
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Control de la fracción de partículas en andamios microporosos de partículas recocidas para cultivo celular 3D
En este artículo
Resumen
Minimizar la variabilidad en la fracción de partículas dentro de andamios granulares facilita la experimentación reproducible. Este trabajo describe métodos para generar andamios granulares con fracciones de partículas controladas para aplicaciones de ingeniería de tejidos in vitro .
Resumen
Los microgeles son los bloques de construcción de los andamios de partículas recocidas microporosas (MAP), que sirven como plataforma tanto para el cultivo celular in vitro como para la reparación de tejidos in vivo . En estos andamios granulares, la porosidad innata generada por el espacio vacío entre los microgeles permite la infiltración y migración celular. El control de la fracción vacía y la fracción de partículas es fundamental para el diseño de andamios MAP, ya que la porosidad es una señal bioactiva para las células. Los microgeles esféricos se pueden generar en un dispositivo microfluídico para controlar el tamaño y la forma y, posteriormente, liofilizar utilizando métodos que evitan la fracturación de la red de polímeros. Tras la rehidratación, los microgeles liofilizados conducen a fracciones de partículas controladas en andamios MAP. La implementación de estos métodos para la liofilización de microgel ha llevado a estudios reproducibles que muestran el efecto de la fracción de partículas en la difusión de macromoléculas y la propagación celular. El siguiente protocolo cubrirá la fabricación, liofilización y rehidratación de microgeles para controlar la fracción de partículas en andamios MAP, así como el recocido de los microgeles a través de reticulación bio-ortogonal para cultivo celular 3D in vitro.
Introducción
Los andamios de partículas recocidas microporosas (MAP) son una subclase de materiales granulares en los que los bloques de construcción de microgel (μgel) están interconectados para formar un andamio poroso a granel. Con la microarquitectura única de estos andamios granulares, la porosidad innata generada por el espacio vacío entre el microgel esférico interconectado admite la infiltración celular acelerada y la migración1. Los bloques de construcción de microgel de los andamios MAP se pueden fabricar a partir de polímeros sintéticos y naturales con modificaciones químicas2. Los métodos descritos aquí destacan específicamente el uso de microgeles compuestos por una columna vertebral de ácido hialurónico (AH) modificada con mangos funcionales de norborneno (NB). El mango funcional NB en el polímero HA admite reacciones químicas de clic para formar microgeles y unirlos para generar andamios MAP 3,4. Se han empleado numerosos esquemas para unir los microgeles (es decir, recocido), como las reacciones enzimáticas1, 5,6 basadas en luz y química de clic sin aditivos 3,7. La química de clic sin aditivos se describe en este trabajo, utilizando la conjugación de demanda inversa de electrones de tetrazina-norborneno Diels-Alder para interconectar los microgeles HA-NB.
Para fabricar andamios MAP, los usuarios primero generan los bloques de construcción de microgel utilizando emulsiones inversas, ya sea en sistemas por lotes o dentro de dispositivos microfluídicos, así como con pulverización electrohidrodinámica, litografía o fragmentación mecánica2. La producción de microgeles esféricos HA-NB ha sido bien descrita y previamente reportada utilizando tanto la emulsión por lotes2 como las técnicas de generación de gotas microfluídicas 8,9,10,11. En este trabajo, se generaron microgeles esféricos HA-NB en una plataforma microfluídica de enfoque de flujo para un tamaño y forma controlados, como se describió anteriormente 8,9,10. Después de la purificación, los microgeles existen en una suspensión acuosa y deben concentrarse para inducir un estado de atascamiento. Cuando se atascan, los microgeles exhiben propiedades de adelgazamiento por cizallamiento, que les permiten funcionar como materiales inyectables que llenan el espacio1. Un método para inducir un estado atascado es secar los microgeles mediante liofilización, o liofilización, y luego rehidratar el producto seco en un volumen controlado12. Alternativamente, el exceso de tampón se puede eliminar de la suspensión de microgel mediante centrifugación sobre un colador o con la extracción manual del tampón del gránulo de microgel, ya sea por aspiración o utilizando un material absorbente. Sin embargo, el uso de la centrifugación para secar los microgeles puede generar un rango muy variable de fracciones de partículas y fracciones vacías al hacer andamios granulares12. Se han descrito técnicas para liofilizar microgeles utilizando 70% IPA para microgeles de polietilenglicol (PEG)13, aceites fluorados para microgeles de gelatina metacriloyl (GelMa) 14 y etanol al 70% para microgeles HA12. Este protocolo destaca los métodos para liofilizar microgeles esféricos de HA utilizando etanol al 70%, un reactivo de laboratorio estándar, para conservar las propiedades originales del microgel durante el proceso de secado. Los microgeles HA liofilizados se pueden pesar y rehidratar en porcentajes de peso definidos por el usuario para controlar las fracciones de partículas finales en andamios MAP12.
El paso final en la formación de andamios MAP se basa en el recocido de los microgeles para crear un andamio voluminoso y poroso1. Al utilizar componentes nativos de la matriz extracelular y emplear esquemas de recocido bioortogonal, los andamios MAP sirven como una plataforma biocompatible tanto para el cultivo celular in vitro como para la reparación de tejidos in vivo 3. A través de estos enfoques, los andamios MAP pueden fabricarse a partir de bloques de construcción HA-NB con fracciones de partículas definidas por el usuario para su empleo en aplicaciones de ingeniería de tejidos12. El siguiente protocolo describe la producción microfluídica de microgeles HA-NB seguida de liofilización y rehidratación para controlar la fracción de partículas en andamios MAP. Por último, los pasos para el recocido de los microgeles se describen utilizando química bio-ortogonal para experimentos de cultivo celular 3D in vitro .
Protocolo
1. Fabricación de dispositivos microfluídicos
- Litografía blanda
NOTA: Este protocolo describe la fabricación del dispositivo de un diseño de dispositivo microfluídico de enfoque de flujo de Wilson et al.9. Sin embargo, este protocolo se puede utilizar con cualquier diseño de dispositivo en una oblea SU-8. La oblea se puede pegar a una placa de Petri, y luego necesita ser silanizada para evitar la adherencia del PDMS a las características de la oblea15.- Mezcle la base de elastómero de polidimetilsiloxano (PDMS) con el agente de curado (consulte la Tabla de materiales) en una proporción de 10:1. Prepare aproximadamente 100 g para cubrir la oblea con ~5 mm PDMS. Vierta la mezcla de PDMS sobre la oblea y desgasifique en un desecador durante aproximadamente 30 minutos. Una vez que todas las burbujas hayan desaparecido, colocar en un horno a 60 °C durante al menos 2 h para curar el PDMS.
- Use un cuchillo para trazar suavemente alrededor del parámetro del dispositivo sin agrietar la oblea; luego, retire cuidadosamente el PDMS de la oblea. Utilice un punzón de biopsia de 1 mm (consulte la Tabla de materiales) para crear los canales de entrada y salida.
NOTA: Sea suave al perforar el dispositivo microfluídico. Los desgarros o rasgaduras alrededor de los canales de entrada o salida pueden causar fugas durante la producción de microgel. - Use cinta adhesiva para eliminar el polvo del dispositivo en el lado de la función. Coloque los dispositivos y las guías de vidrio limpias en una placa caliente a 135 °C durante al menos 15 minutos para eliminar la humedad.
- En una campana extractora, use una pistola de plasma corona (consulte la Tabla de materiales) en lo alto tanto de las guías de vidrio como de los dispositivos (el lado de la característica expuesta) durante aproximadamente 30 s, y luego unirlos rápidamente. Aplique presión suavemente para asegurar un buen sellado entre el dispositivo y el portaobjetos de vidrio. Coloque los dispositivos en un horno a 60 °C durante la noche para asegurar la unión.
2. Producción microfluídica de microgeles de ácido hialurónico (AH) con mangos funcionales de norborneno (NB)
- Síntesis de HA-NB
NOTA: La síntesis de HA-norborneno (HA-NB) se adaptó de Darling et al.3 utilizando HA sódico de 79 kDa con equivalentes molares de 1:1.5:2.5 de unidades de repetición de HA a cloruro de 4-(4,6-dimetoxi-1,3,5-triazin-2-il)-4-metilmorfolinio (DMTMM) a 5-norborneno-2-metilamina (NMA).- Pesar los reactivos. Disuelva el AH a 20 mg/ml en un tampón MES de 200 mM (pH ~6) agitando un vaso de precipitados o matraz en una placa de agitación. Una vez disuelto, añadir el DMTMM a la solución de HA y dejar reaccionar durante aproximadamente 20 min a temperatura ambiente. Por ejemplo, se puede usar 1 g HA + 1.09 g DMTMM + 845 μL NMA.
- Agregue NMA gota a gota a la solución HA/DMTMM. Agregue parafilm a la abertura del recipiente de reacción para minimizar la evaporación y cubra el recipiente de reacción con papel de aluminio. Continúe agitando mientras deja que la reacción continúe durante aproximadamente 24 h.
- Después de 24 h, enfríe 200 etanol a prueba (aproximadamente 10 veces el volumen de reacción). En una placa de agitación, transfiera la reacción gota a gota al etanol enfriado para precipitar el HA-NB y continúe agitando a 200-300 rpm durante 20 min.
- Transfiera la solución a tubos cónicos de 50 ml y luego centrifugar a 5.000 x g durante 10 min. Vierta el exceso de etanol para desecharlo como desecho. En este punto, el producto HA-NB debe ser pellets blancos en los tubos cónicos. Tire al vacío del HA-NB en un desecador para que se seque durante la noche.
- Purificar el HA-NB utilizando tubos de diálisis de celulosa de corte de 12-14 kDa de peso molecular (ver Tabla de materiales). Disolver HA-NB en solución de NaCl 2 M y transferirlo al tubo de diálisis. Ate el tubo y asegúrelo con abrazaderas, si es necesario. Transfiera el tubo de diálisis lleno a un cubo con 5 L de agua ultrapura y dialice el HA-NB contra el agua durante la noche.
- Al día siguiente, retire el agua y vuelva a colocarla con una solución de NaCl 1 M durante 30 min. Retire la solución de NaCl y luego dialice contra agua ultrapura durante 3 días, reemplazando el agua diariamente.
- Filtrar el producto dializado con un filtro accionado por vacío de 0,2 μm y, a continuación, transferir el producto filtrado a tubos cónicos de 50 ml.
- Agregue nitrógeno líquido a un recipiente criogénico y congele rápidamente los tubos HA-NB durante 10 minutos. Luego, retire los tubos cónicos con fórceps y retire rápidamente la tapa y la cúbrala con un pañuelo de papel de laboratorio (consulte la Tabla de materiales). Asegure el tejido con una banda elástica y transfiéralo a un recipiente o cámara de liofilización (consulte la Tabla de materiales) y liofilizar. Conservar el producto liofilizado a -20 °C.
PRECAUCIÓN: El nitrógeno líquido es una sustancia peligrosa. Use el equipo de protección personal adecuado cuando trabaje con nitrógeno líquido. - Cuantificar la modificación del norborneno disolviendo el HA-NB a 10 mg/mL enD2Oy analizando mediante RMN de protones (Figura 1A)16.
- Para determinar la cantidad de funcionalización, primero calibre el pico de disolventeD2Oa 4,8 PPM. Integrar el pico para los protones metilo HA (δ2.05) y calibrar la integración a 3.0. A continuación, integre los picos para los grupos norborneno colgantes en δ6.33 y δ6.02 (protones de vinilo, endo). Normalizar la integración de estos picos al número correspondiente de protones para determinar el grado medio de modificación3.
- Preparación del precursor del microgel HA-NB
- Prepare un tampón HEPES de 50 mM (pH 7,5) y filtre estéril el tampón utilizando un filtro accionado por vacío de 0,2 μm. Utilizando el tampón HEPES, preparar las respectivas existencias de 50 mM de fenil(2,4,6,-trimetilbenzoil)fosfinato de litio (LAP) fotoiniciador y agente reductor de tris(2-carboxietil)fosfina (TCEP). Mantenga la solución LAP alejada de la luz.
- Preparar los demás componentes precursores de microgel preparando las respectivas existencias de 50 mM de enlazador de di-tiol y péptido RGD en agua destilada estéril. Pesar HA-NB y disolver en tampón HEPES para preparar una cepa de 10 mg/ml.
NOTA: Se podrían utilizar diferentes enlazadores de di-tiol para la reticulación interna de los microgeles según las preferencias del usuario. Tanto un enlazador degradable (es decir, MMP-esciblable) como no degradable (ditiothreitol o DTT) se han incluido en la Tabla de materiales. El péptido RGD se incluye en la formulación de microgel para promover la adhesión celular en andamios MAP, pero este componente podría eliminarse y reemplazarse con un volumen igual de tampón HEPES. - Combine los componentes precursores con concentraciones finales de 9,9 mM LAP, 0,9375 mM TCEP (4 tiol/TCEP), 2,8 mM di-tiol linker, 1 mM péptido RGD y 3,5% en peso (p/v) de HA-NB añadiendo tampón HEPES adicional para alcanzar el volumen final deseado. Mezclar bien el precursor con una pipeta de desplazamiento positivo.
- Con una pipeta P1000, tire lentamente de toda la mezcla. Coloque la punta en el extremo de una jeringa de 1 ml y expulse la punta de la pipeta. Tire del émbolo de la jeringa para cargar la mezcla en la jeringa y, a continuación, agregue un filtro de 0,2 μm en el extremo de la jeringa y filtre en un nuevo tubo de microcentrífuga de 1,5 ml. Centrifugar la solución precursora filtrada para eliminar las burbujas producidas durante el filtrado.
- Nuevamente, usando una pipeta P1000, tire lentamente hacia arriba del precursor filtrado teniendo cuidado de no crear burbujas. Si hay burbujas, golpee suavemente la punta para que se desprendan y floten hacia la parte superior.
- Coloque la punta en el extremo de una jeringa de 1 ml y expulse la punta de la pipeta. Mantenga la jeringa vertical y tire del émbolo de la jeringa lentamente hasta que toda la solución precursora esté en la jeringa. Agregue una aguja de punta roma a la jeringa y empuje el precursor a través de la punta de la aguja. Envuelva la jeringa en papel de aluminio para mantenerla alejada de la luz.
- Preparación de la solución de pellizco de microgel
- Prepare 5% v / v Span-80 en aceite mineral blanco pesado y mezcle bien. Desecar para eliminar burbujas. Mantenga la mezcla de surfactante/aceite a temperatura ambiente envuelta en papel de aluminio. Mezclar bien y dessecar antes de cada uso.
- Use una jeringa de 5 ml para extraer la mezcla de aceite/surfactante (minimice las burbujas) hasta que la distancia entre el émbolo y la sujeción sea aproximadamente igual a la distancia de la jeringa precursora. Agregue una aguja roma a la jeringa y empuje el aceite a través de la punta de la aguja.
- Configuración del dispositivo microfluídico
- Agregue una aguja roma a una jeringa de 1 ml y llénela con una solución de tratamiento hidrofóbica sintética (consulte la Tabla de materiales). Haga fluir suavemente la solución a través del dispositivo microfluídico hasta que se acumule en cada entrada/salida. Deje que la solución se seque en el dispositivo en la mesa de trabajo durante aproximadamente 30 minutos, y luego tire del vacío en la salida para eliminar el exceso de solución. Asegure el dispositivo con abrazaderas en un microscopio de mesa.
- Envuelva un tubo cónico de 15 ml con papel de aluminio y colóquelo en un estante para tubos para que sirva como recipiente de recolección de microgel. Utilice un soporte de anillo con una abrazadera para colocar la sonda de luz UV en la abertura del tubo de recolección. Utilice un detector UV (ver Tabla de materiales) para medir la intensidad UV, moviendo la sonda hasta alcanzar 20 mW/cm2 . Apague la luz UV hasta más tarde.
- Corte el tubo a una longitud que llegue desde el dispositivo microfluídico hasta el recipiente de recolección. En un extremo del tubo, corte un ángulo de 45°. Inserte suavemente el extremo angulado del tubo en el canal de salida.
NOTA: Sea cuidadoso al insertar el tubo en el dispositivo microfluídico. Los desgarros o rasgaduras alrededor de los canales de entrada o salida pueden causar fugas durante la producción de microgel. - Asegure las jeringas precursoras y en fase oleosa en una bomba de doble jeringa (consulte la Tabla de materiales). Corte dos piezas más de tubo a una longitud que llegue desde las puntas de la jeringa hasta el dispositivo microfluídico. En un extremo de cada tubo, corte un ángulo de 45°. Asegure cuidadosamente el tubo (extremo romo) en ambas puntas de la jeringa.
- Cambie la configuración de la bomba para la jeringa de 1 ml e incluya el volumen aproximado del precursor. Empuje lentamente la bomba hacia adelante hasta que se aplique suficiente presión a los émbolos de la jeringa para empujar tanto el aceite como el precursor a los extremos del tubo, eliminando el aire del sistema. Deje que la presión se iguale 5-10 minutos antes de pasar al paso 2.4.6.
- Inserte suavemente el extremo angulado del tubo en los canales de entrada del dispositivo microfluídico con la solución precursora de microgel en la entrada frontal y el aceite de pellizco en la entrada posterior. Mueva la bomba hacia adelante en pequeños incrementos hasta que comience el flujo en el dispositivo y comiencen a formarse microgeles esféricos en la región de enfoque de flujo. Encienda la bomba con un caudal de 0,4 μL/min y deje que el dispositivo funcione hasta que se estabilice. Si es necesario, ajuste el caudal ±0,1 μL/min en pequeños incrementos para estabilizar la producción de microgel.
- Una vez que la producción de microgel se estabilice como se muestra en la Figura 1B, reemplace el tubo de recolección con un tubo nuevo y encienda la luz UV. Revise la ejecución periódicamente para asegurarse de que la producción de microgel sea estable durante la duración de la ejecución.

Figura 1: Producción microfluídica de microgeles de ácido hialurónico (AH) con mangos funcionales de norborneno (NB). (A) Aproximadamente el 31% de las unidades de repetición de HA se modificaron con éxito con NB, según lo determinado por el análisis de RMN de protones realizado en óxido de deuterio. 1 Los desplazamientos de RMN H de los norbornenos colgantes a δ6.33 y δ6.02 (protones de vinilo, endo), y δ6.26 y δ6.23 ppm (protones de vinilo, exo) se compararon con el grupo metilo HA δ2.05 ppm para determinar la funcionalización. Reimpreso de Anderson et al.12 con permiso de Elsevier. (B) Esquema del dispositivo microfluídico de enfoque de flujo utilizado para generar μgeles HA-NB. (C) Se utilizaron proyecciones de intensidad máxima de microscopía confocal para visualizar μgeles marcados con fluorescencia (barra de escala = 500 μm). (D) Las distribuciones de frecuencia del diámetro del microgel de ejecuciones independientes en la configuración microfluídica demuestran el control sobre el tamaño del microgel ~ 50 μm o ~ 100 μm dependiendo del dispositivo utilizado. (E) El diámetro del microgel se informa como la media y la desviación estándar para cada tirada independiente. Reimpreso de Wilson et al.9 con permiso de Wiley. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
3. Microgeles purificadores y secadores
- Purificación de microgeles
- Prepare el tampón de lavado de microgel (300 mM HEPES, 50 mM NaCl, 50 mM CaCl 2), así como la solución de surfactante Pluronic F-127 al2% (p/v) en tampón de lavado. Esterilizar las soluciones con un filtro accionado por vacío de 0,2 μm.
- Centrifugar el tubo de recolección de microgel (5,000 x g) durante 5 min. En una campana estéril, aspire cuidadosamente la fase de aceite sobrenadante. Combine los μgels 1:1 con la solución de surfactante Pluronic F-127 al 2% y el vórtice para mezclar bien. Centrifugar (5.000 x g) durante 5 min y aspirar la solución de lavado sobrenadante.
- Agregue el tampón de lavado a un volumen de microgel 4x y el vórtice para mezclar bien. Centrifugar (5.000 x g) la mezcla durante 5 min y aspirar la solución de lavado. Complete de 4 a 8 lavados con el tampón de lavado hasta que el surfactante se elimine del sistema (es decir, no queden burbujas).
- Etiquetado fluorescente de microgeles HA-NB
NOTA: La síntesis interna de una tetrazina marcada con fluorescencia se basa en dos reacciones de adición de tiol-Michael catalizadas por bases en serie que han sido bien descritas y previamente reportadas3. Para este trabajo, Alexa Fluor-488 se conjugó con tetrazina para el etiquetado de μgels modificados con norborneno. El producto liofilizado (harina de Alexa 488-Tet) se disolvió en dimetilformamida a 1 mg/ml y se almacenó a -20 °C.- Para etiquetar fluorescentemente los μgels, primero prepare una solución de trabajo de Alexa Fluor 488-Tet diluyendo el stock de 1 mg / ml 1:14 en 1x PBS estéril. En una campana estéril, combine los μgels con la solución de trabajo (2:1 por volumen).
- Utilizar una pipeta de desplazamiento y mezclar bien. Incubar la mezcla durante 1 h a temperatura ambiente o durante la noche a 4 °C.
- Centrifugar (5.000 x g) y aspirar la solución de tinción. Lave los μgels dos veces con 1x PBS (1:1 por volumen) para eliminar Alexa Fluor 488-Tt sin reaccionar.
NOTA: En este punto, los μgels marcados con fluorescencia pueden ser fotografiados en un microscopio confocal para cuantificar el tamaño del microgel (Figura 1C-E)9. Los métodos para medir el tamaño del microgel han sido descritos exhaustivamente por Roosa et al.17.
- Secado de microgeles HA-NB
- Transfiera μgeles purificados (Figura 2A) a un tubo de tapón de rosca crioseguro utilizando una pipeta de desplazamiento positivo. Añadir etanol al 70% a los μgels purificados al 50% (v/v) y mezclar bien con una pipeta de desplazamiento. Centrífuga durante 5 min a 5.000 x g.
PRECAUCIÓN: El etanol es una sustancia altamente inflamable.
NOTA: El tubo de tapón de rosca crioseguro se puede pesar antes de agregar μgels, y luego se puede pesar nuevamente después de la liofilización para determinar la masa de μgels. Esto se recomienda para minimizar el error cuando se utilizan cantidades inferiores a 1 mg. Asegúrese de que la báscula esté ajustada o calibrada internamente antes de su uso. - Aspirar el líquido sobrenadante y reemplazarlo con etanol al 70% (50% v/v) (Figura 2B). Mezclar bien con una pipeta de desplazamiento. Incubar durante la noche a 4 °C.
NOTA: Los microgeles se pueden almacenar en etanol al 70% a 4 °C antes de la liofilización para su almacenamiento a largo plazo, si es necesario. Los microgeles liofilizados se muestran en la Figura 2C. Se pueden usar otros medios de liofilización en este paso si se desea la formación de criogel (Figura 2D). - Centrifugar brevemente para asegurarse de que los μgels estén en la parte inferior del tubo del tapón de rosca. Agregue nitrógeno líquido a un recipiente criogénico y luego agregue el tubo de μgels a la congelación rápida.
- Después de 5-10 minutos, retire el tubo de μgels con fórceps. Retire rápidamente la tapa y cúbrala con un pañuelo de papel de laboratorio. Asegure el tejido con una banda elástica y transfiéralo a un recipiente o cámara de liofilización.
- Cargue la muestra en el liofilizador siguiendo las instrucciones del fabricante. Liofilizar a 0,066 Torr y -63 °C. Guarde los μgels liofilizados (lyo-μgels) herméticamente sellados a temperatura ambiente.
NOTA: La liofilización se completa cuando se elimina todo el líquido del tubo y queda un producto seco. Los solventes orgánicos pueden disminuir la longevidad de los accesorios de caucho en los sistemas comunes de liofilización.
- Transfiera μgeles purificados (Figura 2A) a un tubo de tapón de rosca crioseguro utilizando una pipeta de desplazamiento positivo. Añadir etanol al 70% a los μgels purificados al 50% (v/v) y mezclar bien con una pipeta de desplazamiento. Centrífuga durante 5 min a 5.000 x g.

Figura 2: Secado de microgeles HA-NB. (A) Proyección de intensidad máxima de μgels en solución acuosa (barra de escala = 100 μm). (B) Los μgeles purificados pueden incubarse 1:1 por volumen en el medio de liofilización de elección y liofilizarse. (C) Proyección de intensidad máxima de lyo-μgels secos (barra de escala = 100 μm). (D) Los microgeles se resuspenden después de la liofilización. Se recomienda EtOH (70%) para conservar las propiedades originales de los μgeles durante todo el proceso de liofilización; sin embargo, otros medios como el alcohol isopropílico (IPA), el agua y el acetonitrilo (MeCN) se pueden usar indistintamente para facilitar la formación de criogel (barra de escala = 100 o 50 μm como se indica). (E) Medición del diámetro del microgel HA-NB antes (gris) y después de la liofilización (verde) en EtOH al 70% mostrada como distribuciones de frecuencia para tres poblaciones de microgel. Reimpreso de Anderson et al.12 con permiso de Elsevier. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
4. Fabricación de andamios MAP
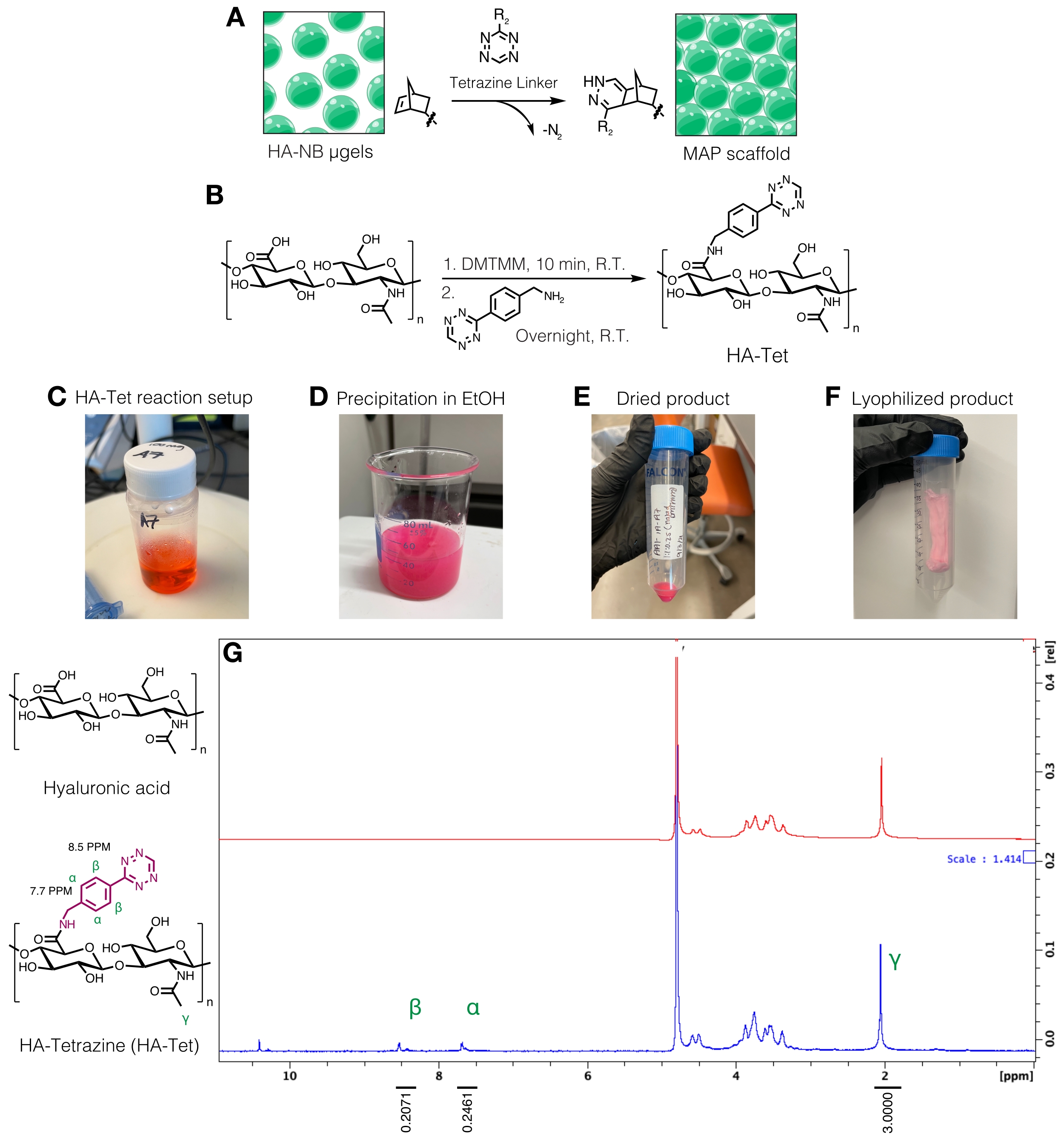
- Síntesis del enlazador de tetrazina
NOTA: Los enlazadores de tetrazina se pueden utilizar para interconectar μgeles que contienen grupos norborneno libres (Figura 3A). El procedimiento de síntesis de HA-tetrazina (HA-Tet) se adaptó de Zhang et al.18 utilizando HA sódico de 79 kDa con equivalentes molares de 1:1:0,25 de unidades de repetición de HA a DMTMM a tetrazina-amina (Figura 3B)12.- Pesar los reactivos. Disuelva el AH a 20 mg/ml en un tampón MES de 200 mM (pH ~6) agitando un vaso de precipitados o matraz en una placa de agitación. Una vez disuelto, añadir el DMTMM a la solución de HA y dejar reaccionar durante aproximadamente 20 min a temperatura ambiente. Por ejemplo, se pueden usar 100 mg HA + 72.8 mg DMTMM + 14.14 mg de tetrazina-amina.
- Disuelva la tetrazina-amina a 15 mg/ml en un tampón MES de 200 mM y agregue gota a gota a la solución de HA/DMTMM. Consulte la Figura 3C para la configuración de la reacción HA-Tet.
- Agregue parafilm a la abertura del recipiente de reacción para minimizar la evaporación y cubra el recipiente de reacción con papel de aluminio. Continúe agitando mientras deja que la reacción continúe durante aproximadamente 24 h.
- Después de 24 h, enfríe 200 etanol a prueba (aproximadamente 10 veces el volumen de reacción). En una placa de agitación, transfiera la reacción gota a gota al etanol enfriado para precipitar el HA-Tet (Figura 3D) y continúe agitando durante 20 min.
- Transfiera la solución a tubos cónicos de 50 ml y luego centrifugar a 5.000 x g durante 10 min. Vierta el exceso de etanol para desecharlo como desecho. Tire de la aspiradora del HA-Tet en un desecador para que se seque durante la noche. Un ejemplo del producto seco en este paso del protocolo se puede encontrar en la Figura 3E.
- Purificar el HA-Tet mediante diálisis. Disolver HA-Tet en solución de NaCl 2 M y transferir a un tubo de diálisis de celulosa con un límite de peso molecular de 12-14 kDa. Transfiera el tubo de diálisis lleno a un cubo con 5 L de agua ultrapura y dialice el HA-Tet contra el agua durante la noche.
- Al día siguiente, retire el agua y vuelva a colocarla con una solución de NaCl 1 M durante 30 min. Retire la solución de NaCl y luego dialice contra agua ultrapura durante 3 días, reemplazando el agua diariamente.
- Filtrar el producto dializado con un filtro accionado por vacío de 0,2 μm y, a continuación, transferir el producto HA-Tet filtrado a tubos cónicos de 50 ml.
- Congele rápidamente los tubos cónicos en nitrógeno líquido durante 10 minutos y luego retire los tubos cónicos con fórceps. Retire rápidamente la tapa y cúbrala con un pañuelo de papel de laboratorio. Asegure el tejido con una banda elástica y transfiéralo a un recipiente o cámara de liofilización y liofilizar. Conservar el producto liofilizado (Figura 3F) a -20 °C.
- Cuantificar la modificación de la tetrazina disolviendo el HA-Tet a 10 mg/mL enD2Oy analizando vía RMN de protones (Figura 3G)16.
- Para determinar la cantidad de funcionalización, primero calibre el pico de disolventeD2Oa 4,8 PPM. Integrar el pico para los protones metilo HA (δ2.05) y calibrar la integración a 3.0. A continuación, integre los picos para los grupos tetrazina colgantes en δ8.5 (2H) y δ7.7 (2H) (protones aromáticos). Normalizar la integración de estos picos al número correspondiente de protones para determinar el grado medio de modificación12.
- Interconexión de lyo-μgels para formar andamios MAP para la caracterización
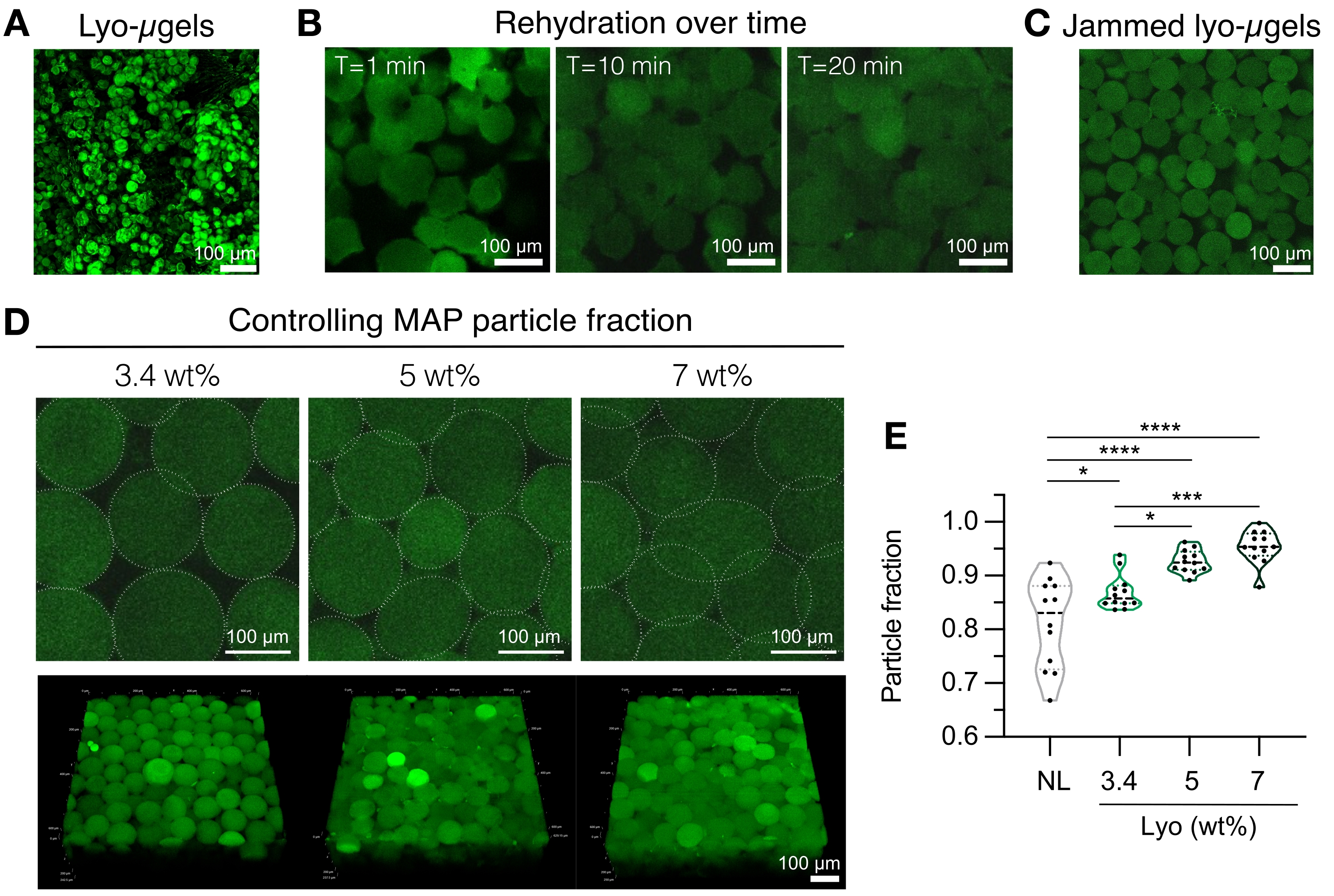
- Prepare los componentes del andamio MAP (es decir, μgels, HA-Tet, volumen de rehidratación). Pesar los lyo-μgels (Figura 4A) y reconstituir en el 84% del volumen final de MAP de 1x PBS. Deje que los microgeles se hinchen durante aproximadamente 20 minutos (Figura 4B, C). El % en peso MAP utilizado para la rehidratación se puede elegir en función de la preferencia del usuario por la fracción de partículas final (consulte la Figura 4D, E).
- Disuelva el HA-Tet en 1x PBS a la concentración elegida (ver NOTA a continuación).
NOTA: Cambiar tanto la fracción de embalaje (a través de % en peso MAP) como la concentración de HA-Tet alterará las propiedades mecánicas del andamio a granel. Por ejemplo, un andamio MAP de 3,4 % en peso reticulado con 0,02 mg/ml de HA-Tet (relación de recocido de 2,6 mol Tet:mol HA-NB) genera andamios MAP con aproximadamente 700 Pa módulo de almacenamiento de corte12. - Utilice una pipeta de desplazamiento para combinar el HA-Tet y los lyo-μgels y mezcle bien. En este punto, la mezcla se puede transferir a través de una pipeta de desplazamiento a portaobjetos de vidrio, placas de pozo o un recipiente de la elección del usuario. Deje que los μgels se reanuden a 37 °C durante 25 min, y luego use una espátula para transferir los andamios MAP a placas de pocillos llenas de 1x PBS. Mantenga los andamios MAP en 1x PBS hasta que estén listos para la caracterización.
- Cálculo de la fracción de partículas del andamio MAP
- Para mejorar la calidad de imagen, transfiera el andamio MAP a un cubreobjetos de vidrio utilizando una espátula. Andamios MAP de imagen en un microscopio confocal utilizando el láser para la excitación y emisión de FIFC. Imagen de andamios MAP en un objetivo 20x y obtener una pila Z que atraviesa 250-300 μm en la dirección Z con un tamaño de paso de 2,5 μm. Tome nota de la calibración de μm/píxel de la imagen.
- Importe la imagen Z-stack en el software de análisis (consulte Tabla de materiales). Seleccione el botón Agregar nuevas superficies . Marque la casilla Segmentar solo una región de interés y, a continuación, seleccione el botón de flecha azul Siguiente: Región de interés.
- Defina una región de interés, realizando un seguimiento de las dimensiones X, Y y Z del volumen que se está analizando. Seleccione el botón de flecha azul Siguiente: Canal de origen.
NOTA: Las dimensiones X e Y están en unidades de píxeles, mientras que la dimensión Z es el número de pasos. Una altura Z recomendada para la región de interés debe incluir un mínimo de dos μgels. - Utilice la lista desplegable Canal de origen para seleccionar el canal FIFC. Marca la casilla situada junto a Suavizar e introduce un detalle de superficie de 2,50 μm. En Umbral, seleccione Intensidad absoluta y, a continuación, seleccione el botón de flecha azul Siguiente: Umbral.
- Utilice el valor de umbral sugerido para el canal FIFC. Gire la proyección 3D para evaluar la calidad de renderizado y ajustarla según sea necesario. Seleccione Siguiente: Clasificar superficies.
NOTA: El botón Atrás se puede utilizar para editar pasos anteriores en el proceso, como Z-dimension, según sea necesario. - Compruebe si Número de vóxeles es 10.0 y, a continuación, seleccione el botón verde de flecha doble Finalizar: Ejecute todos los pasos de creación y finalice el asistente.
NOTA: Los parámetros de representación de volumen se pueden almacenar para el análisis por lotes, de modo que se apliquen los mismos ajustes para analizar todos los andamios. - Para exportar los datos, seleccione la pestaña Estadísticas y, a continuación, la pestaña Detallado . Utilice el segundo cuadro desplegable para seleccionar la variable Volumen. Seleccione el botón de disquete Exportar estadísticas en la pestaña Mostrar a archivo y guárdelo como un archivo de hoja de cálculo (.xls) cuando se le solicite.
- Abra el archivo y utilice la función SUMA en la columna A Volumen para determinar el volumen total (μm3) de los μgels en la región de interés.
- Convierta las dimensiones de la región de interés que se analizó de píxeles a μm. Utilice la calibración μm/píxel de la imagen del paso 4.3.1 para convertir las dimensiones X e Y. Multiplique la dimensión Z (número de pasos) por el tamaño del paso de la imagen para convertir la dimensión Z a μm. Calcula el volumen de la región de interés (μm3) multiplicando las dimensiones X, Y y Z.
- Para determinar la fracción de partículas del andamio, divida el volumen total de los μgels en la región de interés (que se encuentra en el paso 4.3.8) por el volumen de la región de interés (que se encuentra en el paso 4.3.9).

Figura 3: Síntesis del enlazador de tetrazina para la fabricación de andamios de partículas recocidas microporosas (MAP). (A) Esquema de μgels HA-NB interconectados con un enlazador de tetrazina para formar andamios MAP. (B) Esquema de reacción para la síntesis de HA-Tet. (C) La reacción HA-Tet se configuró y se permitió reaccionar durante la noche seguida de (D) precipitación de HA-Tet en etanol. (E) Una vez purificado y secado, el HA-Tet fue rehidratado y liofilizado para producir (F) un producto seco de color rosa claro. (G) El análisis de RMN de protones muestra una modificación exitosa del 11% de las unidades de repetición de AH. Reimpreso de Anderson et al.12 con permiso de Elsevier. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Rehidratación de microgeles liofilizados para la fabricación de andamios MAP. (A) Proyección de intensidad máxima de lyo-μgels secos (barra de escala = 100 μm). (B) Después de la liofilización, se demuestra que la rehidratación de los lyo-μgels toma aproximadamente 20 min (barra de escala = 100 μm). (C) Los Lyo-μgels pueden rehidratarse a un % variable en peso de MAP para producir μgeles atascados (barra de escala = 100 μm). (D) El aumento del % en peso de MAP al rehidratar lyo-μgels altera la fracción de partículas en los andamios MAP, como se muestra en las rodajas Z individuales de andamios MAP y proyecciones de volumen (barra de escala = 100 μm). (E) Usando estos andamios wt% MAP definidos por el usuario, se pueden lograr fracciones de partículas únicas (NL = μgels no liofilizados). Se realizó un ANOVA unidireccional con Tukey HSD en las muestras (n = 3), con significación reportada en p < 0,05 (*), p < 0,01 (**), p < 0,005 (***) y p < 0,001 (****). Reimpreso de Anderson et al.12 con permiso de Elsevier. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
5.3D cultivo celular en andamios de mapas
- Preparar dispositivos de cultivo celular
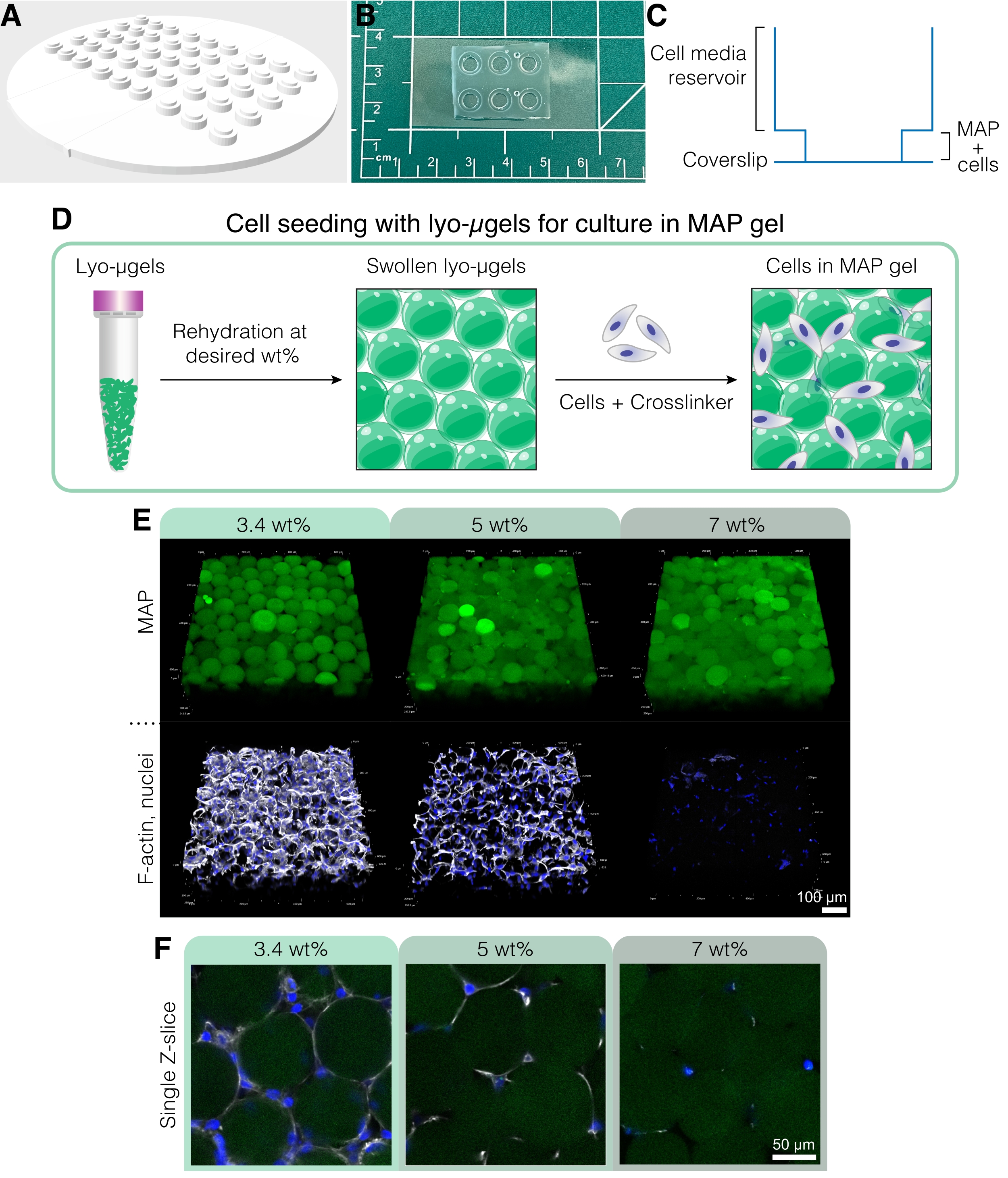
- Para crear un dispositivo de cultivo celular personalizado para estos experimentos (Figura 5A-C), use una impresora 3D para imprimir un molde negativo utilizando el archivo CAD que se encuentra en el Archivo de codificación suplementario 1.
NOTA: Las dimensiones del dispositivo de cultivo celular son las siguientes: 94,9 mm x 94,9 mm x 4,8 mm con una altura total del pozo de 2,6 mm. El diámetro de los pozos interiores y exteriores es de 4 mm y 6 mm, respectivamente. - Mezcle la base de elastómero de polidimetilsiloxano (PDMS) con el agente de curado en una proporción de 10:1 en masa. Vierta la mezcla de PDMS en una placa de Petri de plástico grande y desgasifique en un desecador durante aproximadamente 30 minutos o hasta que todas las burbujas hayan desaparecido.
- Una vez que todas las burbujas hayan desaparecido, coloque cuidadosamente el molde impreso en 3D en el PDMS para minimizar la formación de nuevas burbujas. Colocar en el horno a 60 °C durante al menos 2 h para curar el PDMS.
- Use un cuchillo o una hoja de afeitar para trazar suavemente alrededor del parámetro del dispositivo de cultivo y luego retire cuidadosamente el molde. Use un punzón de biopsia de 4 mm para extraer cualquier PDMS del fondo de los pocillos. Corte los dispositivos para que quepan en un cubreobjetos de vidrio.
NOTA: Los dispositivos de cultivo celular también se pueden unir a portaobjetos de vidrio, pero los cubreobjetos de vidrio mejoran las imágenes de la muestra. - Use cinta adhesiva para eliminar el polvo de la parte inferior de los dispositivos de cultivo. Coloque los cubreobjetos de vidrio limpios y los dispositivos de cultivo (de abajo hacia arriba) en una placa caliente a 135 °C durante al menos 15 minutos para eliminar la humedad.
- En una campana extractora, use una pistola de plasma corona en lo alto tanto en el cubreobjetos de vidrio como en la parte inferior del dispositivo durante 30 s, y luego una rápidamente las superficies tratadas. Aplique presión suavemente para asegurar un buen sellado entre el dispositivo de cultivo y el cubreobjetos de vidrio.
- Repita el paso 5.1.6 para todos los dispositivos y, a continuación, colóquelo en un horno a 60 °C durante la noche para asegurar la unión. Autoclave los dispositivos a esterilizar antes de su uso in vitro.
- Para crear un dispositivo de cultivo celular personalizado para estos experimentos (Figura 5A-C), use una impresora 3D para imprimir un molde negativo utilizando el archivo CAD que se encuentra en el Archivo de codificación suplementario 1.
- Cultivo celular en andamios MAP
- Prepare los componentes del andamio MAP (es decir, μgels, HA-Tet, volumen de medios) en función de la fracción de partículas deseada (consulte la Figura 4D-E). Pesar los lyo-μgels en una campana estéril y reconstituir en el 84% del volumen final de MAP de los medios celulares en función del % de peso elegido. Deje que los μgels se hinchen durante aproximadamente 20 minutos.
NOTA: Estos métodos requieren que el usuario pese el producto de lio-microgel para la rehidratación. Para masas pequeñas (1 mg o menos), se sugiere pesar primero el criotubo antes de agregar y liofilizar μgels, y luego volver a pesar el tubo después de la liofilización para determinar la masa del producto para minimizar el error. - Disuelva el HA-Tet en medios celulares en un 16% del volumen final de MAP.
NOTA: Los siguientes pasos para preparar las celdas para la siembra en andamios MAP se pueden modificar dependiendo del tipo de celda que se utilice. En este protocolo, se cultivaron células mesenquimales de ratón D1 en el Medio de Águila Modificada (DMEM) de Dulbecco suplementado con 1% de penicilina-estreptomicina (pen-estreptococo) y 10% de suero fetal bovino (FBS) (ver Tabla de materiales). Se deben seguir los protocolos estándar de cultivo celular adherente para estas células, manteniéndose los cultivos a 37 °C y 5% deCO2 en vasos de cultivo tratados con cultivo de tejidos. - Una vez que las células mesenquimales del ratón D1 hayan alcanzado una confluencia del 70% -80%, aspire los medios y lave las células con 1x PBS. Levante las células agregando suficiente volumen de tripsina-EDTA al 1% para cubrir la superficie del vaso de cultivo de tejidos. Incubar a 37 °C durante 1-3 min, y luego apagar la tripsinización añadiendo medios DMEM suplementados con pen-estreptococo al 1% y FBS al 10% a 2 veces el volumen de tripsina-EDTA.
- Centrifugar la suspensión celular a 100 x g durante 5 min a temperatura ambiente para granular las células. Aspirar el medio sobrenadante y resuspender las células en medios DMEM de 1 ml suplementados con 1% pen-estreptococo y 10% FBS.
- Asegúrese de que la suspensión celular esté bien mezclada y luego transfiera 20 μL a un nuevo tubo de microcentrífuga. Añadir 20 μL de solución azul de tripano y mezclar bien. Use 20 μL de esta mezcla para contar las células usando un hemocitómetro o un contador celular automatizado con portaobjetos de cámara de conteo celular.
- Transfiera el número de células necesarias para sembrar 10.000 células/μL MAP a un nuevo tubo de microcentrífuga. Centrifugar a 100 x g durante 5 min a temperatura ambiente para granular las células. Aspire cuidadosamente el medio sobrenadante de la bolita celular sin aspirar las células.
- Añadir los μgels y la reticulación al pellet de la célula con una pipeta de desplazamiento. Mezclar bien con una pipeta de desplazamiento y, a continuación, sembrar 10 μL de la mezcla por pocillo. Al emplatar, pipetear con movimientos circulares para distribuir uniformemente la mezcla en el pozo.
- Deje que los μgels se recojan a 37 °C en la incubadora de células durante 25 minutos antes de agregar medios celulares para llenar los pocillos (~ 50 μL de medios por pocillo). Mantenga las referencias culturales 3D a 37 °C y cambie los soportes según sea necesario. Para evitar aspirar el andamio al cambiar de medio, estabilice la punta de la pipeta a lo largo de la cresta del pocillo superior.
NOTA: Al agregar o quitar líquido de los pocillos de cultivo, apoye el extremo de la punta de la pipeta en la repisa sobre el andamio MAP para minimizar la posibilidad de interrumpir o aspirar el andamio del pozo. - En los momentos deseados, fijar las muestras retirando el medio y añadiendo 50 μL de paraformaldehído al 4% por pocillo durante 30 min a temperatura ambiente. Lave las muestras 3 veces con 50 μL de 1x PBS o tampón preferido. En este punto del protocolo, se pueden seguir métodos estándar para la inmunofluorescencia o la tinción de fluorescencia, utilizando 50 μL por pocillo como el volumen de trabajo.
NOTA: Estos métodos de fijación y tinción celular describen específicamente el uso de tinciones fluorescentes; sin embargo, la inmunotinción con conjugaciones de anticuerpos primarios y/o secundarios también se puede realizar en estos andamios siguiendo las instrucciones del fabricante utilizando 50 μL como volumen de trabajo por pocillo. - Imágenes de células en andamios MAP en un microscopio confocal utilizando un objetivo 20x y obtienen una pila Z que atraviesa 200-250 μm en la dirección Z con un tamaño de paso de 2,5 μm. Un ejemplo de tinción fluorescente con DAPI (tinción nuclear diluida 1:1000 en Triton-X al 0,15% en 1x PBS) y faloidina-647 (tinción de actina F diluida 1:40 en Triton-X al 0,15% en 1x PBS) se muestra en la Figura 5E, F con células D1 fijas cultivadas en andamios MAP durante 3 días.
NOTA: El tratamiento con plasma de las superficies de vidrio da como resultado un aumento de la hidrofilicidad, que se ha demostrado que mejora la adhesión celular. Es probable que se observe que las células se extienden a lo largo del fondo de los pocillos de cultivo celular, pero no deben incluirse en los recuentos celulares o en la cuantificación del volumen celular para evaluar la respuesta celular en andamios MAP.
- Prepare los componentes del andamio MAP (es decir, μgels, HA-Tet, volumen de medios) en función de la fracción de partículas deseada (consulte la Figura 4D-E). Pesar los lyo-μgels en una campana estéril y reconstituir en el 84% del volumen final de MAP de los medios celulares en función del % de peso elegido. Deje que los μgels se hinchen durante aproximadamente 20 minutos.

Figura 5: Cultivo celular en andamios MAP. (A) El molde para crear pozos de cultivo celular se puede imprimir en 3D y fundir con PDMS. Todo el molde tiene 95 mm de diámetro, los pozos grandes tienen 6 mm de diámetro y los pozos interiores pequeños tienen 4 mm de diámetro. (B) Una vez fundidos con PDMS, los dispositivos de cultivo celular se unen con plasma a cubreobjetos para mejorar las capacidades de microscopía. (C) La sección transversal de un pocillo de cultivo celular representa el reservorio para medios celulares (~50 μL) y un reservorio más pequeño para sembrar andamios MAP con células (~10 μL). (D) El proceso de siembra de células en andamios MAP se basa primero en la rehidratación de lyo-μgels al peso deseado por el usuario, seguido de la mezcla con células y el reticulante para interconectar los μgels. (E) Las células pueden encapsularse en andamios MAP (verde) con % de MAP variado. Las imágenes representativas son del día 5 del cultivo celular D1 en andamios MAP (barra de escala = 100 μm). (F) Los cortes Z individuales muestran diferencias en el crecimiento celular en andamios que comprenden diferentes % en peso de MAP (barra de escala = 50 μm). Reimpreso de Anderson et al.12 con permiso de Elsevier. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Resultados
El objetivo de este protocolo es demostrar la preparación de andamios de partículas recocidas microporosas (MAP) con un esquema de reticulación bio-ortogonal, así como fracciones de partículas controladas para cultivo celular 3D. Primero, HA se modificó con grupos colgantes de norborneno para ser utilizados tanto en la formación de microgel como en la interconexión para formar andamios MAP. Usando estos métodos, aproximadamente el 31% de las unidades de repetición de HA se modificaron con éxito con un mango fu...
Discusión
Se ha demostrado que la producción microfluídica de microgeles HA-NB genera microgeles con un rango de distribución de tamaño más estrecho que la producción por lotes de emulsiones 3,9. Los microgeles descritos en este protocolo se formularon utilizando un reticulante MMP (Ac-GCRDGPQGIWGQDRCG-NH2) para apoyar la degradación del material. Sin embargo, los microgeles HA-NB también pueden ser reticulados utilizando un enlazador de di-tiol alterna...
Divulgaciones
ARA y TS han presentado una patente provisional sobre esta tecnología.
Agradecimientos
Los autores desean agradecer a los Institutos Nacionales de Salud, los Institutos Nacionales de Trastornos Neurológicos y Accidentes Cerebrovasculares (1R01NS112940, 1R01NS079691, R01NS094599) y el Instituto Nacional de Alergias y Enfermedades Infecciosas (1R01AI152568). Este trabajo se realizó en parte en la Instalación de Instrumentación de Materiales Compartidos de la Universidad de Duke (SMIF), miembro de la Red de Nanotecnología del Triángulo de Investigación de Carolina del Norte (RTNN), que cuenta con el apoyo de la Fundación Nacional de Ciencias (número de premio ECCS-2025064) como parte de la Infraestructura Nacional Coordinada de Nanotecnología (NNCI). Los autores desean agradecer al ex postdoctorado del laboratorio, el Dr. Lucas Schirmer, así como a Ethan Nicklow por su ayuda en la generación del dispositivo impreso en 3D para experimentos de cultivo celular.
Materiales
| Name | Company | Catalog Number | Comments |
| 1 mL Luer-Lok syringe sterile, single use, polycarbonate | BD | 309628 | |
| 5 mL Luer-Lok syringe sterile, single use, polycarbonate | BD | 309646 | |
| Alexa Fluor 488 C5 maleimide | Invitrogen | A10254 | For synthesis of fluorescently-labeled tetrazine |
| Alexa Fluor 647 Phalloidin | Invitrogen | A22287 | For staining cell culture samples |
| Aluminum foil | VWR | 89107-726 | |
| Biopsy punch with plunger, 1.0 mm | Integra Miltex | 69031-01 | |
| Biopsy punch, 4 mm | Integra Miltex | 33-34 | |
| Blunt needle, 23 G 0.5", Non-Sterile, Capped | SAI Infusion Technologies | B23-50 | |
| Bottle-top vacuum filter, 0.22 μm | Corning | CLS430521 | |
| Calcium chloride | VWR | 1B1110 | For microgel washing buffer |
| Capillary-piston assemblies for positive-displacement pipettes, 1000 μL max. volume | Rainin | 17008609 | |
| Capillary-piston assemblies for positive-displacement pipettes, 25 μL max. volume | Rainin | 17008605 | |
| Capillary-piston assemblies for positive-displacement pipettes, 250 μL max. volume | Rainin | 17008608 | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10228 | |
| Countess II FL Automated Cell Counter | Invitrogen | AMQAF1000 | |
| Centrifuge tube, 15 mL | CELLTREAT | 667015B | |
| Centrifuge tube, 50 mL | CELLTREAT | 229421 | |
| Chloroform, ACS grade, Glass Bottle | Stellar Scientific | CP-C7304 | For synthesis of fluorescently-labeled tetrazine |
| Corona plasma gun, BD-10A High Frequency Generator | ETP | 11011 | |
| CryoTube Vials, Polypropylene, Internal Thread with Screw Cap | Nunc | 368632 | |
| D1 mouse mesenchymal cells | ATCC | CRL-12424 | Example cell line for culture in MAP gels |
| DAPI | Sigma-Aldrich | D9542 | For staining cell culture samples |
| Deuterium oxide, 99.9 atom% D | Sigma-Aldrich | 151882 | For NMR spectroscopy |
| Dialysis tubing, regenerated cellulose membrane, 12-14 kDa molecular weight cut-off | Spectra/Por | 132703 | For purifying HA-NB and HA-Tet |
| Diethyl ether | VWR | BDH1121-4LPC | For synthesis of fluorescently-labeled tetrazine |
| Dimethylformamide | Sigma-Aldrich | 277056 | For synthesis of fluorescently-labeled tetrazine |
| 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM) | TCI-Chemicals | D2919 | For modifying HA |
| Dithiothreitol (DTT) | Thermo Scientific | R0861 | Non-degradable dithiol linker (substitute for MMP-cleavable peptide) |
| Dulbecco's Modified Eagle's Medium (DMEM), high glucose, w/ 4500 mg/L glucose, L-glutamine, sodium pyruvate, and sodium bicarbonate, liquid, sterile-filtered, suitable for cell culture | Sigma-Aldrich | D6429-500ML | For D1 cell culture |
| EMS Paraformaldehyde, Granular | VWR | 100504-162 | For making 4% PFA |
| Ethanol absolute (200 proof) | KOPTEC | 89234-850 | |
| Fetal bovine serum (FBS) | ATCC | 30-2020 | For D1 cell culture |
| Heating Plate | Kopf Instruments | HP-4M | |
| Hemacytometer with coverglass | Daigger Scientific | EF16034F | |
| 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) | Sigma-Aldrich | H3375 | |
| Sodium hyaluronate, 79 kDa average molecular weight, produced in bacteria Streptococcus zooepidemicus, pharmaceutical grade, microbial contamination <100 CFU/g, bacterial endotoxins <0.050 IU/mg | Contipro | N/A | 79 kDa average molecular weight was used for HA-Tet synthesis, but these methods could be adapted for other molecular weights. |
| IMARIS Essentials software package | Oxford Instruments | N/A | Microscopy image analysis software |
| Infusion pump, dual syringe | Chemyx | N/A | |
| Kimwipe | Kimberly-Clark | 34120 | |
| Laboratory stand with support lab clamp | Geyer | 212100 | |
| Liquid nitrogen | Airgas | NI 180LT22 | |
| Lithium Phenyl(2,4,6-trimethylbenzoyl)phosphinate | TCI-Chemicals | L0290 | |
| Lyophilizer | Labconco | N/A | Labconco FreeZone 6 plus has been discontinued, but other lab grade console freeze dryers could be used for this protocol. |
| Methyltetrazine-PEG4-maleimide | Kerafast | FCC210 | For synthesis of fluorescently-labeled tetrazine |
| 2-(4-Morpholino)ethane Sulfonic Acid (MES) | Fisher Scientific | BP300-100 | For modifying HA |
| Micro cover glass, 24 x 60 mm No. 1 | VWR | 48393-106 | |
| Microfluidic device SU8 master wafer | FlowJem | Custom design made either in-house in clean room or outsourced | |
| Mineral oil, heavy | Sigma-Aldrich | 330760 | |
| MMP-cleavable dithiol crosslinker peptide (Ac-GCRDGPQGIWGQDRCG-NH2) | GenScript | N/A | |
| 5-Norbornene-2-methylamine | TCI-Chemicals | 95-10-3 | For HA-NB synthesis |
| Packing tape | Scotch | 3M 1426 | |
| Parafilm | Bemis | PM996 | |
| PEG(thiol)2 | JenKem Technology USA | A4001-1 | For synthesis of fluorescently-labeled tetrazine |
| Penicillin-Streptomycin, 10,000 units/mL | Thermo Fisher Scientific | 15140122 | For D1 cell culture |
| Petri dish, polystyrene, disposable, Dia. x H=150 x 15 mm | Corning | 351058 | |
| Pluronic F-127 | Sigma-Aldrich | P2443 | For washing HMPs |
| Phosphate buffered saline (PBS) 1x | Gibco | 10010023 | |
| RainX water repellent glass treatment | Grainger | 465D20 | Synthetic hydrophobic treatment solution for microfluidic device treatment |
| RGD peptide (Ac-RGDSPGERCG-NH2) | GenScript | N/A | |
| Rubber bands | Staples | 112417 | |
| Sodium chloride | Chem-Impex | 30070 | For dialysis |
| Span 80 for synthesis | Sigma-Aldrich | 1338-43-8 | |
| Sylgard 184 Silicone Elastomer | Electron Microscopy Science | 4019862 | polydimethylsiloxane (PDMS) elastomer for making microfluidic devices and tissue culture devices |
| Syringe filter, Whatman Uniflo, 0.2 μm PES, 13 mm diameter | Cytvia | 09-928-066 | |
| Tetraview LCD digital microscope | Celestron | 44347 | |
| Tetrazine-amine HCl salt | Chem-Impex | 35098 | For HA-Tet synthesis |
| Triethylamine | Sigma-Aldrich | 471283 | For synthesis of fluorescently-labeled tetrazine |
| Tris(2-carboxyethyl)phosphine (TCEP) | Millipore Sigma | 51805-45-9 | |
| Triton X-100 | VWR | 97063-864 | |
| Trypan blue solution, 0.4% | Thermo Fisher Scientific | 15250061 | |
| Trypsin EDTA (0.25%), Phenol red | Fisher Scientific | 25-200-056 | For lifting adherent cells to seed in MAP gels |
| Tygon ND-100-80 Non-DEHP Medical Tubing, Needle Gauge=23, Wall Thickness=0.020 in, Internal diameter = 0.020, Outer diameter = 0.060 in | Thomas Scientific | 1204G82 | |
| UV curing system controller, LX500 LED | OmniCure | 010-00369R | |
| UV curing head, LED spot UV | OmniCure | N/A | |
| UV light meter, Traceable | VWR | 61161-386 | |
| Vacuum dessicator | Bel-Art | 08-594-15C | |
| X-Acto Z Series Precision Utility Knife | Elmer's | XZ3601W |
Referencias
- Griffin, D. R., Weaver, W. M., Scumpia, P. O., Di Carlo, D., Segura, T. Accelerated wound healing by injectable microporous gel scaffolds assembled from annealed building blocks. Nature Materials. 14 (7), 737-744 (2015).
- Daly, A. C., Riley, L., Segura, T., Burdick, J. A. Hydrogel microparticles for biomedical applications. Nature Reviews Materials. 5 (1), 20-43 (2020).
- Darling, N. J., et al. Click by click Microporous Annealed Particle (MAP) scaffolds. Advanced Healthcare Materials. 9 (10), 1901391 (2020).
- Truong, N. F., et al. Microporous annealed particle hydrogel stiffness, void space size, and adhesion properties impact cell proliferation, cell spreading, and gene transfer. Acta Biomaterialia. 94, 160-172 (2020).
- Pfaff, B. N., et al. Selective and improved photoannealing of Microporous Annealed Particle (MAP) scaffolds. ACS Biomaterials Science & Engineering. 7 (2), 422-427 (2021).
- Sideris, E., et al. Particle hydrogels based on hyaluronic acid building blocks. ACS Biomaterials Science & Engineering. 2 (11), 2034-2041 (2016).
- Caldwell, A. S., Campbell, G. T., Shekiro, K. M. T., Anseth, K. S. Clickable microgel scaffolds as platforms for 3D cell encapsulation. Advanced Healthcare Materials. 6 (15), 1700254 (2017).
- Qazi, T. H., et al. Anisotropic rod-shaped particles influence injectable granular hydrogel properties and cell invasion. Advanced Materials. 34 (12), 2109194 (2022).
- Wilson, K. L., et al. Stoichiometric post modification of hydrogel microparticles dictates neural stem cell fate in microporous annealed particle scaffolds. Advanced Materials. 34 (33), 2201921 (2022).
- Muir, V. G., Qazi, T. H., Shan, J., Groll, J., Burdick, J. A. Influence of microgel fabrication technique on granular hydrogel properties. ACS Biomaterials Science & Engineering. 7 (9), 4269-4281 (2021).
- Highley, C. B., Song, K. H., Daly, A. C., Burdick, J. A. Jammed microgel inks for 3D printing applications. Advanced Science. 6 (1), 1801076 (2018).
- Anderson, A. R., Nicklow, E., Segura, T. Particle fraction as a bioactive cue in granular scaffolds. Acta Biomaterialia. 150, 111-127 (2022).
- Pruett, L., Ellis, R., McDermott, M., Roosa, C., Griffin, D. R. Spatially heterogeneous epidermal growth factor release from microporous annealed particle (MAP) hydrogel for improved wound closure. Journal of Materials Chemistry B. 9 (35), 7132-7139 (2021).
- Sheikhi, A., et al. Microengineered emulsion-to-powder technology for the high-fidelity preservation of molecular, colloidal, and bulk properties of hydrogel suspensions. ACS Applied Polymer Materials. 1 (8), 1935-1941 (2019).
- Brower, K., White, A. K., Fordyce, P. M. Multi-step variable height photolithography for valved multilayer microfluidic devices. Journal of Visualized Experiments. (119), e55276 (2017).
- JoVE. Nuclear Magnetic Resonance (NMR) Spectroscopy. JoVE Science Education Database. Organic Chemistry. JoVE. , (2022).
- Roosa, C., et al. Microfluidic synthesis of microgel building blocks for microporous annealed particle scaffold. Journal of Visualized Experiments. (184), e64119 (2022).
- Zhang, H., Dicker, K. T., Xu, X., Jia, X., Fox, J. M. Interfacial bioorthogonal crosslinking. ACS Macro Letters. 3 (8), 727-731 (2014).
- Welzel, P. B., et al. Cryogel micromechanics unraveled by atomic force microscopy-based nanoindentation. Advanced Healthcare Materials. 3 (11), 1849-1853 (2014).
- Plieva, F., Huiting, X., Galaev, I. Y., Bergenståhl, B., Mattiasson, B. Macroporous elastic polyacrylamide gels prepared at subzero temperatures: control of porous structure. Journal of Materials Chemistry. 16 (41), 4065-4073 (2006).
- Rommel, D., et al. Functionalized microgel rods interlinked into soft macroporous structures for 3D cell culture. Advanced Science. 9 (10), 2103554 (2022).
- Kurt, E., Segura, T. Nucleic acid delivery from granular hydrogels. Advanced Healthcare Materials. 11 (3), 2101867 (2021).
- Isaac, A., et al. Microporous bio-orthogonally annealed particle hydrogels for tissue engineering and regenerative medicine. ACS Biomaterials Science & Engineering. 5 (12), 6395-6404 (2019).
- Truong, N. F., Lesher-Pérez, S. C., Kurt, E., Segura, T. Pathways governing polyethylenimine polyplex transfection in Microporous Annealed Particle scaffolds. Bioconjugate Chemistry. 30 (2), 476-486 (2019).
- Koh, J., et al. Enhanced in vivo delivery of stem cells using microporous annealed particle scaffolds. Small. 15 (39), 1903147 (2019).
- Li, F., et al. Cartilage tissue formation through assembly of microgels containing mesenchymal stem cells. Acta Biomaterialia. 77, 48-62 (2018).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados