
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Ablaciones de volumen profundas y espacialmente controladas utilizando un microscopio de dos fotones en la gastrula del pez cebra
En este artículo
Resumen
El desarrollo embrionario requiere una coordinación a gran escala del movimiento celular. La ablación láser mediada por excitación de dos fotones permite la ablación tridimensional controlada espacialmente de grandes grupos de células profundas. Además, esta técnica puede sondear la reacción de las células que migran colectivamente in vivo a las perturbaciones en su entorno mecánico.
Resumen
La morfogénesis implica muchos movimientos celulares para organizar las células en tejidos y órganos. Para un desarrollo adecuado, todos estos movimientos deben estar estrechamente coordinados, y la evidencia acumulada sugiere que esto se logra, al menos en parte, a través de interacciones mecánicas. Probar esto en el embrión requiere perturbaciones físicas directas. Las ablaciones con láser son una opción cada vez más utilizada que permite aliviar las restricciones mecánicas o aislar físicamente dos poblaciones celulares entre sí. Sin embargo, muchas ablaciones se realizan con un láser ultravioleta (UV), que ofrece una resolución axial limitada y penetración en el tejido. Aquí se describe un método para extirpar volúmenes profundos, significativos y espacialmente bien definidos utilizando un microscopio de dos fotones. Las ablaciones se demuestran en una línea de pez cebra transgénica que expresa la proteína verde fluorescente en el mesendodermo axial y se utiliza para cortar el mesendodermo axial sin afectar el ectodermo suprayacente o la célula de la yema subyacente. El comportamiento celular se monitorea mediante imágenes en vivo antes y después de la ablación. El protocolo de ablación se puede utilizar en diferentes etapas de desarrollo, en cualquier tipo de célula o tejido, a escalas que van desde unas pocas micras hasta más de cien micras.
Introducción
Las interacciones célula-célula juegan un papel vital en el desarrollo. Las células proporcionan señales que sus vecinas directas, o células más lejanas, pueden percibir, influyendo así en su destino y / o comportamiento. Muchas de estas señales son de naturaleza química. Por ejemplo, en los eventos de inducción bien caracterizados, un grupo celular produce moléculas difusibles que afectan el destino de otra población celular1. Otras señales, sin embargo, son mecánicas; las células ejercen fuerzas y restricciones sobre sus vecinos, que los vecinos perciben y a los que responden2.
Una forma de estudiar la importancia de estas interacciones célula-célula in vivo es eliminar algunas células y observar el desarrollo posterior. Desafortunadamente, las técnicas disponibles para eliminar o destruir células son limitadas. Las células se pueden extraer quirúrgicamente3,4, utilizando agujas o alambres pequeños, pero tales tratamientos son invasivos, no muy precisos y generalmente se realizan bajo un estereomicroscopio, lo que impide la obtención de imágenes inmediatas bajo un microscopio. Además, apuntar a las células profundas implica perforar un agujero en los tejidos suprayacentes, creando perturbaciones no deseadas. Los fotosensibilizadores codificados genéticamente, como KillerRed, se han utilizado para inducir la muerte celular a través de la iluminación con luz5. Los fotosensibilizadores son cromóforos que generan especies reactivas de oxígeno tras la irradiación de la luz. Su principal limitación es que requieren iluminaciones de luz largas (alrededor de 15 min), lo que puede ser difícil de lograr si las células se mueven, y que inducen la muerte celular a través de la apoptosis, que no es inmediata.
Finalmente, las ablaciones con láser han sido desarrolladas y ampliamente utilizadas en los últimos 15 años6,7,8,9,10,11,12. Un rayo láser se enfoca en la célula / tejido objetivo. Induce su ablación a través del calentamiento, la fotoablación o la ablación inducida por plasma; el proceso involucrado depende de la densidad de potencia y el tiempo de exposición13. La mayoría de los protocolos de ablación utilizan láseres UV por su alta energía. Sin embargo, la luz UV es absorbida y dispersada por los tejidos biológicos. Por lo tanto, apuntar a las células profundas requiere una alta potencia láser, que luego induce daños en tejidos más superficiales y fuera del plano. Esto limita el uso de láseres UV a estructuras superficiales y explica su resolución axial relativamente baja. La óptica no lineal (la llamada microscopía de dos fotones) utiliza propiedades no lineales de la luz para excitar un fluoróforo con dos fotones de aproximadamente media energía en el dominio infrarrojo. Cuando se aplica a las ablaciones, esto tiene tres ventajas principales. En primer lugar, la luz infrarroja está menos dispersa y menos absorbida que la luz UV por los tejidos biológicos14, lo que permite llegar a estructuras más profundas sin aumentar la potencia láser requerida. En segundo lugar, el uso de un láser pulsado de femtosegundo proporciona densidades de potencia muy altas, creando una ablación a través de la inducción plasmática, que, contrariamente al calentamiento, no se difunde espacialmente15. En tercer lugar, la densidad de potencia que induce la formación de plasma se alcanza solo en el punto focal. Gracias a estas propiedades, las ablaciones con láser de dos fotones se pueden utilizar para apuntar con precisión a las células profundas sin afectar el entorno del tejido circundante.
Las migraciones colectivas son un excelente ejemplo de procesos de desarrollo en los que las interacciones célula-célula son fundamentales. Las migraciones colectivas se definen como migraciones celulares en las que las células vecinas influyen en el comportamiento de una célula16. La naturaleza de estas interacciones (químicas o mecánicas) y cómo afectan la migración celular puede variar mucho y, a menudo, no se entiende por completo. La capacidad de eliminar células y observar cómo esto afecta a las demás es fundamental para desentrañar aún más estos procesos colectivos. Hace unos años, establecimos, utilizando enfoques quirúrgicos, que la migración del polster durante la gastrulación del pez cebra es una migración colectiva17. El polster es un grupo de células que constituye las primeras células interiorizantes en el lado dorsal del embrión18. Estas células, marcadas en verde en la línea transgénica Tg(gsc:GFP), se encuentran en lo profundo del embrión, debajo de varias capas de células epiblásticas. Durante la gastrulación, este grupo lidera la extensión del mesodermo axial, migrando desde el organizador embrionario hasta el polo animal19,20,21,22,23 (Figura 1A). Establecimos que las células requieren contacto con sus vecinas para orientar su migración en la dirección del polo animal. Sin embargo, una mejor comprensión de las bases celulares y moleculares de esta migración colectiva implica la eliminación de algunas células para ver cómo esto influye en las restantes. Por lo tanto, desarrollamos ablaciones de volúmenes grandes y profundos utilizando una configuración de microscopía de dos fotones. Aquí, demostramos el uso de este protocolo para cortar el polster en su medio y observar las consecuencias en la migración celular mediante el seguimiento de núcleos marcados con Histone2B-mCherry.
Protocolo
Todo el trabajo con animales fue aprobado por el Comité ético N 59 y el Ministère de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche bajo el número de expediente APAFIS#15859-2018051710341011v3. Algunos de los pasos que se describen a continuación son específicos de nuestro equipo y software, pero podrían adaptarse fácilmente a diferentes equipos.
1. Preparación de la inyección
- Preparar 75 ml de solución de agarosa al 1% en medio embrionario (EM).
- Coloque el molde de inyección en una placa de Petri de 90 mm y vierta aproximadamente 50 ml de agarosa, suficiente para que el molde flote. Deje que la agarosa se solidifique y retire el molde de inyección.
- Prepare un plato recubierto de agarosa vertiendo 1 ml de agarosa en una placa de Petri de 30 mm.
- Preparar 4 μL de solución de ARNm de Histone2B-mCherry de 30 ng/μL diluyendo la solución madre en agua libre de RNasa y mantenerla en hielo.
NOTA: Tenga cuidado de usar guantes mientras manipula el ARNm para evitar la degradación mediada por la RNasa. - Extraiga una aguja de inyección de un capilar usando el extractor de micropipetas.
2. Preparación embrionaria
- Una vez que los peces hayan puesto huevos, recolecte, enjuague y coseche en una placa de Petri de 90 mm en EM. Coloque los embriones en una incubadora de 28,5 °C.
- Espere 20 minutos para que la primera celda se haga visible.
- Transfiera 30 embriones a la placa de inyección llena de EM. Apriete los embriones en los surcos con fórceps ligeramente romas y orientelos con el polo animal hacia arriba.
- Usando una punta de microcargador, llene una aguja de inyección con 2 μL de solución de ARNm. Inserte la aguja en el soporte capilar colocado en un micromanipulador conectado con tubos de politetrafluoroetileno (PTFE) a un inyector de aire.
- Debajo del estereomicroscopio, rompa cuidadosamente la punta de la aguja.
- Inyecte la solución de ARNm en los embriones en etapa de 1 célula insertando la aguja en la célula.
NOTA: El volumen inyectado es aproximadamente un tercio del volumen celular. - Coloque de nuevo los embriones inyectados en la incubadora de 28,5 °C.
3. Preparación del microscopio de dos fotones
NOTA: En este protocolo se utilizan dos láseres. Uno se utiliza para obtener imágenes de GFP (a 920 nm) y realizar ablaciones (a 820 nm). Se le conocerá como el láser verde / ablación. El otro se utiliza a 1160 nm para obtener imágenes de mCherry. Se le conocerá como el láser rojo.
- Ajuste el láser verde/ablación a 820 nm (longitud de onda de ablación) y el láser rojo a 1160 nm (excitación mCherry).
- Usando espejos móviles en la trayectoria óptica, alinee los rayos láser verdes / ablación y rojos tanto a la entrada como a la salida del cabezal de escaneo.
NOTA: Esto aumenta el enfoque del rayo láser y minimiza el volumen focal para la excitación y la ablación. - Mida la potencia máxima del láser verde/ablación a 820 nm bajo el objetivo. Para hacerlo, coloque el medidor de potencia debajo del objetivo, cierre la cámara negra, ajuste la potencia del láser verde / ablación al 100% y abra las persianas. Calcule el porcentaje de potencia láser necesaria para alcanzar los 300 mW.
- Retroceda el láser verde/ablación a 920 nm (excitación GFP) y ajuste la potencia del láser al 7%. Ajuste la potencia del láser rojo al 15%.
- Activar detectores epi-PhotoMultiplier Tubes (PMT) para líneas verdes y rojas; establezca la sensibilidad PMT de la línea verde y roja en 65.
- Establezca el campo de visión en 400 x 400 μm, la resolución de imagen en 512 x 512 píxeles y la frecuencia de escaneo en 800 Hz.
- Seleccione el modo de imagen de lapso de tiempo 3D . Luego, cree una carpeta y active Guardar automáticamente los datos después de cada adquisición.
- Ensamble la cámara de calentamiento y colóquela a 28 °C. Espere al menos 10 minutos para que la cámara y el objetivo se calienten.
4. Montaje del embrión
- Bajo un estereomicroscopio de fluorescencia, identifique embriones al 70% epiboly que expresen GFP.
NOTA: Seleccione embriones con una señal brillante en el mesodermo axial y sin fluorescencia de fondo para una mejor calidad de imagen. - Transfiera de tres a cuatro embriones seleccionados en el plato recubierto de agarosa (paso 1.3) utilizando una pipeta pasteur de plástico y descorarlos cuidadosamente con fórceps finos.
NOTA: Los embriones descoronados son muy delicados y estallarán al contacto con el aire o el plástico. - Vierta 1 ml de agarosa al 0,2% en 1x penicilina-estreptomicina EM en un vial de vidrio pequeño. Coloque el vial en un calentador de bloque seco precalentado a 42 °C.
NOTA: Los siguientes pasos deben realizarse rápidamente para permitir la orientación del embrión antes de las series de agarosa. - Transfiera un embrión decoronado en el vial de vidrio de agarosa al 0,2% utilizando una pipeta de vidrio pulida al fuego. Tenga cuidado de no agregar demasiado EM en la agarosa para evitar diluirla. Deseche el EM restante de la pipeta y aspire el embrión de nuevo junto con suficiente agarosa para cubrir el portaobjetos del plato de fondo de vidrio antes de que el embrión caiga de la pipeta.
- Sopla la agarosa y el embrión en el portaobjetos de vidrio del plato. Tenga cuidado de no dejar que el embrión toque el aire o el lado de plástico del plato. A continuación, llene la cámara alrededor del portaobjetos de vidrio con agarosa.
- Use una pestaña para orientar el embrión de modo que la región objetivo esté en la parte superior (Figura 1B).
NOTA: Al orientar embriones, tenga cuidado de tocar solo el blastodermo, no la yema muy frágil. La agarosa se establecerá en alrededor de 1 minuto, dependiendo de la temperatura ambiente. - Espere ~ 5 minutos para que la agarosa se cuaje por completo, y luego agregue unas gotas de penicilina-estreptomicina EM.
5. Localización del embrión e imágenes previas a la ablación
- Coloque el plato con fondo de vidrio debajo del objetivo en la cámara calentada. Sumergir el objetivo en penicilina-estreptomicina EM y cerrar la cámara calentada.
- Mueva el control deslizante para establecer la trayectoria de la luz hacia los oculares. Luego, usando oculares, lámparas fluorescentes y control de etapas, encuentre un embrión y fije el enfoque en la superficie del embrión.
- Apague la lámpara de fluorescencia, ajuste la trayectoria de la luz a los PMT y cierre la cámara negra.
NOTA: Tenga cuidado de apagar todas las fuentes de luz en la cámara negra, ya que podría dañar los PMT. - Comience a tomar imágenes en vivo y localice el mesodermo axial. Ajuste las potencias del láser verde/ ablación y rojo para tener una buena señal (es decir, entre 1.000 y 20.000 fotones por píxel para las áreas de expresión GFP). Use el canal rojo para mover el escenario a la parte superior del embrión y establezca esta posición como Z = 0.
- Elija un paso de tiempo de 1 min y un paso Z de 2 μm. Un curso Z de 110 μm es suficiente para abarcar todo el polster y se adquiere en menos de 1 min con estos ajustes. Coloque la primera rebanada 15 μm por encima del mesodermo axial (en el ectodermo más superficial).
NOTA: El polster se mueve a lo largo de una línea curva, de modo que la porción inferior de la pila Z debe ajustarse 30 μm más profunda que la posición más profunda del polster para acomodar su movimiento durante la imagen de lapso de tiempo (Figura 1E). - Graba 10-15 minutos de película previa a la ablación.

Figura 1: Resultado exitoso de las ablaciones con láser. (A) Esquema de un embrión gastrulante al 70% de epibolia en vista dorsal; pAM: mesodermo axial posterior; la flecha negra marca la dirección de la migración de polster; El cuadrado negro indica un campo de visión típico para las ablaciones en el polster. (B) Esquema de montaje de embriones para el corte de polster. Vista lateral. El embrión está montado de tal manera que el plano del polster es perpendicular al eje óptico. (C) supervivencia y (D) morfología de embriones control y ablacionados a las 24 h post-fecundación. La barra de escala es de 300 μm. (E) Secuencia de tiempo de la ablación con láser en el polster de un embrión Tg(gsc:GFP) que expresa Histone2B-mCherry. Las vistas con el canal verde solo son proyecciones máximas. El primer plano muestra el área ablacionada que contiene restos celulares. Las vistas con canales verdes y rojos (mostrados como magenta) son cortes XY y XZ antes y después de la ablación (el rayo verde representa la ablación). Las rodajas de XZ muestran que los tejidos suprayacentes (núcleos magenta sin expresión de GFP) no se han visto afectados por la ablación de las estructuras subyacentes. La caja discontinua amarilla corresponde al ROI seleccionado para el tratamiento de ablación con láser. La barra de escala es de 50 μm en vistas grandes y 25 μm en primer plano. Haga clic aquí para ver una versión más grande de esta figura.
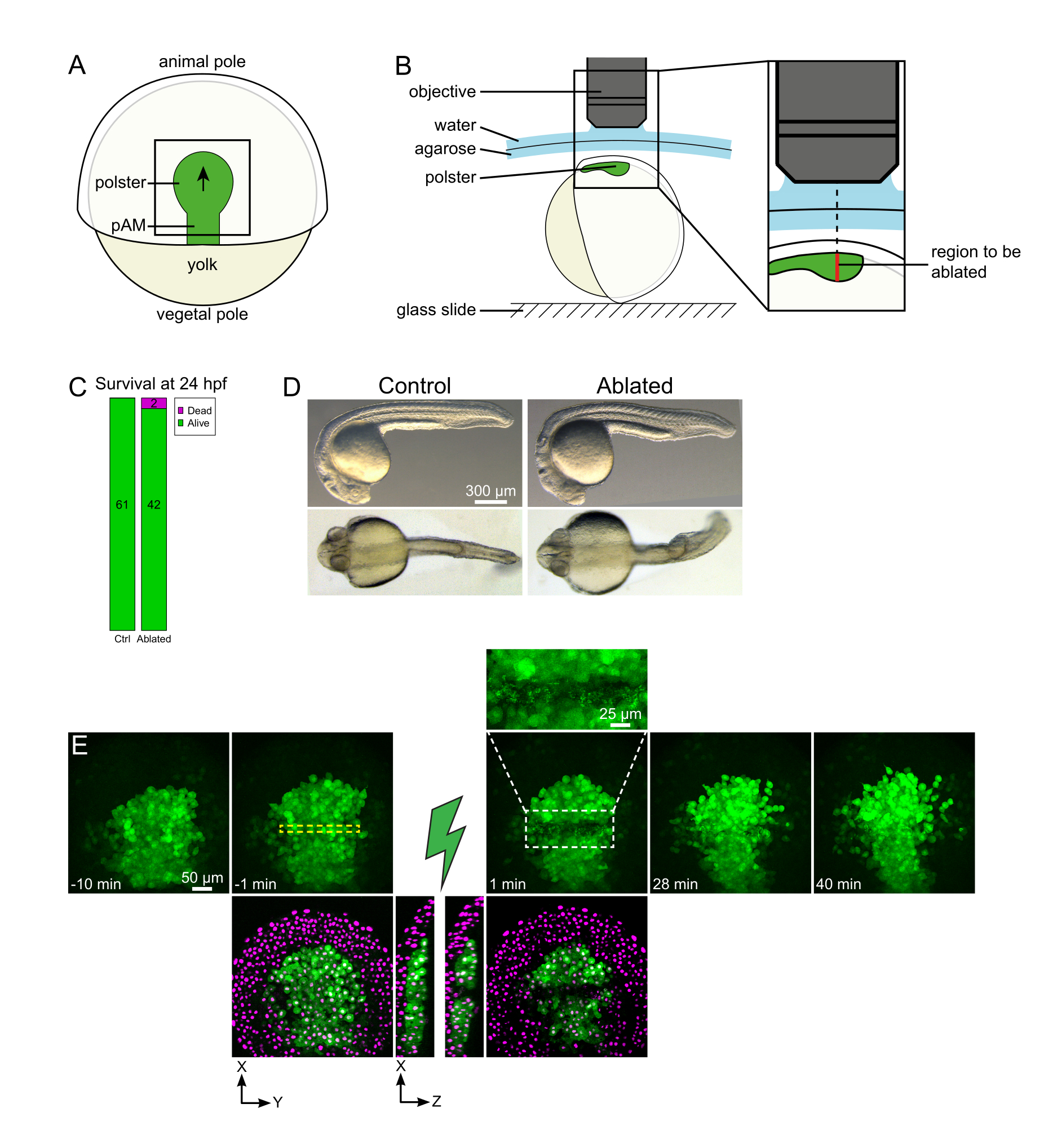{kind=link}
6. Ubicación del objetivo y ablación con láser
- Localice el contorno del polster en imágenes en vivo y, utilizando la herramienta Región de interés del modulador electroóptico (EOM ROI), dibuje un rectángulo grande de 20 píxeles (15 μm) que abarque el ancho del polster. Coloque este rectángulo en el centro del polster (Figura 1E).
- Observe la posición axial de los planos más altos y más bajos que contienen celdas de polster. Las ablaciones se realizarán cada 10 μm entre estos dos planos. Tenga cuidado de que el ROI no se superponga a la celda de yema en ninguno de estos planos.
- Coloque el escenario en la posición Z más baja del intervalo. Las ablaciones deben realizarse de abajo hacia arriba a medida que los escombros absorben la luz.
- Establezca la longitud de onda del láser verde/ablación en 820 nm y establezca el porcentaje de potencia para obtener una potencia de salida de 300 mW (paso 3.3).
- Establezca la frecuencia de imagen en 200 Hz.
- Establezca la MOE de imágenes láser verdes/de ablación en 0 y seleccione el modo ROI-Treat .
- Encienda la MOE y configure el tratamiento para que comience inmediatamente (después de 0 fotogramas).
- Establezca el modo de imagen en Timelapse y desactive el guardado automático.
- Establezca el paso de tiempo en modo rápido.
- Establezca el número de tramas de tratamiento y el número de tramas en el valor correspondiente a la profundidad de destino (Tabla 1).
| Profundidad (μm) | Marcos de tratamiento |
| -30 | 1 |
| -35 | 1-2 |
| -40 | 1-2 |
| -45 | 2 |
| -50 | 2-3 |
| -55 | 3 |
| -60 | 3-4 |
| -65 | 4 |
| -70 | 4 |
| -75 | 4-5 |
| -80 | 4-5 |
| -85 | 5 |
| -90 | 5 |
| -95 | 5-6 |
| -100 | 6 |
| -105 | 6 |
Tabla 1: Número sugerido de marcos de tratamiento con láser en función de la profundidad celular objetivo en el embrión (0 es la superficie del embrión).
- Comience a crear imágenes. La adquisición es negra ya que el obturador de PMT se cierra durante el tratamiento de la MOE.
- Suba el escenario a la siguiente posición Z de la lista (paso 6.2).
- Repita los pasos 6.10 a 6.12 hasta que se alcance la parte superior del polster.
7. Verificación e imágenes posteriores a la ablación
- Ajuste el láser verde/ablación a 920 nm y 5% de potencia. Establezca la MOE de imagen láser verde/ablación en 100 y seleccione el modo de campo completo .
- Establezca la frecuencia de imagen en 800 Hz. Desactive la MOE.
- Revisa toda la pila en modo en vivo para verificar si todos los aviones han sido ablacionados. Si este no es el caso, vuelva al paso 6.2.
NOTA: La ablación a veces induce un desplazamiento vertical de los tejidos vecinos, por lo que la pila Z podría tener que ser redefinida. - Establezca el modo de imagen en Timelapse 3D y vuelva a activar el guardado automático. Graba 40-60 minutos de película post-ablación.
- Verifique, en la película posterior a la ablación, si las células objetivo fueron ablacionadas de manera efectiva. La recuperación de fluorescencia, o células objetivo que ocupan espacio y evitan que las células seguidoras se muevan, indican que las células objetivo solo fueron fotoblanqueadas y no ablacionadas (Figura 1E y Figura 2A).

Figura 2: Resultados negativos de las ablaciones con láser. (A) Ejemplos típicos de posibles fallos en la ablación con láser. Las vistas XY grandes son proyecciones máximas, la vista XZ es una sección reconstruida. El área tratada con láser se encuentra entre las dos puntas de flecha blancas. Tres planos focales se resaltan en la sección reconstruida y se muestran a la derecha. Corresponden a tres tipos diferentes de fallas. El plano 1 muestra que las células por encima del polster han sido ablacionadas. Esto se puede identificar por la presencia de desechos autofluorescentes en este plano focal (ver primer plano) sobre el polster (ver posición del plano 1 en la sección reconstruida). Esto probablemente resulta de una definición incorrecta de la región a ablacionar. El plano 2 muestra células que han sido blanqueadas pero no ablacionadas. Se pueden identificar como la señal de baja fluorescencia que aún revela contornos celulares intactos (ver de cerca). El plano 3 muestra células intactas, que apenas han sido blanqueadas por el tratamiento con láser. Esto podría deberse a una definición incorrecta de la región a ablacionar o a un tratamiento deficiente. En las situaciones representadas en los planos 2 y 3, es posible volver a aplicar el tratamiento de ablación a las células diana no ablacionadas. La barra de escala es de 50 μm en vistas grandes y 20 μm en primeros planos. (B) Un ejemplo típico de burbujas (marcadas por asteriscos blancos) formadas por cavitación debido a un tratamiento con láser demasiado intenso. Tales burbujas no se limitan a un plano Z, a veces incluso abarcando toda la altura del polster, deformando los tejidos vecinos. La barra de escala es de 50 μm. Haga clic aquí para ver una versión más grande de esta figura.
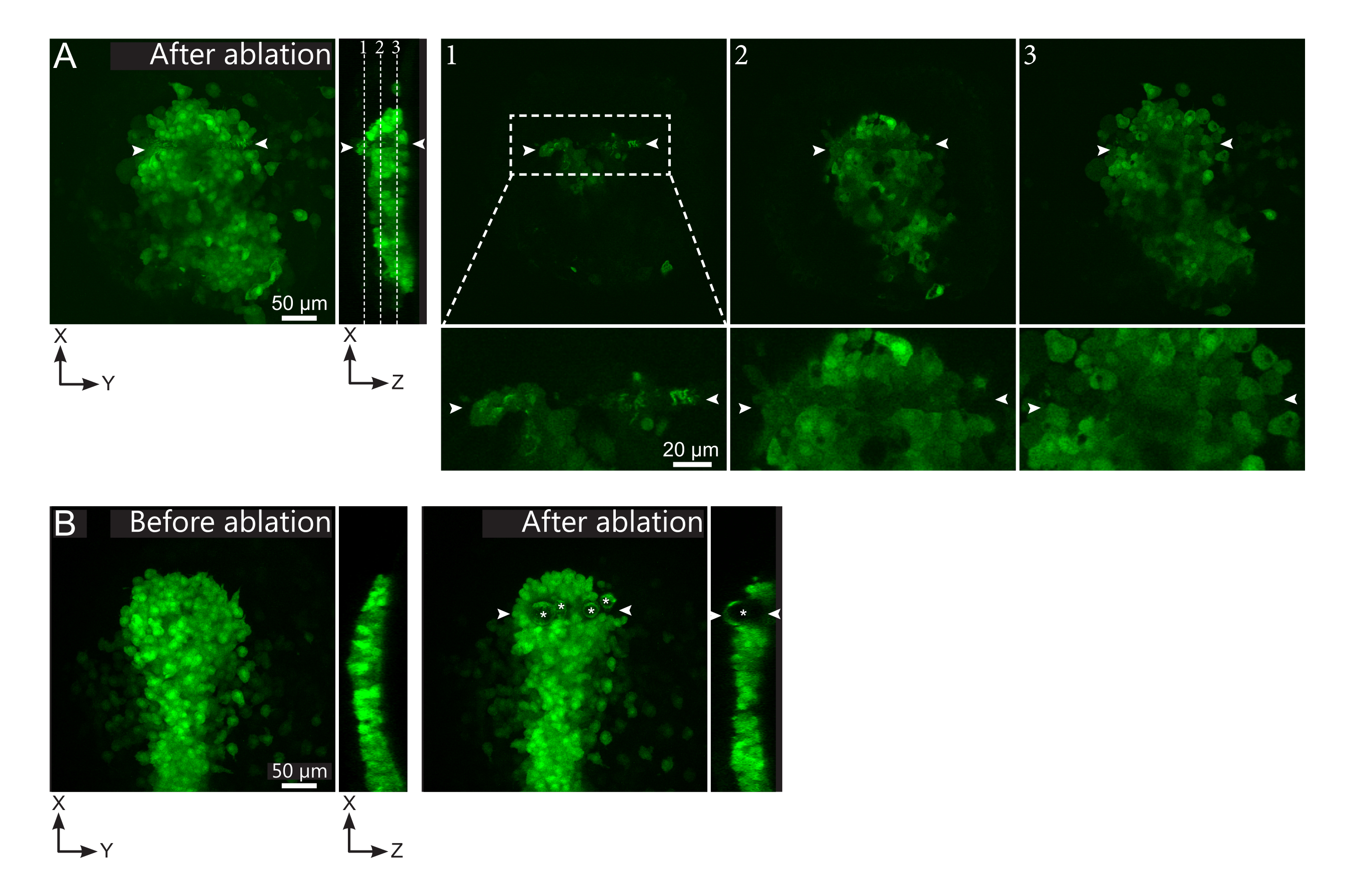{kind=link}
8. Análisis de datos
- Abra series de lapso de tiempo con el software de análisis de imágenes y establezca el tamaño de píxel correcto.
- En la función Spot , establezca el tamaño del objeto en 10 μm, ya que este es el tamaño promedio del núcleo durante la gastrulación. Luego, ejecute la función Spot para detectar y rastrear los núcleos.
NOTA: La detección puede mejorarse ligeramente teniendo en cuenta la resolución axial más baja, ajustando una forma elipsoidal de 12 μm de largo a lo largo del eje Z. - Utilice filtros para eliminar falsos positivos. En la línea Tg(Gsc:GFP), las células del eje embrionario y algunas células endodérmicas están marcadas en verde. Por lo tanto, el filtrado en intensidad verde permite una selección rápida de estas células (Figura 3A).
- Establezca la distancia máxima entre puntos consecutivos en un valor compatible con la velocidad de las celdas.
NOTA: Tenga cuidado de considerar el intervalo de tiempo entre dos fotogramas. Las células de Polster migran a 2,8 ± 0,8 μm/min. Por lo tanto, permitir 4 μm de desplazamiento máximo durante un paso de tiempo de 1 minuto elimina la mayoría de las pistas artefactuales. - Permitir brechas en uno o dos puntos de tiempo proporciona pistas continuas más largas, pero puede introducir errores de seguimiento. Si un núcleo no se detecta correctamente en un punto único, considere la posibilidad de volver a ejecutar la detección de puntos con diferentes parámetros / filtros.
- Compruebe visualmente las pistas y, si es necesario, corríjalas.
- Exporte los resultados como un archivo .xlsx. Procese el archivo utilizando rutinas de hoja de cálculo publicadas24 (Figura 3B) y rutinas personalizadas en software de análisis de datos (disponible a pedido).

Figura 3: El aislamiento de la mitad anterior del polster afecta la direccionalidad celular. (A) Reconstrucciones 3D de un embrión Tg(gsc:GFP) que expresa Histone2B-mCherry (mostrado en magenta), antes y después de una ablación con láser cortando el polster en su centro. Los núcleos pertenecientes a la mitad anterior del polster están marcados con un punto magenta y rastreados a lo largo del tiempo antes y después de la ablación (ver Película S1). La barra de escala es de 50 μm. (B) Como medida de la persistencia de la migración, dirección autocorrelación de las células pertenecientes a la parte anterior del polster antes y después de la ablación. Las células muestran un movimiento continuo antes de la ablación, que disminuye drásticamente después de la ablación, lo que indica la pérdida de la migración orientada al colectivo. La autocorrelación de la dirección también se midió en las células que forman la mitad anterior del polster de un embrión no ablacionado, como control. Los sobres del gráfico indican un error estándar. Haga clic aquí para ver una versión más grande de esta figura.
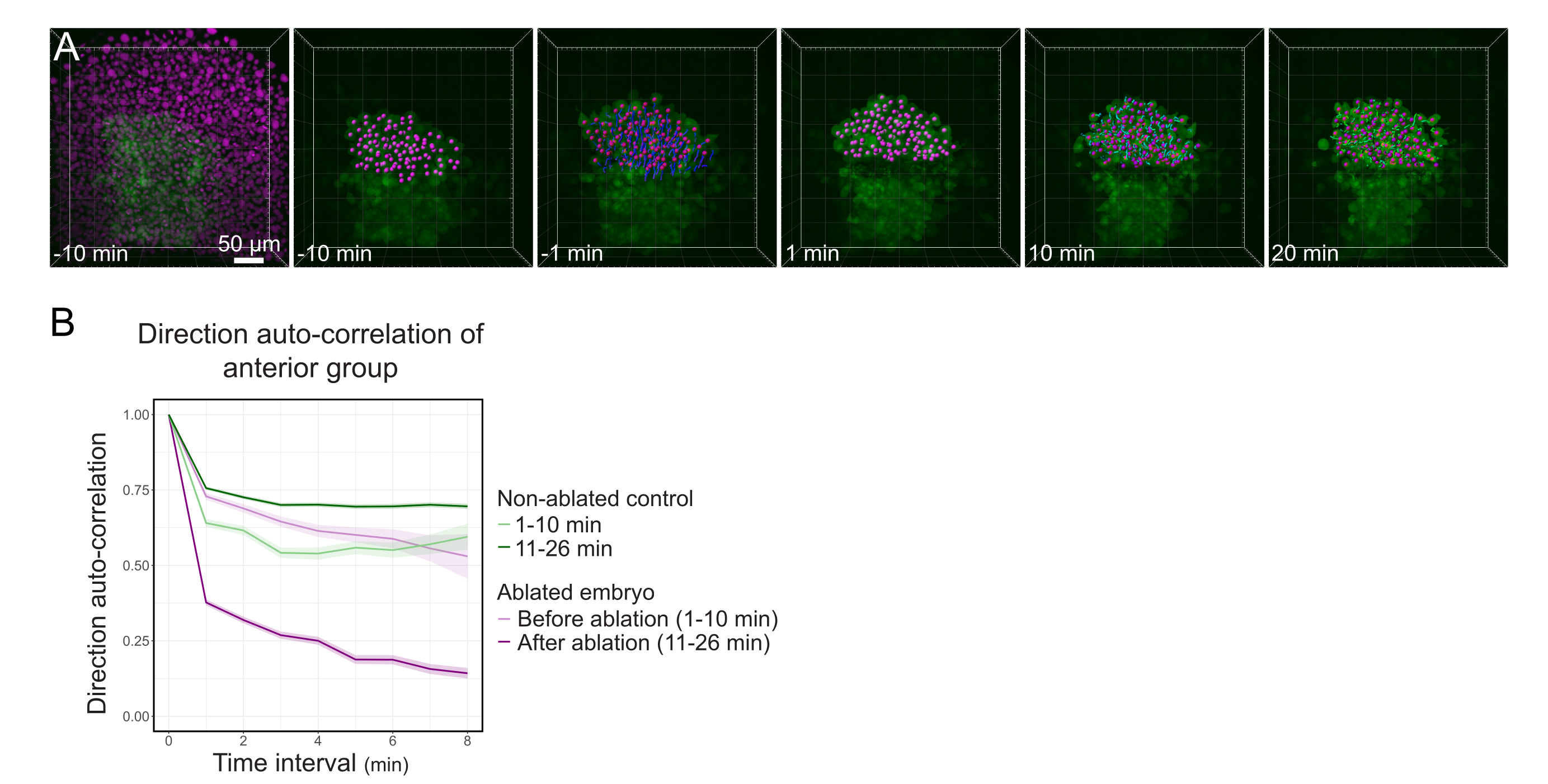{kind=link}
Resultados
Para cortar el polster en su medio, se montó un embrión Tg(gsc:GFP), inyectado con ARNm Histone2B-mCherry en la etapa epibólica del 70%, como se describe en el paso 4. El polster se identificó por expresión de GFP, y el embrión se montó de modo que el plano del polster sea perpendicular al eje óptico (Figura 1B). Inclinar el embrión lejos de esta posición complicará el procedimiento. La luz tendrá que atravesar más tejidos para llegar a los planos de ablación, y los pl...
Discusión
Aquí, describimos un protocolo que utiliza óptica no lineal para realizar ablaciones de volumen profundas y espacialmente bien definidas. El paso más crítico del protocolo es encontrar condiciones de tratamiento que proporcionen suficiente energía para permitir las ablaciones, pero no demasiada energía, para evitar el exceso de escombros o cavitación. La cantidad de energía entregada en el sitio objetivo depende principalmente de: (1) la potencia de salida del láser, (2) la calidad de la alineación del láser, ...
Divulgaciones
Los autores no declaran intereses contrapuestos.
Agradecimientos
Agradecemos a Emilie Menant por el cuidado de los peces, a la Instalación de Bioimagen Politécnica, en particular a Pierre Mahou, por la asistencia con imágenes en vivo en sus equipos, en parte con el apoyo de la Région Ile-de-France (interDIM) y la Agence Nationale de la Recherche (ANR-11-EQPX-0029 Morphoscope2, ANR-10-INBS-04 France BioImaging). Este trabajo fue apoyado por las subvenciones ANR 15-CE13-0016-1, 18-CE13-0024, 20-CE13-0016, y el programa de investigación e innovación Horizonte 2020 de la Unión Europea en el marco del acuerdo de subvención Marie Skłodowska-Curie No 840201, el Ministère de l'Enseignement Supérieur et de la Recherche y el Centre National de la Recherche Scientifique.
Materiales
| Name | Company | Catalog Number | Comments |
| 25x water immersion objective | Olympus | XLPLN25XWMP2 | |
| Agarose | PanReac AppliChem | A8963,0500 | |
| Data analysis software : Matlab | Math Works | ||
| Electro-optic modulator (EOM) | ConOptics | 350-80LA | |
| Embryo Medium (EM) solution | Westerfield, M. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio), 5th Edition. University of Oregon Press, Eugene (Book). (2000). | ||
| Environmental chamber chamber | Okolab | H201-T-UNIT-BL | |
| EOM driver | ConOptics | 302RM | |
| Fluorescence source | Lumencor | SOLA | |
| Glass bottom dishes | MatTek | P35G-0-10-C | |
| Glass capillaries | Harvard Apparatus | 300085 | Outside diameter 1.0 mm, inside diameter 0.58 mm |
| Glass pipettes | Volac | D810 | Tip should be fire polished |
| Green/ablation laser | Spectra Physics | Mai Tai HP DeepSee | |
| Histone2B-mCherry mRNA | Synthesized from pCS2-H2B-mCherry plasmid (Dumortier& al. 2012) | ||
| Image analysis software: IMARIS | Bitplane | ||
| ImSpector software | Abberior Instruments Development Team | ||
| Injection mold | Adapative Science Tools | I-34 | |
| Microloader tips | Eppendorf | 5242956003 | |
| Micromanipulator | Narishige | MN-151 | |
| Micropipette puller | Sutter | P-1000 | |
| mMESSAGE mMACHINE SP6 Transcription Kit | Invitrogen | AM1340 | |
| Penicillin-Streptomycin | Thermofisher | 15140-122 | 10 000 units penicillin and 10 mgstreptomycin per ml |
| Photomultiplier tube (PMT) | Hammamatsu | H7422-40 | |
| PicoPump (Air injector) | World Precision Instrument | PV820 | |
| Red laser | Spectra Physics | OPO/Insight DeepSee | |
| RNAse free water for injection | Sigma | W3500 | |
| Spreadsheet software: Excel | Microsoft | ||
| Stereomicroscope | Nikon | SMZ18 | |
| Tg(gsc:GFP) zebrafish line | Doitsidou, M. et al. Guidance of primordial germ cell migration by the chemokine SDF-1. Cell. 111 (5), 647–59, doi: doi.org/10.1016/S0092-8674(02)01135-2 (2002). | ||
| TriM Scope II microscope | La Vision Biotech |
Referencias
- Slack, J. M. W. Embryonic induction. Mechanisms of Development. 41 (2-3), 91-107 (1993).
- Fernandez-Sanchez, M. -. E., Brunet, T., Röper, J. -. C., Farge, E. Mechanotransduction's impact on animal development, evolution, and tumorigenesis. Annual Review of Cell and Developmental Biology. 31, 373-397 (2015).
- Shih, J., Fraser, S. E. Characterizing the zebrafish organizer: microsurgical analysis at the early-shield stage. Development. 122 (4), 1313-1322 (1996).
- Selleck, M. A. J. Culture and microsurgical manipulation of the early avian embryo. Methods in Cell Biology. 51 (51), 1-21 (1996).
- Bulina, M. E., et al. A genetically encoded photosensitizer. Nature Biotechnology. 24 (1), 95-99 (2006).
- Fang-Yen, C., Gabel, C. V., Samuel, A. D. T., Bargmann, C. I., Avery, L. Laser microsurgery in Caenorhabditis elegans. Methods in Cell Biology. 107, 177-206 (2012).
- Colombelli, J., Grill, S. W., Stelzer, E. H. K. Ultraviolet diffraction limited nanosurgery of live biological tissues. Review of Scientific Instruments. 75 (2), 472-478 (2004).
- Smutny, M., Behrndt, M., Campinho, P., Ruprecht, V., Heisenberg, C. -. P. UV laser ablation to measure cell and tissue-generated forces in the zebrafish embryo in vivo and ex vivo. Methods in Molecular Biology. 1189, 219-235 (2015).
- Behrndt, M., et al. Forces driving epithelial spreading in zebrafish gastrulation. Science. 338 (6104), 257-260 (2012).
- Volpe, B. A., Fotino, T. H., Steiner, A. B. Confocal microscope-based laser ablation and regeneration assay in zebrafish interneuromast cells. Journal of Visualized Experiments: JoVE. (159), (2020).
- Bonnet, I., et al. Mechanical state, material properties and continuous description of an epithelial tissue. Journal of the Royal Society, Interface. 9 (75), 2614-2623 (2012).
- Rauzi, M., Lenne, P. F., Lecuit, T. Planar polarized actomyosin contractile flows control epithelial junction remodelling. Nature. 468 (7327), 1110-1115 (2010).
- Niemz, M. H. . Laser-Tissue Interactions. Encyclopedia of Biomaterials and Biomedical Engineering, Second Edition - Four Volume Set. , (2019).
- Smith, A. M., Mancini, M. C., Nie, S. Bioimaging: second window for in vivo imaging. Nature Nanotechnology. 4 (11), 710-711 (2009).
- Rauzi, M., Lenne, P. -. F. Cortical forces in cell shape changes and tissue morphogenesis. Current Topics in Developmental Biology. 95, 93-144 (2011).
- Theveneau, E., David, N. B. Migrations cellulaires collectives. Medecine/Sciences. 30 (8-9), 751-757 (2014).
- Dumortier, J. G., Martin, S., Meyer, D., Rosa, F. M., David, N. B. Collective mesendoderm migration relies on an intrinsic directionality signal transmitted through cell contacts. Proceedings of the National Academy of Sciences of the United States of America. 109 (42), 16945-16950 (2012).
- Solnica-Krezel, L., Stemple, D. L., Driever, W. Transparent things: cell fates and cell movements during early embryogenesis of zebrafish. BioEssays. 17 (11), 931-939 (1995).
- Montero, J. -. A., Kilian, B., Chan, J., Bayliss, P. E., Heisenberg, C. -. P. Phosphoinositide 3-kinase is required for process outgrowth and cell polarization of gastrulating mesendodermal cells. Current Biology. 13 (15), 1279-1289 (2003).
- Ulrich, F., et al. Slb/Wnt11 controls hypoblast cell migration and morphogenesis at the onset of zebrafish gastrulation. Development. 130 (22), 5375-5384 (2003).
- Kai, M., Heisenberg, C. -. P., Tada, M. Sphingosine-1-phosphate receptors regulate individual cell behaviours underlying the directed migration of prechordal plate progenitor cells during zebrafish gastrulation. Development. 135 (18), 3043-3051 (2008).
- Smutny, M., et al. Friction forces position the neural anlage. Nature Cell Biology. 19 (4), 306-317 (2017).
- Johansson, M., Giger, F. A., Fielding, T., Houart, C. Dkk1 controls cell-cell interaction through regulation of non-nuclear β-Catenin pools. Developmental Cell. 51 (6), 775-786 (2019).
- Gorelik, R., Gautreau, A. Quantitative and unbiased analysis of directional persistence in cell migration. Nature Protocols. 9 (8), 1931-1943 (2014).
- Grill, S. W., Howard, J., Schäffer, E., Stelzer, E. H. K., Hyman, A. A. The distribution of active force generators controls mitotic spindle position. Science. 301 (5632), 518-521 (2003).
- Desprat, N., Supatto, W., Pouille, P. -. A. A., Beaurepaire, E., Farge, E. Tissue deformation modulates twist expression to determine anterior midgut differentiation in Drosophila embryos. Developmental Cell. 15 (3), 470-477 (2008).
- Farhadifar, R., Röper, J. -. C., Aigouy, B., Eaton, S., Jülicher, F. The influence of cell mechanics, cell-cell interactions, and proliferation on epithelial packing. Current Biology. 17 (24), 2095-2104 (2007).
- Willier, B. H., Oppenheimer, J. M. . Foundations of Experimental Embryology. , (1964).
- Ashby, W. J., Zijlstra, A. Established and novel methods of interrogating two-dimensional cell migration. Integrative Biology: Quantitative Biosciences from Nano to Macro. 4 (11), 1338-1350 (2012).
- Bosze, B., et al. Pcdh18a regulates endocytosis of E-cadherin during axial mesoderm development in zebrafish. Histochemistry and Cell Biology. 154 (5), 463-480 (2020).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados