antes del montaje de la muestra (consulte los pasos 6 y 7 del protocolo).
6. Montaje y obtención de imágenes de muestras de tejido aclaradas para microscopía confocal
- Despegue un lado de la capa protectora de un aislador de silicona adhesivo.
- Pegue un cubreobjetos de microscopio (22 mm x 40 mm, 0,25 mm de grosor) en el lado despegado del aislador de silicona para formar un espacio a prueba de líquidos para la muestra.
- Despegue el otro lado de la capa protectora del aislador de silicona adhesivo.
- Coloque la muestra para la obtención de imágenes en el centro del aislador de silicona y, a continuación, agregue el reactivo CUBIC-2 o el medio de adquisición de imágenes RI-2, según corresponda, hasta que la superficie del líquido esté tan alta como el borde del aislador.
- Para minimizar el atrapamiento de burbujas de aire dentro del aislador de silicona, alinee y coloque suavemente el segundo cubreobjetos desde un lado con una pinza EM. Limpie el exceso de líquido. Presione suavemente el cubreobjetos alrededor de los pocillos de muestra con la parte posterior de las pinzas para sellar el adhesivo. Guarde las muestras montadas horizontalmente en la oscuridad.
NOTA: Las muestras se pueden obtener semanas o meses después de ser montadas; Sin embargo, la calidad de las imágenes generalmente disminuye con el tiempo. - Coloque el portaobjetos montado en la platina del microscopio y localice la muestra utilizando luz blanca y un objetivo de aumento más bajo (2-10x).
- Configure el perfil de adquisición de fluorescencia en función de los fluoróforos individuales elegidos.
NOTA: Se recomienda adquirir individualmente canales de fluoróforos separados. Esto da como resultado un tiempo de adquisición más largo, pero reduce la superposición espectral y la adquisición de señales de fluorescencia no específicas. Un perfil de fluoróforo común puede incluir DAPI (450 nm), Alexa488, Alexa594 y Alexa647 (o combinaciones relacionadas) para minimizar la superposición espectral durante la adquisición de imágenes. - Elija un objetivo de ampliación adecuado para visualizar las regiones de interés. Utilice objetivos de aumento más bajos (2-10x) para obtener imágenes de mayor volumen o de tejido completo con resolución de una sola célula y utilice objetivos de aumento más altos (20-63x) para una visualización de mayor resolución de los detalles subcelulares en el tejido aclarado. Haga coincidir el índice de refracción de los objetivos, los medios de imagen y el tejido lo más cerca posible para minimizar la introducción de distorsiones ópticas durante la adquisición de imágenes.
- Elija un tamaño de paso para la adquisición de la pila Z. Para objetivos de aumento más bajos (2-10x), seleccione un tamaño de paso de ~3-5 μm para detectar la fluorescencia de una celda individual en múltiples cortes Z continuos para el modelado 3D, al tiempo que reduce el tiempo total de adquisición y el tamaño total del archivo. Para objetivos de aumento más altos (20-63x), seleccione un tamaño de paso de ~1 μm o menos para minimizar la pérdida de información subcelular entre cortes Z individuales.
- Amplíe el campo de visión para visualizar toda la región del tejido que se va a visualizar en las dimensiones X e Y con la menor cantidad posible de área desocupada. Establezca las coordenadas de adquisición de la etapa Z superior e inferior que abarcan toda la región de interés que se va a fotografiar.
- Adquiera las imágenes de la pila Z. Guarde y exporte el archivo para su posprocesamiento utilizando cualquier software de análisis de imágenes. Para ciertos paquetes de software, convierta los archivos a tipos de archivo específicos (por ejemplo, .tiff, .ome-tiff, .jpeg, etc.). Realice la conversión utilizando cualquier software de adquisición de imágenes de microscopio o software gratuito de análisis de imágenes (por ejemplo, ImageJ/Fiji).
7. Montaje y obtención de imágenes de las muestras en la cámara o cubeta LSFM
- Llene la cámara de imágenes con CUBIC Reagent-2 o RI-2 según el protocolo específico utilizado. Evite la formación de burbujas durante la transferencia del líquido. Retire el exceso de burbujas con una pipeta.
- Sumerja la muestra en la cámara de imágenes y restrinja el movimiento de la muestra.
NOTA: Dependiendo del microscopio específico utilizado, esto puede incluir la inclusión de la muestra en agarosa, la suspensión de la muestra de un gancho o adaptador de puercoespín, la impresión 3D de un soporte de muestra o la fijación de la muestra con adhesivo a un plato de plástico. - Coloque el objetivo en la solución de imagen, enfocado en la muestra. Deje la muestra montada en la cámara de imágenes durante varias horas o toda la noche para permitir el equilibrio completo de las soluciones y los tejidos en la cubeta.
- Adquiera la pila Z de la región de interés (consulte los pasos 6.7-6.11 para la adquisición de imágenes).
NOTA: Este enfoque puede permitir la obtención de imágenes de volúmenes de tejido superiores a 1cm3 con resolución de una sola célula.
8. Reconstrucción de superficies y cuantificación de células con el software de análisis de imágenes Imaris
NOTA: Estos pasos son específicos del software de análisis de imágenes Imaris, pero se pueden realizar pasos similares de procesamiento de imágenes utilizando otros paquetes de software (por ejemplo, ImageJ/Fiji, Aivia, Arivis, Amira, etc.).
- Utilice el convertidor de archivos Imaris para convertir el archivo de imagen Z-stack al formato nativo de Imaris .ims. Esto facilitará una conversión de archivos más rápida al tiempo que minimizará los errores de conversión y los posibles problemas de software una vez abiertos.
NOTA: Algunos LSFM más recientes permiten al usuario guardar archivos directamente en el formato .ims. - Arrastre el archivo .ims que se va a analizar al área Arena del software Imaris. Ajuste el contraste o la intensidad de cada canal de color mediante el panel Ajuste de pantalla . Haga clic en el icono Agregar nuevas superficies en la parte superior izquierda.
- Haga clic en Siguiente: Canal de origen (el icono azul con una flecha apuntando a la derecha). Elija el canal de origen de la superficie que se va a construir. No cambie los demás parámetros.
- Haga clic en Siguiente: Umbral (el icono azul con una flecha apuntando hacia la derecha).
- Para ajustar el umbral (intensidad absoluta), arrastre la línea del umbral hacia la izquierda o hacia la derecha. Habilite Dividir objetos táctiles e introduzca el diámetro medio de la celda en micras como estándar de división para que el sistema produzca muchos puntos como origen para cada superficie individual.
- No incluya señales fluorescentes que sean demasiado pequeñas o demasiado brillantes, ya que pueden representar posibles tinciones o artefactos del microscopio. Incluya solo los puntos que tienen tamaños e intensidades de fluorescencia aceptables cambiando el diámetro promedio de la celda en consecuencia.
NOTA: El diámetro promedio de la célula variará para tejidos o tipos de células específicos, pero generalmente residirá entre 5 y 15 μm.
- No incluya señales fluorescentes que sean demasiado pequeñas o demasiado brillantes, ya que pueden representar posibles tinciones o artefactos del microscopio. Incluya solo los puntos que tienen tamaños e intensidades de fluorescencia aceptables cambiando el diámetro promedio de la celda en consecuencia.
- Haga clic en Siguiente: Clasificar superficies (el icono azul con una flecha apuntando a la derecha etiquetado). Ajuste las superficies que se incluirán arrastrando la línea de umbral hacia la izquierda o hacia la derecha. Asegúrese de que las superficies se aproximen exactamente a la señal de fluorescencia bruta, al tiempo que separa la señal de fluorescencia de las células individuales.
- Haga clic en Finalizar: Ejecutar todos los pasos de creación y finalizar el asistente (el icono verde con dos flechas apuntando a la derecha etiquetado). La superficie está oficialmente construida.
- Haga clic en el sexto icono etiquetado como Estadísticas en el panel izquierdo para ver el número de celdas, en esta circunstancia, el número de superficies para el canal de color específico analizado.
- Asegúrese de que las cuatro variables Número de componentes desconectados por punto de tiempo, Número de superficies por punto de tiempo, Número total de componentes desconectados y Número total de superficies tengan el mismo número, que es el recuento de celdas de ese canal de color.
Resultados
La limpieza de tejidos consiste en tratar los tejidos preservados con cócteles químicos para extraer biomoléculas opacas del tejido mientras se mantiene la arquitectura del tejido. Estas soluciones de limpieza de tejidos hacen coincidir el índice de refracción del tejido con el medio de imagen circundante para minimizar las distorsiones ópticas, mejorar la relación señal-ruido en lo profundo de los tejidos y minimizar la autofluorescencia de fondo. Se utilizaron dos protocolos basados en agua para la limpieza óptica de tejidos, CUBIC3 y CLARITY9, para eliminar muestras preservadas de hu-ratón, primates no humanos y tejido humano infectadas por VIH/VIS antes de la tinción de inmunofluorescencia y la obtención de imágenes con microscopía de fluorescencia confocal y de lámina de luz.
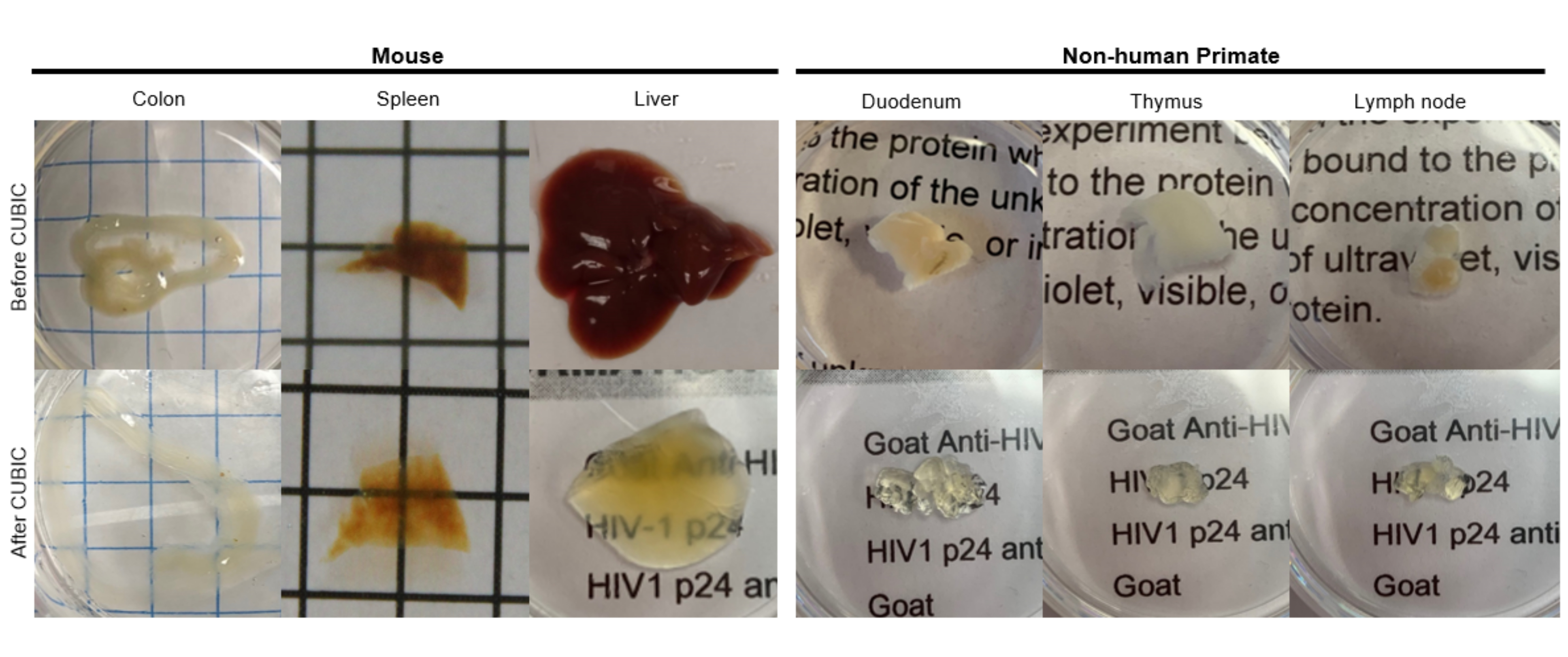
Para el protocolo CUBIC, los tejidos fijados se lavaron con PBS para eliminar los fijadores y se sumergieron en CUBIC Reagent-1, una solución tamponada básica de aminoalcoholes que eluye cromóforos como el hemo, lo que resulta en la decoloración y deslipidación del tejido (Figura 1, arriba). Los volúmenes de tejido más pequeños (~mm 3) se pueden decolorar después de 3 días de tratamiento con CUBIC Reagent-1, pero los volúmenes de tejido más grandes (~ cm3) o los tejidos con una gran cantidad de hemo (como el hígado, el bazo o el corazón) requieren tiempos de incubación y volúmenes de solución más largos (>1 mes y ~ 50 mL), así como un intercambio frecuente de la solución cada 2-3 días. Después de la decoloración, los tejidos se lavaron y se colocaron en CUBIC Reagent-2, una solución que contiene sacarosa con un índice de refracción de aproximadamente 1,48-1,49, que coincide con el índice de refracción del tejido y aumenta la transmitancia de la luz. Los tejidos aclarados se inmunoteñieron y se montaron en una solución de reactivo CUBIC-2 antes de la obtención de imágenes con un microscopio confocal o de lámina de luz. Se obtuvieron imágenes de los efectos del procedimiento de limpieza CUBIC para varios tejidos de ratón hu y NHP de varios tamaños y concentraciones de cromóforos (Figura 2). El aclarado óptico hizo que los tejidos fueran visiblemente transparentes a simple vista, lo que permitió que las líneas de cuadrícula y el texto de las hojas de papel se vieran "a través" del tejido. Es posible que los tejidos ricos en cromóforos, como el bazo, el hígado, la médula ósea y el corazón, no se decoloren por completo, pero siguen siendo adecuados para la inmunotinción y la obtención de imágenes (Figura 2 y Figura 5).
Para el protocolo CLARITY, los tejidos fijados se lavaron con PBS para eliminar los fijadores y luego se incubaron durante la noche a 4 °C en una solución de acrilamida al 40% con un iniciador térmico para formar enlaces covalentes entre las proteínas de la muestra y los monómeros de acrilamida (Figura 1, abajo). Al día siguiente, después de que el tejido se equilibró a temperatura ambiente y luego se calentó en un baño de agua a 37 °C, se inició la polimerización de acrilamida y rápidamente encerró la muestra en un hidrogel. La muestra se trató con una solución de SDS al 8% durante 2-5 días para eliminar los lípidos opacos. Inmediatamente antes de la tinción fluorescente, la muestra se sumergió en una solución de coincidencia de índice de refracción (RIMS) para CLARITY (Imaging Media RI-2) que contenía un medio de gradiente de densidad no iónico del 90%. Para tejidos que contienen grandes cantidades de hemo, se puede agregar un paso de decoloración al final del paso de delipidación 9,11,12. Se comparó la progresión del aclaramiento CUBIC y CLARITY en diferentes secciones de la misma muestra de bazo humano (Figura 3). El aclarado CLARITY produce un gel de poliacrilamida visible que recubre la solución y, por lo general, presenta una decoloración reducida en comparación con el aclarado CUBIC, a menos que se agregue un paso de decoloración adicional 9,12.
Posteriormente, en ambos protocolos, los tejidos limpios e intactos se inmunotiñeron para detectar poblaciones específicas de células inmunitarias. Las muestras se lavaron, se bloquearon con un reactivo que contenía α-FcR para reducir la unión de anticuerpos no específicos y se tiñeron durante 3 días cuando se utilizó un anticuerpo primario conjugado directamente con un fluoróforo. Alternativamente, las muestras se tiñeron durante 3 días con un anticuerpo primario no conjugado, seguido de 3 días adicionales con un anticuerpo secundario conjugado con un fluoróforo. Los tejidos se lavaron de nuevo y luego se incubaron con tinción de DAPI durante la noche a 4 °C para la visualización nuclear. Las muestras se lavaron e incubaron en CUBIC Reagent-2 durante 24-36 h o Imaging Media RI-2 (CLARITY) durante la noche en la oscuridad. Para la microscopía confocal, los tejidos se montaron en un portaobjetos de microscopio en el RIMS apropiado antes de la obtención de imágenes (Figura 4). Para la microscopía de fluorescencia de lámina de luz (LSFM), las muestras se sumergieron completamente con RIMS en una cubeta de imágenes durante la noche anterior a la obtención de imágenes.
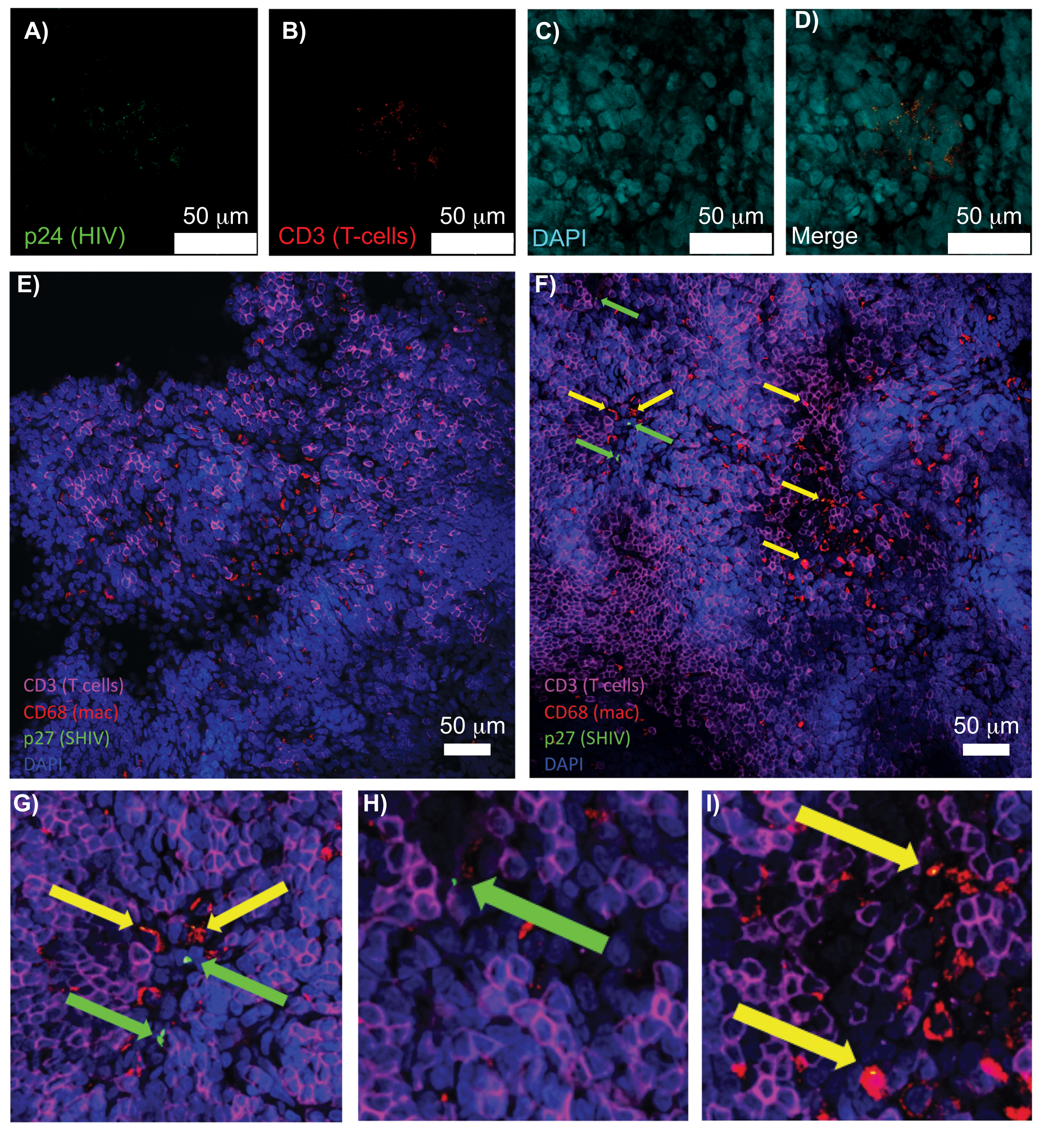
La microscopía confocal de tejidos linfoides intactos, aclarados e inmunoteñidos permitió la visualización simultánea de múltiples señales fluorescentes, incluidos núcleos, marcadores de células inmunitarias y proteínas CA (CÁPSIDE) DEL VIH/SIV (figura 5). Las células productoras de virus se determinaron mediante la colocalización por fluorescencia de los marcadores de células inmunitarias y las proteínas del VIH. El bazo humano infectado por el VIH aclarado y teñido reveló múltiples células T CD3+ colocalizadas con el VIH p24, lo que indica la presencia de células productoras de virus dentro de una región de tejido intacto (Figura 5A-D). Los ganglios linfáticos NHP aclarados e inmunoteñidos con SHIV revelaron las distribuciones de células T CD3+ y macrófagos CD68+ en regiones de tejidos sin virus detectados (Figura 5E), además de regiones con numerosas células productoras de virus (Figura 5F). Estos resultados mostraron que las células productoras de virus de diversas fuentes de tejido se distinguían de otras células dentro de un campo de visión determinado y permitieron la detección de eventos biológicos raros dentro de un entorno tisular complejo.
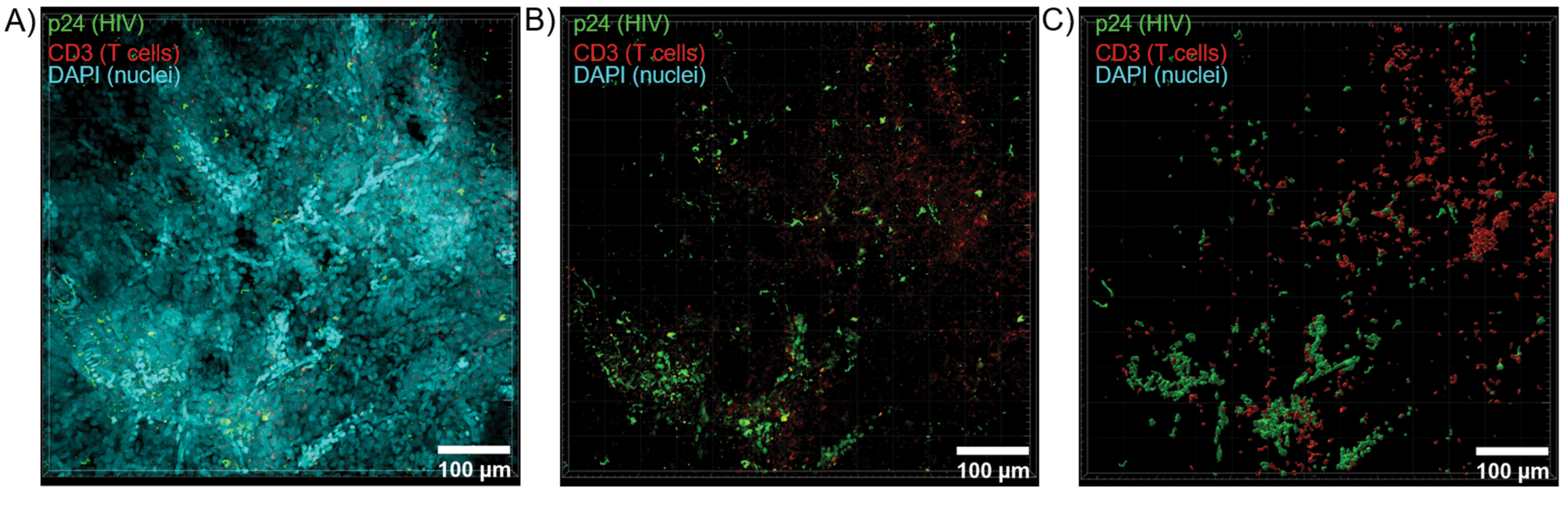
Se aplicó el corte óptico de los tejidos aclarados con un microscopio confocal para generar pilas Z y modelos de superficie 3D, que revelaron la heterogeneidad celular exhibida durante la infección por VIH (Figura 6). Las pilas Z se recombinaron en una imagen de proyección Z utilizando el paquete de software Imaris (Figura 6A) y se eliminó el canal nuclear DAPI para una visualización clara de las células T CD3+ y la fluorescencia de la proteína de la cápside del VIH (p24) en volúmenes enteros de tejido (Figura 6B). La fluorescencia de proyección Z se segmentó automáticamente con el software Imaris para generar un modelo de superficie 3D reconstruido para la visualización espacial y la cuantificación de la señal de fluorescencia en toda la pila Z (Figura 6C). El análisis del modelo de superficie 3D reveló 546 células T CD3+ y 218 células productoras de VIH p24. De forma acumulativa, la adquisición de inmunofluorescencia a partir de tejidos linfoides aclarados e infectados por el VIH permitió la generación de modelos 3D de la composición celular dentro del tejido y la cuantificación automatizada de las poblaciones de células inmunitarias dentro de los volúmenes de tejido.
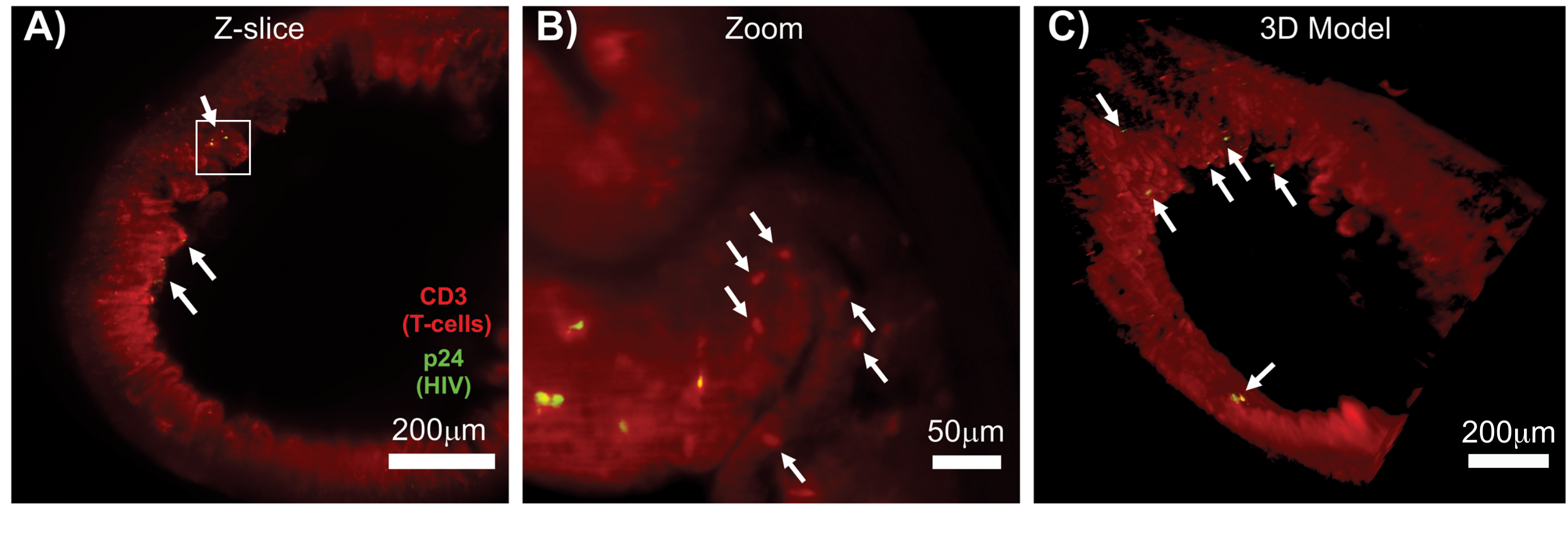
La LSFM de tejidos linfoides intactos, aclarados e inmunoteñidos permitió obtener imágenes de inmunofluorescencia (IF) de mayor volumen de la distribución de las células inmunitarias y productoras de virus en los tejidos linfoides (Figura 7). La inmunotinción del tejido del colon de un ratón hu infectado por el VIH para las células T hCD3+ y el VIH p24 reveló focos de células productoras de virus dispersas en grandes regiones de tejido sin evidencia de infección (Figura 7A). Una vista ampliada de un foco de células productoras de virus reveló múltiples células productoras de virus muy cerca de las posibles células objetivo (Figura 7B). Se utilizó la autofluorescencia tisular (neblina roja) para visualizar la arquitectura completa del tejido y distinguir poblaciones específicas de células inmunitarias dentro del tejido que se tiñeron más brillantemente que la autofluorescencia (óvalos rojos). Un modelo 3D de todo el volumen de la pila Z de LSFM mostró la distribución espacial de los focos de células productoras de virus dentro de una región de tejido intacto y permitió el mapeo de las ubicaciones de producción de virus en relación con la arquitectura general del tejido (Figura 7C). Sorprendentemente, los focos de células productoras de virus a menudo se intercalaban entre grandes regiones de tejido sin evidencia de producción de virus. Estos resultados pueden permitir la cuantificación de los parámetros de distribución del virus y la densidad de células infectadas dentro de diferentes tejidos y en diferentes momentos de infección o respuesta a diferentes tratamientos.

Figura 1: Flujo de trabajo de la limpieza de tejidos, la inmunotinción y la obtención de imágenes típicas de CUBIC y CLARITY. Los tiempos de aclarado CUBIC (arriba) y CLARIDAD (abajo) pueden variar ampliamente según el tamaño y el tipo de tejido. Para el aclarado de CLARITY, se requiere un paso de incubación adicional con medios de índice de refracción coincidente antes de la inmunotinción para verificar que el tejido esté limpio. La inmunotinción suele durar 3 días cuando los anticuerpos primarios se conjugan con fluoróforos y 6 días si se requieren anticuerpos secundarios fluorescentes. Las muestras se pueden obtener con un confocal o LSFM. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Limpieza CUBIC de muestras de tejido de hu-mouse y NHP. Dependiendo de las diferentes densidades de hemo y lípidos de las muestras de tejido, el tiempo necesario para limpiar cada tipo de tejido varía. Por ejemplo, el colon y el duodeno suelen requerir períodos relativamente cortos (~7 días), mientras que el bazo y el hígado pueden tardar más en volverse transparentes (~30 días). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Comparación longitudinal de los métodos de limpieza de tejidos en muestras humanas. CUBIC (paneles superiores) y CLARITY (paneles inferiores) eliminaron el bazo de una persona infectada por el VIH en tratamiento antirretroviral. Ambos métodos limpiaron adecuadamente el tejido para el día 32 para la inmunotinción y la obtención de imágenes. El paso de decoloración para el método CUBIC reduce visiblemente la autofluorescencia causada por la presencia de hemo contenido en las muestras de bazo. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

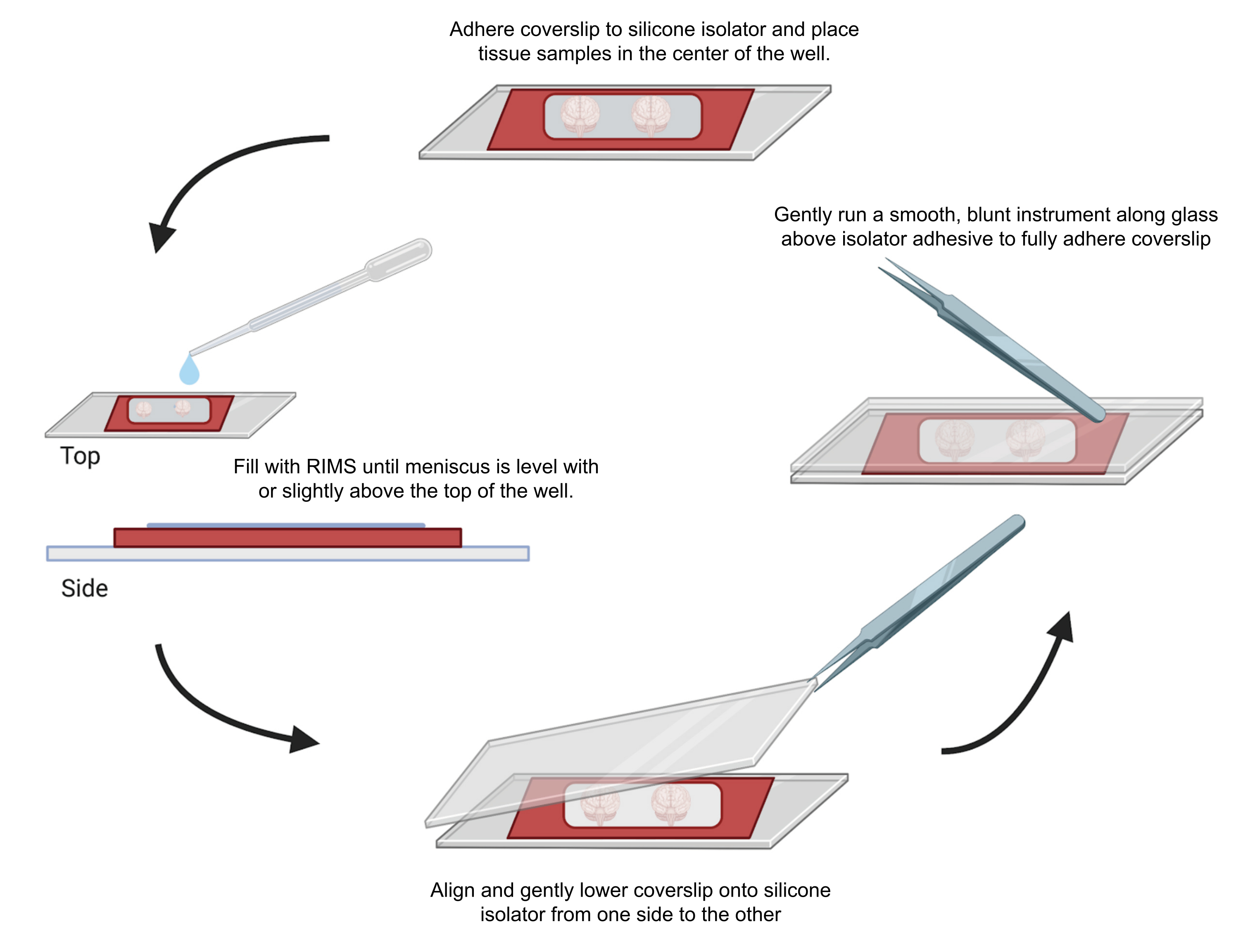
Figura 4: Montaje de muestras para microscopía confocal. Las muestras se montaron entre cubreobjetos separados con aisladores de silicona adhesivos de 0,5-1 mm. Se adhirieron aisladores de silicona al primer cubreobjetos y se colocó tejido en el centro del pocillo (parte superior). El pozo se llenó con RIMS hasta que el menisco estuvo al nivel o ligeramente por encima de la parte superior del pozo (izquierda). El segundo cubreobjetos se bajó cuidadosamente en su lugar de un lado a otro, evitando burbujas (parte inferior). Los cubreobjetos se adhirieron completamente al aislador de silicona pasando suavemente un instrumento romo alrededor del perímetro del pozo (derecha). Las muestras se tomaron en un microscopio confocal estándar. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Microscopía confocal de bazo humano claro e intacto y ganglios linfáticos NHP. (A-D) El tejido humano infectado por el VIH se tiñó para el VIH-1 p24 (verde), las células T hCD3+ (rojo) y los núcleos (cian). (E) Corte Z confocal de ganglio linfático CÚBICO aclarado de un NHP infectado con SHIV 8 semanas después de la infección inmunoteñido para células T CD3+ (magenta), macrófagos CD68+ (mac/rojo), SHIV p27 (verde) y núcleos (azul). El campo de visión contiene células T, macrófagos y otros tipos de células, pero no hay evidencia de células productoras de SHIV (verde). F) Cortes Z confocales de una región adyacente del mismo ganglio linfático que muestran diferencias en la densidad y el número de células, junto con la presencia de células T CD3+ productoras de virus (flechas verdes) y macrófagos CD68+ (flechas amarillas). (G-I) Vista ampliada de las regiones seleccionadas de la tinción de p27 desde (F). Las barras de escala son de 50 μm. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 6: Volumen de la pila Z y superficie reconstruida en 3D a partir de bazo humano infectado por el VIH. (A) Imagen de proyección Z de una pila Z de 600 μm x 600 μm x 100 μm de tejido de bazo humano infectado por el VIH teñido para VIH-1 p24 (verde), células T hCD3+ (rojo) y núcleos (cian). (B) La misma imagen de proyección Z sin tinción nuclear DAPI. (C) Modelo de superficie 3D reconstruido de fluorescencia CD3 (rojo) y p24 (verde) de todo el volumen de la pila Z. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 7: LSFM y reconstrucción en 3D de volúmenes superficiales de tejidos infectados por el VIH (A) corte en Z (1.000 μm x 1.000 μm) de colon de un ratón hu infectado por el VIH inmunoteñido para células T CD3+ (rojo) y VIH p24 (verde). La neblina roja opaca representa la autofluorescencia del tejido, mientras que las puntas rojas distintivas indican células T. Las vellosidades son visibles alrededor de la periferia, apuntando hacia el lumen central, con varios focos de producción activa de virus (flechas blancas) dispersos en grandes áreas que no contienen virus. El recuadro indica la región de interés aproximada para el panel B. (B) Región ampliada del tejido que muestra células T productoras de virus individuales hCD3+ (amarillo) en las proximidades de células T no infectadas (rojo). La imagen se rotó y se cambió a un corte Z cercano para mostrar un foco de células p24 positivas en un solo plano Z. La autofluorescencia roja de fondo muestra la arquitectura general del tejido, además de la tinción específica de células T hCD3+ (puntos rojos; flechas blancas). (C) Modelo 3D de la superficie del volumen completo (1.000 μm x 1.000 μm x 200 μm) generado con el software Imaris girado para mostrar focos de infección por VIH (amarillo) en distintas ubicaciones del intestino. Las flechas blancas indican focos individuales dentro del volumen. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
Los tejidos linfoides de interés deben recolectarse rápidamente después de la autopsia y colocarse inmediatamente en tampones fijadores preenfriados para evitar la necrosis tisular (tejido oscuro o negro) que puede afectar negativamente la tinción y la imagen. Después de recolectar los tejidos deseados, sumerja inmediatamente los tejidos en paraformaldehído (PFA) al 4%-8% congelado durante la noche para su fijación, que también inactiva los patógenos potenciales asociados con las muestras. El 4% de PFA es óptimo para la fijación de muestras de LM, mientras que el 8% de PFA puede preservar adecuadamente los tejidos tanto para LM como para EM. Seguir estos procedimientos y almacenar las muestras en un fijador a 4 °C en la oscuridad puede preservar eficazmente los tejidos para la obtención de imágenes de LM durante varios años. Una advertencia es que el almacenamiento a largo plazo en el fijador puede conducir a la introducción de artefactos de tinción, especialmente el enmascaramiento de antígenos, que es causado por la reticulación de proteínas adyacentes a la proteína de interés, lo que puede ocluir la accesibilidad de los anticuerpos de tinción al epítopo13,14. Si los tejidos contienen proteínas fluorescentes expresadas endógenamente, tome medidas para evitar exponer los tejidos a la luz siempre que sea posible durante todo el protocolo. Por lo general, las proteínas fluorescentes endógenas mantendrán la fluorescencia durante 6-12 meses después de la fijación, pero las muestras de tejido individuales pueden variar durante períodos de tiempo más largos o más cortos. Si la fluorescencia endógena se pierde debido a la degradación de la proteína, las proteínas fluorescentes a menudo se pueden detectar utilizando un anticuerpo primario específico para la proteína de interés. La perfusión es otra opción para fijar rápidamente los tejidos antes de la eliminación12; sin embargo, debido a las preocupaciones al trabajar con patógenos como el VIH, se eligió la ruta de la necropsia de tejidos seguida de la inmersión en un fijador helado para preparar las muestras de la manera más segura posible.
Una ventaja de los protocolos de limpieza a base de agua descritos es que generalmente son más suaves que los protocolos basados en orgánicos, que a veces pueden dañar tejidos que son más frágiles, como el hígado. Los protocolos de limpieza a base de agua generalmente requerirán más tiempo para lograr una limpieza completa de la muestra (semanas frente a días) en comparación con los protocolos de limpieza a base de orgánicos. Los protocolos CLARITY y CUBIC se pueden llevar a cabo más rápidamente utilizando la perfusión para eliminar simultáneamente todos los órganos dentro de un roedor11,12; sin embargo, esta no era una opción factible para las autopsias de NHP y humanas. Las muestras procesadas con CLARITY tienden a mostrar cierta expansión de volumen, mientras que CUBIC reveló una influencia reducida en el volumende muestra 9. Aunque generalmente son más rápidos, muchos protocolos de limpieza de tejidos de base orgánica hacen que los tejidos sufran una contracción15, lo que puede dificultar la detección de detalles unicelulares o subcelulares dentro de los tejidos densos de células, como los ganglios linfáticos y el bazo. La expansión inducida por el aclaramiento puede aumentar efectivamente la resolución de las imágenes, lo que facilita la observación de aspectos que serían difíciles de observar en el tamaño original del tejido. Alternativamente, la contracción del tejido puede disminuir efectivamente el tamaño total de la muestra, lo que puede hacer posible la obtención de imágenes de todo el órgano sin disección. Un beneficio de los protocolos CLARITY y CUBIC es que preservan la fluorescencia de las proteínas fluorescentes expresadas endógenamente en el tejido, mientras que permanecen susceptibles a la tinción de inmunofluorescencia11,12. La inmunotinción se puede llevar a cabo utilizando métodos de limpieza de tejidos acuosos u orgánicos; Sin embargo, la experiencia personal mostró una mayor proporción de compatibilidad de anticuerpos utilizando protocolos basados en agua en comparación con protocolos basados en orgánicos. Los investigadores deben considerar qué método de limpieza de tejidos utilizar en función de los tejidos de los que se obtienen imágenes y de las cuestiones biológicas abordadas (p. ej., imágenes de órganos completos frente a imágenes de regiones específicas de interés). No existe una técnica universal de aclarado de tejidos que permita un análisis sólido llave en mano para todas las preguntas de imágenes de gran volumen, y los métodos disponibles presentan distintas ventajas y desventajas según la aplicación biológica.
A la hora de realizar la tinción de anticuerpos, hay que tener en cuenta numerosos aspectos. Debido a que las muestras de CLARITY están incrustadas en hidrogel de acrilamida, tienden a requerir tiempos más largos para la incubación12. El tiempo necesario para la incubación de anticuerpos también depende del volumen y el grosor de cada muestra. La mayoría de las muestras descritas aquí tenían ~2-3 milímetros de grosor, y 3 días fueron suficientes para una tinción completa en todo el tejido. Si el objetivo es obtener imágenes de todo el cerebro de un ratón, el tiempo de incubación de los anticuerpos puede durar 1 semanao más. La elección de un método de limpieza de tejido acuoso frente a uno orgánico para la obtención de imágenes de inmunofluorescencia puede depender de la compatibilidad de los anticuerpos. En general, para CUBIC o CLARITY, la tasa de aciertos para los anticuerpos que funcionan en células y tejidos cultivados es de ~70%. Ya sea que se utilice un método de limpieza de tejido acuoso u orgánico, es necesario evaluar la compatibilidad y efectividad de todos los anticuerpos con el método específico utilizado. Como se muestra en esta sección del protocolo, la inmunotinción para las muestras procesadas CUBIC y CLARITY se lleva a cabo una vez finalizado el aclarado. Por el contrario, este paso tiene lugar antes del procedimiento de autorización para algunos protocolos de base orgánica, seguido de la posfijación.
Es de vital importancia que los tejidos estén completamente inmersos en un medio de imagen que coincida con su índice de refracción. De lo contrario, se introducirán aberraciones esféricas durante la obtención de imágenes y se distorsionará la luz capturada durante la adquisición de la imagen. Se debe tener cuidado de eliminar todas las burbujas de aire del medio de imagen al montar muestras tanto para confocal como para LSFM, ya que las burbujas pueden interrumpir el camino de la luz hacia o lejos de la muestra. Las burbujas se pueden eliminar manualmente con una pipeta antes del montaje final de la muestra. Para obtener imágenes de muestras más gruesas con un microscopio confocal, se pueden colocar varios espaciadores de silicona uno encima del otro para acomodar tejidos de más de 0,5 mm de grosor. Una recomendación es equilibrar todos los tejidos en RIMS durante varias horas o toda la noche mientras se montan en el microscopio sin movimiento adicional de la muestra. El equilibrio completo del tejido y los medios de imagen evitará la mezcla de soluciones con índices de refracción no coincidentes que pueden generar aberraciones durante la obtención de imágenes. Es importante recordar que cuando se montan muestras de tejido aclarado, no existe un único método de montaje llave en mano para obtener imágenes de todas las muestras en todos los microscopios. Este protocolo analiza las opciones de montaje de muestras que funcionaron de manera óptima en un contexto, pero existen numerosos enfoques para el montaje de muestras según el microscopio individual utilizado y la pregunta biológica abordada. Estos enfoques pueden incluir, entre otros, la incrustación de la muestra en agarosa, la suspensión de la muestra de un gancho o una línea de plástico con índice de refracción, el uso de un adaptador de puercoespín, la impresión 3D de un soporte de muestra o la fijación de la muestra con adhesivo a un plato de plástico.
Los microscopios confocales pueden funcionar bien para obtener imágenes de volúmenes de tejido ~ 1 mm,3-1 cm,3. En el caso de los microscopios confocal, utilice un objetivo de 2 a 10x para localizar inicialmente las regiones de interés y adquirir pilas Z de mayor volumen o de tejido completo con resolución de una sola célula. Cambie a objetivos de 20-63x para adquirir imágenes de mayor resolución de regiones específicas de interés con información subcelular. El objetivo ideal para la obtención de imágenes de tejidos aclarados CUBIC y CLARITY es un objetivo específico de CLARITY/Scale que coincida con precisión con el índice de refracción del tejido y la solución de imagen. Si no se dispone de este tipo de objetivo, lo óptimo es obtener imágenes de muestras con un objetivo de inmersión en glicerol o aceite (por ejemplo, LD LCI Plan-Apochromat 25 x 0,8 NA Imm Corr DIC M27 objetivo de inmersión múltiple: distancia de trabajo = 0,57 mm) en lugar de un objetivo de aire. Esto minimizará la introducción de distorsiones ópticas debido a índices de refracción no coincidentes durante la captura de imágenes. Los objetivos de 20-25x pueden equilibrar la adquisición de imágenes de mayor volumen con la obtención de detalles de tinción de células individuales en un entorno de tejido complejo. Es importante destacar que la mayoría de los microscopios confocales contienen módulos que permiten el mosaico 3D de los volúmenes de imágenes. Idealmente, este tipo de adquisición de imágenes puede generar pilas Z de mayor volumen que contengan información subcelular.
Las imágenes LSFM pueden permitir la visualización en 3D de poblaciones celulares específicas en el contexto de grandes volúmenes de tejido (>1 cm3) e incluso órganos completos. Durante los últimos 10 años, la limpieza de tejidos combinada con LSFM se centró en gran medida en comprender la conectividad cerebral dentro de los roedores; Sin embargo, las aplicaciones más recientes incluyen la visualización de paisajes metastásicos tumorales16, la distribución celular dentro de los compartimentos anatómicos 9,17 y la dispersión de patógenos18. En comparación con las células cultivadas, la mayoría de los eventos biológicos en los tejidos no son uniformes y la LSFM puede ser particularmente hábil para visualizar y cuantificar la heterogeneidad tisular espacial de estos eventos (por ejemplo, replicación del virus, señalización inmunitaria, distribución celular, etc.).
Los conjuntos de datos 3D adquiridos a través de confocal o LSFM se pueden posprocesar con numerosas plataformas de análisis de imágenes. El paquete de software Imaris se puede utilizar para la construcción de superficies, la generación de animación 3D y la cuantificación de celdas; Sin embargo, existen numerosos sistemas de análisis de imágenes que permiten un postprocesamiento y análisis de imágenes eficientes. ImageJ/Fiji freeware19 es una atractiva plataforma alternativa de procesamiento de imágenes accesible para la mayoría de los laboratorios, pero no existe un software de análisis único que sobresalga en todas las formas de análisis y visualización de imágenes. Muchos paquetes de software de análisis de imágenes pueden ser prohibitivamente caros si no están disponibles a través de instalaciones de uso compartido. Por último, un aspecto crítico de LSFM o grandes conjuntos de datos 3D confocales en mosaico es la gestión de datos. Estas plataformas de imágenes pueden generar archivos masivos (>1 Tb) que requieren estaciones de trabajo informáticas de gama alta para la visualización y cuantificación de datos. En última instancia, este flujo de trabajo de imágenes puede agilizar la adquisición y cuantificación de poblaciones celulares espacialmente distintas dentro de tejidos completos y es ampliamente aplicable a la mayoría de las fuentes de tejidos y sistemas biológicos.
Divulgaciones
Los autores no tienen conflictos de intereses que revelar.
Agradecimientos
Gracias a las instalaciones centrales del Instituto de Biología Genómica de la Universidad de Illinois en Urbana-Champaign por el uso de los microscopios de fluorescencia confocal y de lámina de luz. Gracias a las increíbles personas de la cohorte "The Last Gift" para muestras de tejido humano, que fue financiada por las siguientes subvenciones: I147821, DA051915, AI131385 y P30 AI036214. Gracias a Nancy Haigwood y Ann Hessell por las muestras de tejido NHP infectadas con SHIV.
Materiales
| Name | Company | Catalog Number | Comments |
| Acrylamide Solution (in 0.1 M PBS, 40 mL in total) | |||
| 40% Acrylamide: 4 mL | Bio-Rad | 1610144 | |
| VA-044 Thermal Initiator: 0.1g | Fujifilm | 011-19365 | |
| CLARITY Blocking solution (in 0.1 M PBS, 5 mL in total) | |||
| Fetal bovine serum (FBS): 200 µL | Atlas Biologicals | F-0500-D | |
| Rat anti-human or anti-mouse FcR: 50 µL | Miltenyi | 130-092-575(mouse)/130-059-901(human) | |
| Sodium azide (from stock solution): 5 µL | Sigma | 71289-50G | |
| Tween-20: 5 µL | Fisher Scientific | BP337-500 | |
| CLARITY wash solution (in 0.1M PBS, 50 mL in total) | |||
| Sodium azide (from stock solution): 50 µL | Sigma | 71289-50G | |
| Tween-20: 50 µL | Fisher Scientific | BP337-500 | |
| CUBIC Blocking solution (in 0.1M PBS, 5 mL in total) | |||
| Fetal bovine serum (FBS): 200 µL | Atlas Biologicals | F-0500-D | |
| Rat anti-human or anti-mouse FcR: 50 µL | Miltenyi | 130-092-575(mouse)/130-059-901(human) | |
| Sodium azide (from stock solution): 5 µL | Sigma | 71289-50G | |
| Triton X-100: 5 µL | VWR | M143-1L | |
| CUBIC Reagent-1 (in 0.1M PBS, 50 mL in total) | |||
| N, N, N’, N’-tetrakis (2-hydroxypropyl) ethylenediamine: 12.5 g | Aldrich | 122262 | |
| Triton X-100: 7.5 g | VWR | M143-1L | |
| Urea: 12.5 g | Fisher chemical | U15-500 | |
| CUBIC Reagent-2 (in 0.1M PBS, 50 mL in total) | |||
| Sucrose: 25 g | Sigma | S1888-500G | |
| Sodium azide (in powder form): 10 g | Sigma | 71289-50G | |
| Sodium azide stock solution (in DI H2O, 50 mL in total) | Sigma | 71289-50G | |
| Triethanolamine: 5 g | Sigma | 90270-500mL | |
| Triton X-100: 50 µL | VWR | M143-1L | |
| Urea: 12.5 g | Fisher chemical | U15-500 | |
| CUBIC wash solution (in 0.1M PBS, 50 mL in total) | |||
| Sodium azide (from stock solution): 50 µL | Sigma | 71289-50G | |
| Triton X-100: 50 µL | VWR | M143-1L | |
| DAPI staining solution (0.5 µg/mL) | |||
| DAPI stock solution: 1 µL | |||
| Wash solution: 10 mL | |||
| DAPI stock solution (5 mg/mL) | |||
| DAPI powder: 5 mg | Sigma-Aldrich | D9542-1MG | |
| DMSO (100%): 1 mL | ThermoFisher | D12345 | |
| Imaging Media RI-2 (in dH2O) | |||
| 90% Histodenz | Sigma | D2158-100G | |
| 0.01% Sodium azide | Sigma | 71289-50G | |
| 0.02 Sodium Phosphate Buffer, pH 7.5 | Sigma-Aldrich | S9638-250G | |
| 0.1% Tween-20 | Fisher Scientific | BP337-500 | |
| Primary antibodies (in blocking solution without rat anti-mouse FcR, 2 mL in total) | |||
| Goat anti-HIV p24: 10 µL (1:200) | Creative Diagnostics | DPATB-H81692 | |
| Mouse anti-human CD68: 10 µL(1:200) | Dako | M0876 | |
| Rabbit anti-human CD3: 10 µL (1:200) | Dako | A0452 | |
| 8% SDS Solution (in 0.1 M PBS, 50 mL in total) | |||
| SDS powder: 4 g | Sigma-Aldrich | L3771-500G | |
| Secondary antibodies (in blocking solution without rat anti-mouse FcR, 2 mL in total) | |||
| Donkey anti-goat conjugated with AlexaFluor647: 2 µL | Invitrogen | A21447 | |
| Donkey anti-mouse conjugated with AlexaFluor594: 2 µL | Invitrogen | A21203 | |
| Donkey anti-rabbit conjugated with AlexaFluor488: 2 µL | Invitrogen | A21206 |
Referencias
- Spalteholz, W., Barker, L. F., Mall, F. P. Hand-Atlas of Human Anatomy. , J.B. Lippincott Co. Philadelphia. Second edition in English (1907).
- Jacob, T., Gray, J. W., Troxell, M., Vu, T. Q. Multiplexed imaging reveals heterogeneity of PI3K/MAPK network signaling in breast lesions of known PIK3CA genotype. Breast Cancer Research and Treatment. 159 (3), 575-583 (2016).
- Kieffer, C., Ladinsky, M. S., Ninh, A., Galimidi, R. P., Bjorkman, P. J. Longitudinal imaging of HIV-1 spread in humanized mice with parallel 3d immunofluorescence and electron tomography. eLife. 6, 23282(2017).
- Chung, K., Deisseroth, K. CLARITY for mapping the nervous system. Nature Methods. 10 (6), 508-513 (2013).
- Compton, A. A., Schwartz, O. They might be giants: Does syncytium formation sink or spread HIV infection. PLoS Pathogens. 13 (2), 2-8 (2017).
- Symeonides, M., et al. HIV-1-induced small T cell syncytia can transfer virus particles to target cells through transient contacts. Viruses. 7 (12), 6590-6603 (2015).
- Sharova, N., Swingler, C., Sharkey, M., Stevenson, M. Macrophages archive HIV-1 virions for dissemination in trans. The EMBO Journal. 24 (13), 2481-2489 (2005).
- Colby, D. J., et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nature Medicine. 24 (7), 923-926 (2018).
- Ladinsky, M. S., et al. Mechanisms of virus dissemination in bone marrow of HIV-1-infected humanized BLT mice. eLife. 8, 46916(2019).
- Buettner, M., Bode, U. Lymph node dissection--understanding the immunological function of lymph nodes. Clinical and Experimental Immunology. 169 (3), 205-212 (2012).
- Treweek, J. B., et al. Whole-body tissue stabilization and selective extractions via tissue-hydrogel hybrids for high-resolution intact circuit mapping and phenotyping. Nature Protocols. 10, 1860-1896 (2015).
- Tainaka, K., et al. Whole-body imaging with single-cell resolution by tissue decolorization. Cell. 159, 911-924 (2014).
- Sompuram, S. R., Vani, K., Bogen, S. A. A molecular model of antigen retrieval using a peptide array. American Journal of Clinical Pathology. 125 (1), 91-98 (2006).
- Scalia, C. R., et al. Antigen masking during fixation and embedding, dissected. The journal of histochemistry and Cytochemistry: Official Journal of the Histochemistry Society. 65 (1), 5-20 (2017).
- Jing, D., et al. Tissue clearing of both hard and soft tissue organs with the PEGASOS method. Cell Research. 28 (8), 803-818 (2018).
- Guldner, I. H., et al. An Integrative platform for three-dimensional quantitative analysis of spatially heterogeneous metastasis landscapes. Scientific Reports. 6, 24201(2016).
- Muntifering, M., et al. Clearing for deep tissue imaging. Current Protocols in Cytometry. 86 (1), 38(2018).
- DePas, W. H., et al. Exposing the three-dimensional biogeography and metabolic states of pathogens in cystic fibrosis sputum via hydrogel embedding, clearing, and rRNA labeling. mBio. 7 (5), 00796(2018).
- Schindelin, J., et al. Fiji: An open-source platform for biological-image. Nature Methods. 9 (7), 676-682 (2012).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados