
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Producción de vectores de virus adenoasociados en pilas celulares para estudios preclínicos en modelos animales grandes
En este artículo
Resumen
Aquí proporcionamos un procedimiento detallado para la producción a gran escala de vectores AAV de grado de investigación utilizando células HEK 293 adherentes cultivadas en pilas celulares y purificación por cromatografía de afinidad. Este protocolo produce consistentemente >1 x 1013 genomas vectoriales / ml, proporcionando cantidades vectoriales apropiadas para estudios con animales grandes.
Resumen
Los vectores de virus adenoasociados (AAV) se encuentran entre los vectores de terapia génica clínicamente más avanzados, con tres terapias génicas AAV aprobadas para humanos. El avance clínico de nuevas aplicaciones para AAV implica la transición de modelos de animales pequeños, como ratones, a modelos animales más grandes, incluidos perros, ovejas y primates no humanos. Una de las limitaciones de la administración de AAV a animales más grandes es el requisito de grandes cantidades de virus de alto título. Si bien el cultivo celular en suspensión es un método escalable para la producción de vectores AAV, pocos laboratorios de investigación tienen el equipo (por ejemplo, biorreactores) o saben cómo producir AAV de esta manera. Además, los títulos de AAV a menudo son significativamente más bajos cuando se producen en células HEK 293 en suspensión en comparación con las células HEK293 adherentes. Aquí se describe un método para producir grandes cantidades de AAV de alto título utilizando pilas de celdas. También se describe un protocolo detallado para la titulación de AAV, así como métodos para validar la pureza del vector. Finalmente, se presentan resultados representativos de la expresión de transgenes mediada por AAV en un modelo de oveja. Este protocolo optimizado para la producción a gran escala de vectores AAV en células adherentes permitirá a los laboratorios de biología molecular avanzar en las pruebas de sus nuevas terapias AAV en modelos animales más grandes.
Introducción
La terapia génica que utiliza vectores de virus adenoasociados (AAV) ha logrado grandes avances en las últimas tres décadas1,2. Las mejoras demostradas en una amplia gama de enfermedades genéticas, incluida la ceguera congénita, la hemofilia y las enfermedades del sistema musculoesquelético y nervioso central, han llevado la terapia génica AAV a la vanguardia de la investigación clínica3,4. En 2012, la Agencia Europea de Medicamentos (EMA) aprobó Glybera, un vector AAV1 que expresa lipoproteína lipasa (LPL) para el tratamiento de la deficiencia de LPL, lo que la convierte en la primera autorización de comercialización para un tratamiento de terapia génica en Europa o estados Unidos5. Desde entonces, dos terapias génicas AAV adicionales, Luxturna6 y Zolgensma7,han recibido la aprobación de la FDA, y se espera que el mercado se expanda rápidamente en los próximos 5 años con hasta 10-20 terapias génicas esperadas para 20258. Los datos clínicos disponibles indican que la terapia génica AAV es una modalidad segura, bien tolerada y eficaz, lo que la convierte en uno de los vectores virales más prometedores, con más de 244 ensayos clínicos con AAV registrados con ClinicalTrials.gov. El creciente interés en las aplicaciones clínicas que involucran vectores AAV requiere métodos de producción robustos y escalables para facilitar la evaluación de las terapias AAV en modelos animales grandes, ya que este es un paso crítico en la tubería traslacional9.
Para la producción de vectores AAV, los dos requisitos principales son el genoma AAV y la cápside. El genoma de tipo salvaje (wt)-AAV es ADN monocatenario que tiene aproximadamente 4,7 kb de longitud10. El genoma wt-AAV comprende repeticiones terminales invertidas (ITR) que se encuentran en ambos extremos del genoma, que son importantes para el empaquetado, y los genes rep y cap 11. Los genes rep y cap, necesarios para la replicación del genoma, el ensamblaje de la cápside viral y la encapsulación del genoma en la cápside viral, se eliminan del genoma viral y se proporcionan en trans para la producción de vectores AAV12. La eliminación de estos genes del genoma viral proporciona espacio para transgenes terapéuticos y todos los elementos reguladores necesarios, incluidos el promotor y la señal de poliA. Los RTI permanecen en el genoma del vector para garantizar la replicación adecuada del genoma y la encapsulación viral13,14. Para mejorar la cinética de la expresión transgénica, los genomas vectoriales AAV pueden diseñarse para ser autocomplementarios, lo que mitiga la necesidad de conversión de ADN de cadena simple a doble cadena durante la replicación del genoma AAV, pero reduce la capacidad de codificación a ~ 2.4 kb15.
Más allá del diseño del genoma AAV, la selección del serotipo de la cápside determina el tropismo tisular y celular del vector AAV in vivo2. Además del tropismo tisular, se ha demostrado que diferentes serotipos de AAV muestran diferentes cinéticas de expresióngénica 16. Por ejemplo, Zincarelli et al.17 clasificaron diferentes serotipos de AAV en serotipos de baja expresión (AAV2, 3, 4, 5), serotipos de expresión moderada (AAV1, 6, 8) y serotipos de alta expresión (AAV7 y 9). También clasificaron los serotipos de AAV en expresión de inicio lento (AAV2, 3, 4, 5) o expresión de inicio rápido (AAV1, 6, 7, 8 y 9). Estos tropismos divergentes y la cinética de expresión génica se deben a variaciones de aminoácidos en las proteínas de la cápside, formaciones de proteínas de la cápside e interacciones con los receptores/co-receptores de la célula huésped18. Algunas cápsides AAV tienen características beneficiosas adicionales, como la capacidad de cruzar la barrera hematoencefálica después de la administración intravascular (AAV9) o residen en células musculares de larga vida para una expresión transgénica duradera (AAV6, 6.2FF, 8 y 9)19,20.
Este artículo tiene como objetivo detallar un método rentable para producir vectores AAV de alta pureza, alto título y grado de investigación para su uso en modelos preclínicos de animales grandes. La producción de AAV utilizando este protocolo se logra utilizando la transfección de doble plásmido en células adherentes de riñón embrionario humano (HEK)293 cultivadas en pilas celulares. Además, el estudio describe un protocolo para la purificación por cromatografía de afinidad de sulfato de heparina, que se puede utilizar para serotipos AAV que contienen dominios de unión a heparina, incluidos AAV2, 3, 6, 6.2FF, 13 y DJ21,22.
Hay varios sistemas de envasado disponibles para la producción de vectores AAV. Entre estos, el uso de un sistema de coinfección de dos plásmidos, en el que los genes Rep y Cap y los genes auxiliares de Ad (E1A, E1B55K, E2A, E4orf6 y ARN VA) están contenidos dentro de un plásmido (pHelper), tiene algunas ventajas prácticas sobre el método común de transfección de tres plásmidos (triple), incluido el costo reducido para la producción de plásmidos23,24 . El plásmido del genoma AAV que contiene el casete de expresión transgénica (pTransgene), debe estar flanqueado por RTI, y no debe exceder ~ 4.7 kb de longitud. El título y la pureza del vector pueden verse afectados por el transgén debido a los posibles efectos citotóxicos durante la transfección. La evaluación de la pureza del vector se describe en este documento. Los vectores producidos utilizando este método, que producen un 1 x 1013 vg / ml para cada uno, se evaluaron en ratones, hámsteres y modelos animales ovinos.
Tabla 1: Composición de las soluciones requeridas. Información necesaria, incluidos porcentajes y volúmenes, de los componentes necesarios para diversas soluciones a lo largo del protocolo. Haga clic aquí para descargar esta tabla.
Protocolo
1. Doble transfección plásmida de células HEK293 en pilas celulares
- Descongele un crio-vial de células HEK293 en un baño de perlas a 37 °C.
NOTA: Precaliente dmEM completo a 37 ° C mientras las celdas se descongelan para garantizar que la temperatura fría no choque las células al enchapar. Asegúrese de que las células tengan un número de paso bajo, idealmente menos de 20, para garantizar un crecimiento óptimo y una eficiencia de transfección. Asegúrese de que las células estén certificadas como libres de micoplasma. - Transfiera el contenido del crio-vial gota a gota en un tubo cónico de 15 ml que contenga 10 ml de DMEM completo precalentado y centrífuga las células a 500 x g durante 5 min.
- Aspire el medio y luego vuelva a suspender las células HEK293 en 20 ml de DMEM completo precalentado. Sembrar las células en una placa de 15 cm e incubar a 37 °C, con un 5% de CO2.
- Divida las células de una placa de 15 cm en tres para sembrarlas en la cámara de cultivo celular.
- Una vez que las células estén 80% confluentes, aspire el medio y lave suavemente la placa con 3 ml de PBS para no interrumpir la monocapa. Luego, aspire PBS y agregue 3 ml de tripsina.
- Incubar durante 2 min a 37 °C hasta que las células se levanten de la placa, y luego neutralizar la tripsina agregando 7 mL de DMEM completo a la placa.
- Recoger todos los medios y células en un tubo de 15 ml y granular las células centrifugando a 500 x g durante 5 min.
- Aspire el sobrenadante del tubo de 15 ml y resuspenda el pellet celular en 3 ml de DMEM completo. Añadir 1 ml a cada placa de 15 cm que contenga 20 ml de DMEM completo; balancear suavemente las placas para distribuir las células de manera uniforme, e incubar a 37 °C, con un 5% de CO2.
- Una vez que las células sean 80% confluentes, repita los pasos 1.4.1 y 1.4.2. Recoja el sobrenadante en tubos cónicos de 50 ml e invierta suavemente el tubo para garantizar que las células sean homogéneas.
- Determine la densidad celular mezclando 10 μL de las muestras de células con 10 μL de azul de tripano y agregando la mezcla a una diapositiva de conteo de células para su análisis en el contador de células.
- Mezclar 1 L de DMEM completo precalentado con la suspensión celular necesaria para sembrar la cámara de cultivo celular (superficie de 6360 cm2) con 1 x 104 celdas/cm2. Vierta la mezcla celular en la cámara de cultivo celular y gire suavemente para distribuir uniformemente las células a través de cada monocapa (Figura 1) e incube a 37 ° C, con 5% de CO2.
- Además de la cámara de cultivo celular, placa una placa de 15 cm con 1 x 104 celdas/cm2 como referencia para la confluencia.
- Después de la incubación de ~ 65-h, verifique la placa de referencia para la confluencia, idealmente ~ 80% -90% de confluente.
NOTA: DMEM completo precalentado para agregar a la cámara de cultivo celular a 37 °C.

Figura 1: Maniobra de la pila de células para la siembra y transfección celular. Para la pila de células de siembra, comience por quitar una de las tapas de ventilación y verter 1 L de DMEM completo precalentado con la cantidad necesaria de células HEK293 (A). Distribuya uniformemente las celdas y los medios apretando ambas tapas de ventilación y lleve todos los medios a la esquina de la pila de celdas con una de las tapas de ventilación y colóquela en esa esquina (B), coloque la pila de celdas en su lado (C) y luego gire la pila de celdas 90 ° (D) para que los puertos de ventilación estén arriba (E). Baje suavemente la pila de celdas a su posición horizontal normal y asegúrese de que todas las cámaras de la pila de celdas estén completamente cubiertas de medios (F). Al transfectar, desenrosque ambas tapas de ventilación y vierta lentamente los medios viejos en un contenedor de desechos estériles para que el flujo uniforme no perturbe la monocapa de las células (G). Haga clic aquí para ver una versión más grande de esta figura.
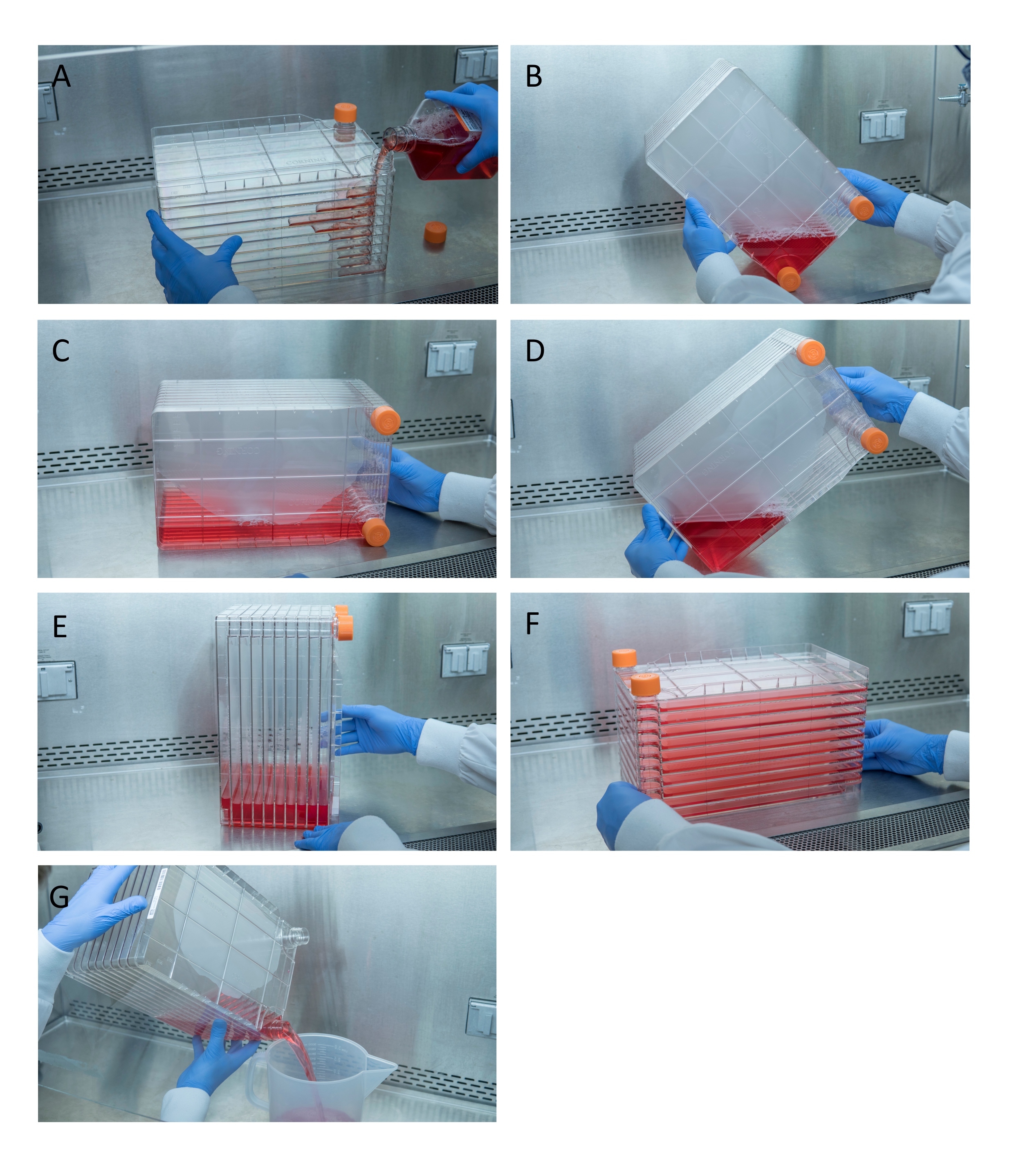{kind=link}
- Preparar la mezcla de polietileneimina (PEI)/ADN a una relación de concentración de 3:1 (p/p).
- Prepare la mezcla de ADN en un tubo cónico de 50 ml agregando 475 μg de pTrangene y 1425 μg de pHelper a 40 ml de medio sérico reducido para crear una proporción de 3: 1 de pHelper: pTrangene.
NOTA: La calculadora de mezcla PEI/DNA se puede encontrar usando la Tabla 2. - Añadir 5,7 ml de PEI (1 g/L) al medio sérico reducido y a la mezcla de ADN a gota. Luego, vórtice brevemente e incube durante 10 minutos a temperatura ambiente.
NOTA: A medida que PEI / DNA se incuba a temperatura ambiente, se volverá ligeramente turbio.
- Prepare la mezcla de ADN en un tubo cónico de 50 ml agregando 475 μg de pTrangene y 1425 μg de pHelper a 40 ml de medio sérico reducido para crear una proporción de 3: 1 de pHelper: pTrangene.
- Después de 8 min de incubación de PEI/ADN, retire los medios de la cámara de cultivo celular.
NOTA: Asegúrese de aflojar ambas tapas naranjas para mantener un flujo suave de medios para evitar el desalojo de las células. - Agregue PEI/DNA a 1 L de DMEM completo precalentado y vierta lentamente la mezcla en el puerto de la cámara de cultivo celular. Distribuya el líquido uniformemente a todas las filas(Figura 1)e incube durante 72 h a 37 °C, con un 5% de CO2.
2 Recolección de AAV y lisis química de las células HEK293 transfectadas
- Agite la cámara de cultivo celular vigorosamente para desalojar las células hasta que el medio aparezca turbio de las células desalojadas y vierta en cuatro tubos de centrífuga de 500 ml.
- Centrifugar los tubos a 18.000 x g durante 30 min a 4 °C para granular las células. Vierta el sobrenadante clarificado en una botella de copoliéster de tereftalato de polietileno (PETG) de 1 L.
NOTA: Si uno no tiene acceso a una centrífuga de alta velocidad, centrífuga a 12.000 x g durante 40 min. Las células peletizadas pueden no ser sólidas a esta velocidad y se deslizarán como sobrenadante de derrame. - Resuspendir los gránulos celulares en tubos centrífugos de 500 ml con 50 ml de tampón de lisis e incubar durante 60 min a 37 °C.
- Centrifugar los tubos a 18.000 x g durante 30 min, y luego transferir el sobrenadante a la misma botella de PETG de 1 L. Deseche los restos celulares granulados.
NOTA: Purificar el sobrenadante clarificado inmediatamente y conservar a 4 °C durante un máximo de 72 h. Para un almacenamiento a más largo plazo, conservar a -80 °C. No conservar a -20 °C.
3 Purificación vectorial AAV mediante cromatografía de afinidad de heparina
- Retire el lisado crudo de -80 °C y deje a 4 °C durante la noche para descongelar. Una vez descongelado, utilice un filtro de 0,22 μM para filtrar el lisado crudo.
- Para pasivar el concentrador centrífugo, agregue 4 ml de tampón de pretratamiento de filtro a un concentrador centrífugo para cada columna de sefalrosa de heparina que se utilice. Pasivar el concentrador centrífugo a temperatura ambiente durante 2-8 h. Configure la pasivación inmediatamente antes de los pasos de purificación.
- Configure el tubo y la bomba (Figura 2).
- Coloque el tubo en una bomba peristáltica y ejecute 20 ml de 1 M de NaOH. A continuación, ejecute 50 ml de agua de grado molecular y luego ejecute 50 ml de DMEM basal.
- Conecte la columna de sefalrosa de heparina de 5 ml al tubo y ejecute 25 ml de DMEM basal para eliminar el conservante.
- Pasar 0,2 μM del lisado crudo filtrado a través de la columna a un caudal de 1-2 gotas/s.

Figura 2: Configuración de la bomba peristáltica para la purificación de AAV. Pase el tubo desde el lisado crudo, a través de la bomba peristáltica, y hacia la columna de la matriz de heparina. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
NOTA: Asegúrese de no introducir burbujas ni permitir que la columna se seque, ya que esto comprometerá la columna y evitará la elución de AAV. Deseche la columna si se seca y use una nueva columna para el resto del lisado crudo.
- Cargue todo el lisado crudo en la columna de heparina y use las siguientes soluciones para lavar la columna.
- Lavar con 50 ml de 1x Hank's Balanced Salt Solutions (HBSS) sin Mg2+ y Ca2+.
- Lavar con 15 ml de N-lauroilsarcosina al 0,5 % en HBSS sin Mg2+ y Ca2+.
- Lavar con 50 ml de HBSS sin Mg2+ y Ca2+.
- Lavar con 50 ml de HBSS con Mg2+ y Ca2+
- Lavar con 50 mL de NaCl/HBSS de 200 mM con Mg2+ y Ca2+.
- Elute 5 x 5 mL (25 mL en total) con 300 mM de NaCl/HBSS con Mg2+ y Ca2+ y etiquete las eluciones como E1-E5 (cada elución es de 5 mL).
- Concentración del virus con un concentrador centrífugo
- Gire el concentrador centrífugo que contiene el tampón de pretratamiento a 900 x g durante 2 min. Descarta el flujo.
- Lavar el filtro concentrador centrífugo con 4 mL de HBSS con Mg2+ y Ca2+ y centrifugar a 1000 x g durante 2 min; descarte el flujo a través.
- Añadir la elución E2 al concentrador centrífugo. Girar a 1000 x g durante 5 min y desechar el flujo.
- Termine de agregar E2 y luego agregue E3 al concentrador centrífugo y gire a 1000 x g durante 5 minutos hasta que el virus concentrado sea de aproximadamente 1 ml.
NOTA: Evite centrifugar el vector de tal manera que el volumen esté por debajo del nivel del filtro. No concentre E1, E4 o E5 en el concentrador centrífugo, ya que contienen muy poco o ningún vector y contienen contaminantes. - Retire el virus concentrado del concentrador centrífugo con una punta filtrada p200 y colóquelo en un tubo de centrífuga estéril de 1,5 ml.
- Enjuague el concentrador centrífugo con 200 μL de HBSS con Mg2+ y Ca2+ para desalojar cualquier AAV restante del filtro. Pipete hacia arriba y hacia abajo vigorosamente varias veces (durante ~ 30 s) para desalojar cualquier virus adherido a la membrana y colóquelo en el tubo centrífugo de 1.5 ml con el resto del virus. Mezclar bien el tubo.
- Alícuota de 5 μL para extracciones de ADN y almacene el vector purificado a -80 °C.
- Lavar la columna con 25 mL de 2 M de NaCl. Además, use 25 ml de Tritón X-100 al 0,1%, precalentado a 37 °C para lavar la columna. A continuación, lave la columna con 50 ml de dH 2 O estéril yluegolave con 25 ml de etanol al 20%.
- Asegúrese de que la membrana de la columna esté completamente saturada en etanol al 20%, ya que esta es la solución de almacenamiento. Selle la columna con tapones provistos y guárdela a 4 °C.
- Guarde el tubo en 1 M NaOH.
NOTA: Si se limpian correctamente, las columnas de sefalrosa de heparina se pueden reutilizar hasta cinco veces.
4 Extracción de ADN genómico AAV
- Prepare la mezcla de reacción mencionada en la Tabla 3 en un tubo de PCR para el tratamiento con DNasa.
| Componente | Volumen |
| Vector AAV purificado | 5 μL |
| Búfer DNasa 10x | 2 μL |
| DNasa | 1 μL |
| ddH2O | 12 μL |
| Volumen final | 20 μL |
Tabla 3: Fórmula de mezcla maestra de tratamiento con DNasa. Componentes y volúmenes recomendados necesarios para el tratamiento con DNasa de vectores virales AAV durante la extracción de ADN.
- Vórtice el tubo de PCR para mezclar y pulsar el tubo de PCR para girar hacia abajo el contenido.
- Usando un termociclador, incubar a 37 °C durante 20 min seguido de 75 °C durante 15 min para inactivar el calor de la DNasa.
- Añadir 5 μL de proteinasa K.
- Usando un termociclador, incubar a 50 °C durante 60 min y luego a 95 °C durante 30 min para inactivar al calor la Proteinasa K.
- Use un kit de limpieza de ADN para eliminar posibles contaminantes.
NOTA: Este paso se realizó utilizando un kit de limpieza de sangre y tejidos disponible comercialmente(Tabla de materiales).- Añadir 200 μL de tampón AL (Kit de limpieza de sangre y tejidos, Tabla de materiales)al tubo de PCR que contiene el vector tratado con DNasa/Proteinasa K.
- Vórtice el tubo de PCR e incube a 56 °C durante 10 min en un termociclador.
- Pipetear el líquido del tubo de PCR en una columna de centrifugado estéril que se encuentra en un tubo de recolección.
- Añadir 200 μL de etanol 100% a la columna y mezclar bien por vórtice.
- Centrifugar a 6.000 x g durante 1 min y desechar el flujo.
- Agregue 500 μL de tampón AW1 (Kit de limpieza de sangre y tejidos, Tabla de materiales)a la columna de centrifugado.
- Centrifugar a 6.000 x g durante 1 min y desechar el flujo.
- Agregue 500 μL de tampón AW2 (Kit de limpieza de sangre y tejidos, Tabla de materiales)a la columna de centrifugado.
- Centrifugar a 15.000 x g durante 3 min y desechar el flujo.
- Coloque la columna de espín en un tubo de centrífuga estéril de 1,5 ml y agregue 200 μL de tampón AE (kit de limpieza de sangre y tejidos, Tabla de materiales)directamente a la membrana de la columna de espín.
- Incubar a temperatura ambiente durante 1 min.
- Centrifugar a 6.000 x gdurante 1 min para eluir el ADN.
- Almacene el ADN a -20 °C.
5 Valoración de genomas de vectores AAV utilizando la reacción en cadena de la polimerasa cuantitativa y una sonda simian Virus 40 (SV40)
NOTA: Realice todo el trabajo de qPCR en una campana de PCR utilizando puntas de pipeta filtradas para evitar la contaminación externa del ADN. Si el genoma AAV no codifica una secuencia de poliA SV40, utilice una sonda contra el ITR descrito en otra parte25. Asegúrese de que el ADN plásmido seleccionado como estándar contenga la secuencia de poliA SV40.
- Preparación estándar de stock
- Diluir el estándar de ADN plásmido stock (plásmido pTransgene que contiene secuencia SV40 poliA) a una concentración final de 10 μg/μL y almacenar a -20 °C en alícuotas de 6 μL.
- Determine el número de copia presente en el estándar de ADN plásmido utilizando la siguiente calculadora en línea26.
NOTA: Utilice un ADN plásmido utilizado para el estándar producido por un proveedor comercial para garantizar la calidad y la concentración correcta. Preparar una gran cantidad de estándar (por ejemplo, 10 ml) para realizar estudios puente cuando se realice la transición a un estándar recién preparado.
- Prepare la siguiente mezcla de reactivos mencionada en la Tabla 4 tanto para las muestras como para el estándar en un tubo centrífugo de 1,5 ml.
NOTA: Prepare un excedente suficiente de mezcla maestra. Consulte la Tabla 5 para ver las secuencias de imprimación/sonda.
| Componente | Volumen |
| Mezcla maestra universal de qPCR (2X) | 10 μL |
| Agua de grado molecular | 4,5 μL |
| Imprimación/sonda de poliA SV40 40x | 0,5 μL |
| Volumen final | 15 μL |
Tabla 4: Mezcla maestra qPCR para la titulación AAV. Componentes y volúmenes recomendados necesarios para la qPCR del ADN extraído de vectores virales AAV.
| Componente | Secuencia |
| Imprimación delantera | 5'-AGCAATAGCATCACAAATTTCACAA-3' |
| Imprimación inversa | 5'-CCAGACATGATAAGATACATTGATGAGTT-3' |
| Sonda | /56-FAM/AGCATTTTT/Zen/TTCACTGCATTCTAGTTGTGGTTTGTC/3IABkFQ |
Tabla 5: Secuencias de imprimación contra la secuencia de ADN poliA SV40. Secuencias de los cebadores y la sonda utilizados para la titulación de qPCR, que se unen a áreas específicas de vectores virales AAV que contienen la secuencia de poliA SV40.
- Pipetear la mezcla maestra hacia arriba y hacia abajo para mezclar.
- Configure la placa de dilución.
- Use una placa transparente de 96 pocillos para preparar diluciones estándar y de muestra, agregue 45 μL de agua de grado molecular a cada pozo en cada otra columna comenzando con la columna 1 (columnas 1, 3, 5, 7, 9 y 11).
- Añadir 5 μL del estándar al pozo A1 y pipeta a mezclar.
- Use una nueva punta de pipeta filtrada para crear una dilución de 1/10 del pozo A1 a B1.
- Continúe una serie de diluciones de 10 veces por la columna hasta llegar a G1.
- No agregue a H1, ya que esto actuará como un control negativo.
- Aplicar la primera muestra (S1) añadiéndola al pozo A3, formando una dilución de 1/10. Pipetear esta mezcla y transferir 5 μLto pozo B3. Deseche la punta de la pipeta después de esta transferencia.
- Con una nueva punta de pipeta, mezcle la solución en el pozo B3 y forme una dilución de 1/100. Transfiera 5 μLof esta mezcla al pozo C3 y deseche la punta después de la transferencia.
- Con una nueva punta de pipeta, pipetee hacia arriba y hacia abajo la solución en el pozo C3 para hacer una dilución de 1/1000. Descarta la propina.
- Continúe diluyendo las muestras sin agregar ninguna muestra a las columnas 2, 4, 6, 8, 10 o 12.
- Una vez que todas las muestras estén diluidas, mezcle el contenido en los pocillos de la columna 1 y luego transfiera 20 μL a la columna 2.
- Repita esto para las columnas 3, 5, 7, 9 y 11 para crear réplicas de cada dilución estándar y muestra. Consulte la Figura 3 para ver el diseño de la placa.
NOTA: Al seguir la configuración de la placa en la Figura 3,las muestras diluidas en las filas G y H solo tendrán diluciones de 1/10 y 1/100.

Figura 3: Diseño de la placa para la titulación qPCR AAV. El azul indica la colocación de la dilución en serie de la norma; verde indica la colocación del control negativo; púrpura indica la colocación de la dilución de las muestras. Cada estándar, negativo o muestra se agrega en replicación. Se ha agregado un ejemplo para la concentración del estándar para mostrar la serie de dilución del estándar, y se han agregado diluciones de muestra a sus respectivos pozos. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
- Valoración por qPCR basada en detección de poliA SV40
- Agregue 15 μL de mezcla maestra de qPCR a cada pocillo de una placa qPCR de 96 pocillos con semifalda blanca.
- Transfiera 5 μL de cada muestra de la placa transparente de 96 pocillos a la placa qPCR de 96 pocillos con semifalda blanca.
- Utilice una pipeta multicanal para garantizar una mezcla adecuada de la mezcla maestra y la muestra de qPCR.
- Selle la placa con una película de sellado y centrífuga la placa qPCR a 1500 x g durante 30 s.
- Ejecute la reacción qPCR en un instrumento de amplificación y detección de PCR en tiempo real basado en placas, utilizando las condiciones sugeridas en la Tabla 6.
| Sección | Ciclos | Hora | Temperatura | Descripción |
| Pre-incubación | 1x | 5 minutos | 95 °C | Desnaturalización del ADN. |
| Amplificación | 38x | 15 s | 95 °C | Amplificación del ADN. Los ajustes se pueden modificar si se utilizan imprimaciones alternativas con diferentes temperaturas de recocido. |
| Años 60 s | 60 °C | |||
| Enfriamiento | 1x | Años 60 s | 40 °C | Enfriamiento de placas. Fin de la carrera. |
Tabla 6: Protocolo de termociclador para la titulación qPCR basada en sonda de hidrólisis. Protocolo de termociclador recomendado para el uso de la titulación qPCR basada en sonda de vectores AAV purificados extraídos de ADN.
NOTA: Para la hoja de trabajo de valoración de QPCR AAV, consulte la Tabla 7.
- Análisis de datos para determinar los números de copia del genoma AAV.
- Rellene las celdas de datos de la hoja de cálculo(Tabla 7A)con los valores de concentración obtenidos de la ejecución de qPCR tanto para diluciones estándar como para muestras.
- Utilice los valores de concentración de la Tabla 7A para producir una curva estándar (Tabla 7B).
NOTA: La curva estándar se mostrará como un logaritmo natural (y = a ln(x) + b) junto con la eficiencia R2. Una curva estándar debe tener una eficiencia cercana al 100 % y R2 cercana a 1,0 (≥0,99). - Complete la eficiencia de la pendiente completando esta calculadora en línea27.
NOTA: Una eficiencia entre el 90% y el 110% es aceptable. Si la eficiencia de qPCR está fuera de este rango, vuelva a ejecutar la qPCR. - Utilice los valores de concentración de la Tabla 7A para promediar las diluciones de cada muestra y determinar la desviación estándar de cada muestra (Tabla 7C).
NOTA: Excluya las diluciones de muestras que estén a más de una desviación estándar del promedio de las diluciones de muestra. - Usando la concentración media de cada dilución, multiplique por el factor de dilución, y luego divida por cinco para obtener los genomas vectoriales (vg)/μL de cada muestra (Tabla 7C).
- Calcular el vg/mL de cada muestra multiplicando la media de las concentraciones de cada muestra por 80.000 (Tabla 7C).
- Promediar el vg/mL de cada dilución para producir el vg/mL final de cada muestra (Tabla 7C).
NOTA: El usuario debe dividir la concentración media de cada dilución por un factor cinco para tener en cuenta los 5 μL cargados en cada pozo para que la qPCR produzca la concentración en vg/μL. El factor de 80.000 explica la transición del valor medio de concentración de cada muestra a vg/mL. En primer lugar, la media del valor de concentración de cada muestra debe multiplicarse por 2 para tener en cuenta los genomas monocatenarios, ya que el conjunto de sondas cebadoras solo cuantifica el ADN monocatenario de sentido positivo (ssDNA), y el genoma AAV existe en una proporción aproximada de 1: 1 entre ssDNA positivo y de sentido negativo25,28. La media del valor de concentración de cada muestra se multiplicará x40 para tener en cuenta la dilución de la muestra de 5 μL de vector purificado (sección 4.1) a 200 μL de ADN extraído (sección 4.6.12). Por último, la media del valor de concentración de cada muestra debe multiplicarse x1000 para convertir de vg/μL a vg/mL.
6 Evaluación de la calidad y pureza del vector
- Control de calidad - Western Blot
- Prepara un gel SDS PAGE al 12%.
- Realizar electroforesis en gel de poliacrilamida.
NOTA: Cargue 6 x 1010 vg de muestras por pozo. - Transfiera las proteínas a la membrana de difluoruro de polivinilideno (PVDF).
- Bloqueo de la membrana de PVDF
- Retire la membrana del aparato de transferencia y enjuague en PBST al 0,1% para eliminar la acrilamida suelta.
- Coloque la membrana en solución de bloqueo durante al menos 1 h a temperatura ambiente o durante la noche a 4 °C.
NOTA: El tampón de bloqueo se puede complementar aún más con suero de cabra al 2%.
- Incubación con anticuerpo primario
- Decantar el tampón de bloqueo y añadir el anticuerpo primario, un anticuerpo monoclonal de ratón anti-AAV a una dilución de 1:200.
- Incubar durante la noche a 4 °C.
- Decantar el anticuerpo primario y lavar cinco veces con PBST al 0,1% durante 5 min a temperatura ambiente con agitación.
- Incubación con anticuerpo secundario
- Decantar la solución de lavado y añadir el anticuerpo secundario conjugado HRP, diluido a un 1:7500 en tampón de bloqueo, e incubar durante 1 h a temperatura ambiente con agitación.
- Decantar el anticuerpo secundario y lavar cinco veces con PBST al 0,1% durante 5 min a temperatura ambiente con agitación.
- Realizar un lavado final con PBS a temperatura ambiente con agitación.
- Detecte las proteínas utilizando sustrato quimioluminiscente mejorado (ECL).
- Imagen del gel para visualizar las proteínas virales (subunidades VP1, VP2 y VP3) (Figura 4).

Figura 4:Western blot que muestra las proteínas de la cápside AAV. Carril A; Escalera MW, Carril B; AAV6.2FF-hIgG01, carril C; AAV6.2FF-hIgG02, carril D; AAV6.2FF-hIgG03 y Lane E; AAV6.2FF-hIgG04. 6 x 1010 vg de cada AAV6.2FF-hIgG se cargó en sus respectivos carriles. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
- Control de pureza - SDS PAGE y Tinción de Coomassie
- Prepare el gel y las muestras de SDS-PAGE como se describe en los pasos 6.1.1 y 6.1.2.
- Fije el gel en solución de fijación durante 1 h o durante la noche con una agitación suave. Cambie la solución de fijación una vez durante la primera hora.
- Manche el gel en solución de tinción durante 2-4 h con agitación suave.
- Desastinúe el gel con una solución de descontención. Reponga la solución de retención varias veces hasta que el fondo del gel esté completamente deformado (4-24 h).
- Guarde el gel inmovilizado en una solución de almacenamiento.
- Imagine el gel para visualizar todas las proteínas teñidas por la solución de tinción de Coomassie.

Figura 5: Gel teñido de coomassie. Carril A; Escalera MW, Carril B; AAV6.2FF-hIgG01, carril C; AAV6.2FF-hIgG02, carril D; AAV6.2FF-hIgG03, carril E; AAV6.2FF-hIgG04, carril F; AAV6.2FF-hIgG05 y Lane G; AAV6.2FF-hIgG06. 6 x 1010 vg de cada AAV6.2FF-hIgG se cargó en sus respectivos carriles. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
- Ensayo alternativo de control de la pureza - HEK293 detección de proteínas de células huésped ELISA
- Realice la detección de proteínas de células huésped HEK293 a través de ELISA según las instrucciones del fabricante.
NOTA: Utilice diluciones de 5 x 10-2 y 1 x 10-3 para muestras de rAAV purificadas. Una vez que TMB se agrega al pozo, incubar lejos de la luz. La regresión lineal no se puede utilizar para analizar los resultados. - Realice un análisis punto a punto, spline cúbico o método de ajuste logístico de cuatro parámetros para interpolar concentraciones de incógnitas y multiplicar por el factor de dilución para determinar la concentración original de la muestra.
- Realice la detección de proteínas de células huésped HEK293 a través de ELISA según las instrucciones del fabricante.
Resultados
La traducción de modelos de roedores pequeños a modelos animales más grandes y la eventual aplicación clínica presenta un desafío significativo debido a la gran cantidad de AAV requerida para transducir animales más grandes y lograr efectos terapéuticos. Para comparar la eficiencia de transducción de la cápside AAV6.2FF diseñada racionalmente, previamente se demostró un aumento de 101 veces en la eficiencia de transducción en células musculares murinas en comparación con AAV63,a rat...
Discusión
La producción de vectores AAV recombinantes (rAAV) descritos en este documento utiliza materiales, reactivos y equipos comunes que se encuentran en la mayoría de los laboratorios e instalaciones de investigación de biología molecular. Este papel permite que el lector produzca rAAV de alta calidad in vitro e in vivo. Sobre todo, este protocolo para la producción de rAAV, en comparación con los protocolos más tediosos que implican la purificación de cloruro de cesio, es eficiente y evita el uso de...
Divulgaciones
Sarah K. Wootton es inventora de una patente estadounidense US10806802B2 para la cápside AAV6.2FF.
Agradecimientos
Amira D. Rghei, Brenna A. Y. Stevens, Sylvia P. Thomas y Jacob G. E. Yates recibieron estipendios para estudiantes de la Facultad de Veterinaria de Ontario, así como becas para graduados de Ontario. Amira D. Rghei recibió un Mitacs Accelerate Studentship. Este trabajo fue financiado por la Subvención del Proyecto de los Institutos Canadienses para la Investigación en Salud (CIHR) (# 66009) y una subvención de Proyectos de Investigación en Salud Colaborativa (NSERC asociada) (# 433339) a SKW.
Materiales
| Name | Company | Catalog Number | Comments |
| 0.22 μm filter | Millipore Sigma | S2GPU05RE | |
| 0.25% Trypsin | Fisher Scientific | SM2001C | |
| 1-Butanol | Thermo Fisher Scientific | A399-4 | CAUTION. Use under a laminar flow hood. Wear gloves |
| 10 chamber cellstack | Corning | 3271 | |
| 1L PETG bottle | Thermo Fisher Scientific | 2019-1000 | |
| 30% Acrylamide/Bis Solution | Bio-Rad | 1610158 | |
| 96-well skirted plate | FroggaBio | FS-96 | |
| Adhesive plate seals | Thermo Fisher Scientific | 08-408-240 | |
| Ammonium persulfate (APS) | Bio-Rad | 161-0700 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Blood and Tissue Clean up Kit | Qiagen | 69506 | Use for DNA clean up in section 4.6 of protocol |
| Bromophenol blue | Fisher Scientific | B392-5 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Cell Culture Dishes | Greiner bio-one | 7000232 | 15 cm plates |
| Culture Conical Tube | Thermo Fisher Scientific | 339650 | 15 mL conical tube |
| Culture Conical Tube | Fisher Scientific | 14955240 | 50 mL conical tube |
| Dulbecco's Modified Eagle Medium (DMEM) with 1000 mg/L D-glucose, L-glutamine | Cytiva Life Sciences | SH30022.01 | |
| ECL Western Blotting Substrate | Thermo Fisher Scientific | 32209 | |
| Ethanol | Greenfield | P016EA95 | Dilute ethyl alcohol(95% vol) to 20% for section 3.7.4 and 70% for section 6.1.1.1 |
| Fetal Bovine Serum (FBS) | Thermo Fisher Scientific | SH30396.03 | |
| Glacial acetic acid | Fisher Scientific | A38-500 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Glycerol | Fisher Scientific | BP229-1 | |
| Glycine | Fisher Scientific | BP381-500 | |
| HBSS with Mg2+ and Ca2+ | Thermo Fisher Scientific | SH302268.02 | |
| HBSS without Mg2+ and Ca2+ | Thermo Fisher Scientific | SH30588.02 | |
| HEK293 cells | American Tissue Culture Collection | CRL-1573 | Upon receipt, thaw the cells and culture as described in manufacturer’s protocol. Once cells have been minimally passaged and are growing well, freeze a subfraction for future in aliquots and store in liquid nitrogen. Always use cells below passage number 30. Once cultured cells have been passaged more than 30 times, it is recommended to restart a culture from the stored aliquots |
| HEK293 host cell protein ELISA kit | Cygnus Technologies | F650S | Follow manufacturer’s instructions |
| Heparin sulfate column | Cytiva Life Sciences | 17040703 | |
| Kimwipe | Thermo Fisher Scientific | KC34120 | |
| L-glutamine (200 mM) | Thermo Fisher Scientific | SH30034.02 | |
| Large Volume Centrifuge Tube Support Cushion | Corning | CLS431124 | Support cushion must be used with large volume centrifuge tubes uless the centrifuge rotor has the approriate V-bottom cushions |
| Large Volume Centrifuge Tubes | Corning | CLS431123-6EA | 500 mL centrifuge tubes |
| MgCl2 | Thermo Fisher Scientific | 7791-18-6 | |
| Microcentrifuge tube | Fisher Scientific | 05-408-129 | 1.5 mL microcentrifuge tube, sterilize prior to use |
| Molecular Grade Water | Cytiva Life Sciences | SH30538.03 | |
| N-Lauroylsarcosine sodium salt | Sigma Aldrich | L5125 | CAUTION. Wear gloves |
| NaCl | Thermo Fisher Scientific | BP35810 | |
| Optimem, reduced serum medium | Thermo Fisher Scientific | 31985070 | |
| Pasteur pipets | Fisher Scientific | 13-678-20D | Sterilize prior to use |
| PBS (10x) | Thermo Fisher Scientific | 70011044 | Dilute to 1x for use on cells |
| Penicillin-Streptomycin Solution | Cytiva Life Sciences | SV30010 | |
| pHelper plasmid | De novo design or obtained from plasmid repository | NA | |
| Pipet basin | Thermo Fisher Scientific | 13-681-502 | Purchase sterile pipet basins |
| Polyethylene glycol tert-octylphenyl ether (Triton X-100) | Thermo Fisher Scientific | 9002-93-1 | CAUTION. Wear gloves |
| Polyethylenimine (PEI) | Polyscience | 24765-1 | Follow manufacturer’s instructions to produce a 1L solution. 0.22μm filter and store at 4°C |
| Polypropylene semi-skirted PCR Plate | FroggaBio | WS-96 | |
| Polysorbate 20 (Tween 20) | Thermo Fisher Scientific | BP337-100 | CAUTION. Wear gloves |
| polyvinylidene difluoride (PVDF) membrane | Cytiva Life Sciences | 10600023 | Use forceps to manipulate. Wear gloves. |
| Primary antibody | Progen | 65158 | |
| Protein Ladder | FroggaBio | PM008-0500 | |
| Proteinase K | Thermo Fisher Scientific | AM2546 | |
| pTrangene plasmid | De novo design or obtained from plasmid repository | NA | Must contain SV40 polyA in genome to be compatible with AAV titration in section 5.0 |
| Pump tubing | Cole-Parmer | RK-96440-14 | Optimize length of tubing and containment of virus in fractions E1-E5 |
| RQ1 Dnase 10 Reaction Buffer | Promega | M6101 | Use at 10x concentration in protocol from section 4.0 |
| RQ1 Rnase-free Dnase | Promega | M6101 | |
| Sample dilutent | Cygnus Technologies | I700 | Must be purchased separately for use with HEK293 host cell protein ELISA kit |
| Secondary antibody, HRP | Thermo Fisher Scientific | G-21040 | |
| Skim milk powder | Oxoid | LP0033B | |
| Sodium dodecyl sulfate (SDS) | Thermo Fisher Scientific | 28312 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Sodium hydroxide (NaOH) | Thermo Fisher Scientific | SS266-4 | |
| SV40 polyA primer probe | IDT | Use sequence in Table X for quote from IDT for synthesis | |
| Tetramethylethylenediamine (TEMED) | Thermo Fisher Scientific | 15524010 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Tris Base | Fisher Scientific | BP152-5 | |
| Trypan blue | Bio-Rad | 1450021 | |
| Ultra-Filter | Millipore Sigma | UFC810024 | Ultra-4 Centrifugal 10K device must be used, as it has a 10000 molecular weight cutoff |
| Universal Nuclease for cell lysis | Thermo Fisher Scientific | 88702 | |
| Universal qPCR master mix | NEB | M3003L | |
| Whatman Paper | Millipore Sigma | WHA1001325 | |
| β-mercaptoethanol | Fisher Scientific | 21985023 | CAUTION. Use under a laminar flow hood. Wear gloves |
| CAUTION: Refer to the Materials Table for guidelines on the use of dangerous chemicals. |
Referencias
- Hastie, E., Samulski, R. J. Adeno-associated virus at 50: A golden anniversary of discovery, research, and gene therapy success-a personal perspective. Human Gene Therapy. 26 (5), 257-265 (2015).
- Wang, D., Tai, P. W. L., Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nature Reviews Drug Discovery. 18 (5), 358-378 (2019).
- Nathwani, A. C., et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. The New England Journal of Medicine. 371 (21), 1994-2004 (2014).
- Kuzmin, D. A., et al. The clinical landscape for AAV gene therapies. Nature Reviews Drug Discovery. 20 (3), 173-174 (2021).
- Ylä-Herttuala, S. Endgame: glybera finally recommended for approval as the first gene therapy drug in the European union. Molecular Therapy: The Journal of the American Society of Gene Therapy. 20 (10), 1831-1832 (2012).
- FDA approves novel gene therapy to treat patients with a rare form of inherited vision loss. FDA News Release. FDA Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-novel-gene-therapy-treat-patients-rare-form-inherited-vision-loss (2020)
- FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. FDA News Release. FDA Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease (2020)
- Statement from FDA Commissioner Scott Gottlieb, MD and Peter Marks, MD Ph.D., Director of the Center for Biologics Evaluation and Research on new policies to advance development of safe and effective cell and gene therapies. FDA Available from: https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-and-peter-marks-md-phd-director-center-biologics (2020)
- Asokan, A., Schaffer, D. V., Samulski, R. J. The AAV vector toolkit: poised at the clinical crossroads. Molecular Therapy: The Journal of the American Society of Gene Therapy. 20 (4), 699-708 (2012).
- Rose, J. A., Hoggan, M. D., Shatkin, A. J. Nucleic acid from an adeno-associated virus: chemical and physical studies. Proceedings of the National Academy of Sciences of the United States of America. 56 (1), 86-92 (1966).
- Lusby, E., Fife, K. H., Berns, K. I. Nucleotide sequence of the inverted terminal repetition in adeno-associated virus DNA. Journal of Virology. 34 (2), 402-409 (1980).
- Masat, E., Pavani, G., Mingozzi, F. Humoral immunity to AAV vectors in gene therapy: challenges and potential solutions. Discovery Medicine. 15 (85), 379-389 (2013).
- Ling, C. Enhanced Transgene Expression from Recombinant Single-Stranded D-Sequence-Substituted Adeno-Associated Virus Vectors in Human Cell Lines In Vitro and in Murine Hepatocytes In Vivo. Journal of Virology. 89 (2), 952-961 (2014).
- Cathomen, T., Stracker, T. H., Gilbert, L. B., Weitzman, M. D. A genetic screen identifies a cellular regulator of adeno-associated virus. Proceedings of the National Academy of Sciences of the United States of America. 98 (26), 14991-14996 (2001).
- McCarty, D. M. Self-complementary AAV vectors; advances and applications. Molecular Therapy. 16 (10), 1648-1656 (2008).
- Aschauer, D. F., Kreuz, S., Rumpel, S. Analysis of transduction efficiency, tropism and axonal transport of aav serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLOS One. 8 (9), 76310 (2013).
- Zincarelli, C., Soltys, S., Rengo, G., Rabinowitz, J. E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Molecular Therapy. 16 (6), 1073-1080 (2008).
- Pillay, S., et al. Adeno-associated virus (AAV) serotypes have distinctive interactions with domains of the cellular AAV receptor. Journal of Virology. 91 (18), (2017).
- Merkel, S. F. Trafficking of adeno-associated virus vectors across a model of the blood-brain barrier; a comparative study of transcytosis and transduction using primary human brain endothelial cells. Journal of Neurochemistry. 140 (2), 216-230 (2017).
- van Lieshout, L. P., et al. A novel triple-mutant AAV6 capsid induces rapid and potent transgene expression in the muscle and respiratory tract of mice. Molecular Therapy. Methods & Clinical Development. 9, 323-329 (2018).
- Wu, Z., Asokan, A., Grieger, J. C., Govindasamy, L., Agbandje-McKenna, M., Samulski, R. J. single amino acid changes can influence titer, heparin binding, and tissue tropism in different adeno-associated virus serotypes. Journal of Virology. 80 (22), 11393-11397 (2006).
- Liu, J., Moon, Y. -. A. Simple purification of adeno-associated virus-DJ for liver-specific gene expression. Yonsei Medical Journal. 57 (3), 790-794 (2016).
- Grimm, D., Kern, A., Rittner, K., Kleinschmidt, J. A. Novel tools for production and purification of recombinant adeno-associated virus vectors. Human Gene Therapy. 9 (18), 2745-2760 (1998).
- Kimura, T., et al. Production of adeno-associated virus vectors for in vitro and in vivo applications. Scientific Reports. 9 (1), 13601 (2019).
- Aurnhammer, C., et al. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Human Gene Therapy Methods. 23 (1), 18-28 (2012).
- . Paramyxoviridae: The viruses and their replication. Fields Virology Available from: https://www.scholars.northwestern.edu/en/publications/paramyxoviridae-the-viruses-and-their-replication (1996)
- Boussif, O., et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proceedings of the National Academy of Sciences of the United States of America. 92 (16), 7297-7301 (1995).
- Kaludov, N., Brown, K. E., Walters, R. W., Zabner, J., Chiorini, J. A. Adeno-associated virus serotype 4 (AAV4) and AAV5 Both require sialic acid binding for hemagglutination and efficient transduction but differ in sialic acid linkage specificity. Journal of Virology. 75 (15), 6884-6893 (2001).
- Burnham, B., et al. Analytical ultracentrifugation as an approach to characterize recombinant adeno-associated viral vectors. Human Gene Therapy Methods. 26 (6), 228-242 (2015).
- Dobnik, D., et al. Accurate quantification and characterization of adeno-associated viral vectors. Frontiers in Microbiology. 10, 1570 (2019).
- Backovic, A., et al. Capsid protein expression and adeno-associated virus like particles assembly in Saccharomyces cerevisiae. Microbial Cell Factories. 11, 124 (2012).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados