
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Visualisation de l’ADN Réparation des protéines Interaction par immunofluorescence
Dans cet article
Résumé
Suite à des dommages à l’ADN, les cellules humaines activent des voies de réparation essentielles pour restaurer l’intégrité de leur génome. Ici, nous décrivons la méthode de l’immunofluorescence indirecte comme un moyen de détecter les protéines de réparation de l’ADN, d’analyser leur recrutement spatial et temporel, et d’aider à interroger l’interaction protéine-protéine sur les sites de dommages à l’ADN.
Résumé
Les cellules de mammifères sont constamment exposées à des produits chimiques, des radiations et des sous-produits métaboliques naturels, qui créent des types spécifiques d’insultes à l’ADN. Les agents génotoxiques peuvent endommager l’épine dorsale de l’ADN, la briser ou modifier la nature chimique des bases individuelles. Suite à l’insulte à l’ADN, les voies de réponse aux dommages à l’ADN (DDR) sont activées et les protéines impliquées dans la réparation sont recrutées. Une pléthore de facteurs sont impliqués dans la détection du type de dommages et l’activation de la réponse de réparation appropriée. L’incapacité d’activer et de recruter correctement des facteurs de DDR peut conduire à l’instabilité génomique, qui sous-tend de nombreuses pathologies humaines, y compris le cancer. Les études sur les protéines DDR peuvent fournir des informations sur la réponse aux médicaments génotoxiques et les mécanismes cellulaires de la résistance aux médicaments.
Il existe deux façons principales de visualiser les protéines in vivo : l’observationdirecte, en taguant la protéine d’intérêt avec une protéine fluorescente et en la suivant par imagerie vivante, ou l’immunofluorescence indirecte sur des échantillons fixes. Bien que la visualisation des protéines étiquetées fluorescentes permette une surveillance précise au fil du temps, le marquage direct dans le terminus N ou C peut interférer avec la localisation ou la fonction des protéines. L’observation des protéines dans leur version endogène non modifiée est préférable. Lorsque les protéines de réparation d’ADN sont recrutées à l’insulte de l’ADN, leur concentration augmente localement et elles forment des groupes, ou « foyers », qui peuvent être visualisés par immunofluorescence indirecte à l’aide d’anticorps spécifiques.
Bien que la détection des foyers protéiques ne fournisse pas de preuve définitive de l’interaction directe, la co-localisation des protéines dans les cellules indique qu’elles se regroupent jusqu’au site des dommages et peuvent éclairer la séquence des événements nécessaires à la formation complexe. Une analyse minutieuse des foyers se chevauchent dans les cellules exprimant des versions sauvages ou mutantes d’une protéine peut fournir des indices précieux sur des domaines fonctionnels importants pour la fonction de réparation de l’ADN. Enfin, la co-localisation des protéines indique des interactions directes possibles qui peuvent être vérifiées par co-immunoprécipation dans les cellules, ou retrait direct à l’aide de protéines purifiées.
Introduction
Les cellules humaines sont constamment exposées à une variété d’agents d’ADN endommageant de diverses origines. Les sources exogènes consistent principalement en l’exposition aux radiations, aux produits chimiques (y compris les agents chimiothérapeutiques et certains antibiotiques) et aux virus, tandis que les principales sources endogènes comprennent des erreurs dans la réplication de l’ADN et le stress oxydatif. Les effets directs de l’exposition génotoxique peuvent aller d’une base modifiée à une rupture potentiellement mortelle à double brin d’ADN (DSB), selon le stress et la dose d’exposition. En fin de compte, les dommages non réparés ou mal réparés à l’ADN peuvent entraîner l’accumulation de mutations, les réarrangements génomiques, l’instabilité du génome et éventuellement conduire à la carcinogenèse1. Les cellules de mammifères ont développé des voies complexes pour reconnaître des types spécifiques de dommages à l’ADN2,3 et les réparer en temps opportun, synchronisés avec la progression du cycle cellulaire.
Le rayonnement ionisant (IR) endommage la double hélice de l’ADN et crée des ruptures à double brin (DSB), l’une des formes les plus nocives de dommages à l’ADN. Le complexe MRN (MRE11, RAD50, NBS1) fonctionne comme un capteur d’ADN se termine et active la protéine kinase ataxie telangiectasia muté (ATM)4,5. Suite à l’activation initiale de l’ATM par les extrémités de l’ADN, ATM déclenche une cascade d’événements DDR sur le site de la pause, initiant avec un événement clé, la phosphorylation de la variante histone H2AX6. La phosphorylation H2AX sur les résidus S139 l’active en γH2AX, couvrant les régions jusqu’à des mégabases autour de la lésion de l’ADN6,7,8,9. Cet événement augmente l’accessibilité de l’ADN, conduisant au recrutement et à l’accumulation d’autres protéines de réparation d’ADN7. Étant donné que γH2AX est abondamment et spécifiquement induit par les DSB environnants, il peut être facilement visualisé à l’aide d’anticorps spécifiques, et est couramment utilisé comme marqueur de substitution pour les DSB dans le domaine de la réparation de l’ADN. Une fois la rupture signalée, les cellules activent leurs voies de réparation de l’ADN et traitent les dommages causés par l’ADN. La protéine MDC1 (mediator de la protéine de point de contrôle des dommages de l’ADN 1) lie directement γH2AX10, interagit avec atm11 et aussi avec NBS112,13. Il contribue à accroître la concentration du complexe MRN à l’ORD et à lancer une boucle de rétroaction positive aux guichets automatiques. γH2AX est rapidement enlevé une fois la rupture réparée, ce qui permet la surveillance du dégagement d’ORD. Suivie d’une microscopie, la diminution du γH2AX au fil du temps fournit une mesure indirecte des ruptures résiduelles et de l’efficacité de la réparation de l’ADN.
Les cellules eucaryotes peuvent réparer les DSB par plusieurs voies, les deux principales étant l’assemblage final non homologue (NHEJ) et la recombinaison homologue (HR) (Figure 1). NHEJ ligates essentiellement ADN double brin se termine sans l’utilisation de l’homologie étendue et fonctionne tout au long du cycle cellulaire14,15. Hr devient prédominant pendant les phases S et G2, et est autrement réprimé, car il nécessite une sœur chromatid comme un modèle homologue pour la réparation14,16. Le choix de la voie entre le NHEJ et le HR dépend non seulement de la proximité physique du chromatid sœur, mais aussi de l’extension de la résection d’extrémité de l’ADN17, qui inhibe le NHEJ.
La réparation d’ADN homologie-dépendante initie par la dégradation nucléolytique du brin de 5' des extrémités de rupture pour produire 3' queues d’ADN à un brin (ssDNA), un processus appelé résection de 5'-3'. Le complexe MRN initie la résection finale de l’ADN et la résection est traitée en combinaison avec BLM/EXO1 (protéine du syndrome de Bloom/exonucléase 1) ou BLM/DNA2 (réplication de l’ADN hélicase/nucléase dépendante de l’ATP)18,19,20,21,22. La résection finale de l’ADN est améliorée par le CtIP (protéine d’interaction CtBP) par son interaction directe avec le complexeMRN 23 et le recrutement de BRCA1 (protéine de susceptibilité de type 1 du cancer du sein)24,25. La protéine de réplication A (RPA) se lie rapidement à l’ADN SsDN exposée et est ensuite déplacée par la protéine recombinase RAD51 pour former un filament de nucléoprotéine qui catalyse la recherche homologue et l’invasion de brin26,27,28.
L’initiation de la résection est une étape critique pour le choix de la voie de réparation. Une fois la résection commencée, les extrémités d’ADN deviennent de pauvres substrats pour la liaison par l’hétérodimer Ku70/Ku80 (composant de la voie NHEJ) et les cellules sont engagées à HR17,29,30. L’hétérodimère Ku70/Ku80 se lie aux extrémités de l’ORD, recrutant des ADN-PKcs et p53 Binding Protein 1 (53BP1)29,30. 53BP1 agit comme un obstacle à la résection en G1, bloquant ainsi hr tout en faisant la promotion de NHEJ31,32, mais il est supprimé d’une manière BRCA1-dépendante dans la phase S, permettant par conséquent la résection de se produire33,34. Par conséquent, 53BP1 et BRCA1 jouent un rôle opposé dans la réparation de l’ORD, avec 53BP1 étant un facilitateur NHEJ tandis que BRCA1 agit permettant des pauses pour réparer par hr.
En laboratoire, la formation d’ORD peut être induite par le rayonnement ionisant (IR). Bien que cet exemple utilise une dose élevée de 4 Gy, 1 Gy et 2 Gy également créer une quantité significative de DSBs, adapté à l’analyse de la formation de foyers par des protéines abondantes. Il est important de noter que le type et la dose de rayonnement utilisé peuvent conduire à différentes lésions dans l’ADN et dans la cellule: tandis que l’IR induit DSBs, il peut également causer des ruptures de brin unique ou la modification de la base (voir35,36 pour une référence sur le transfert d’énergie linéaire irradiation (LET) et le type de dommages à l’ADN). Pour déterminer la cinétique de la formation de foyers induits par rayonnement ionisant (IRIF) et leur dégagement, qui indiquent la réparation des dommages et l’inversion du DDRactivé 8,9,37,38, la formation de foyers peut être surveillée à différents points de temps après rayonnement ionisant. Le moment de l’activation et le dégagement de toutes les protéines majeures de dommages d’ADN est connu39, et beaucoup sont étudiés en tant que marqueurs de substitution des événements principaux. Par exemple, pRPA, qui possède une forte affinité pour l’ADN SsDN est utilisé comme un substitut de la résection de rupture, protéines MRN (MRE11, RAD50, NBS1) et exonucleases peuvent être utilisés pour évaluer l’efficacité de la résection aussi. Bien que RAD51, BRCA1, BRCA2 (protéine de susceptibilité de type 2 du cancer du sein) et PALB2 (partenaire et localisateur de BRCA2) puissent être surveillés pour évaluer l’efficacité des RH, la présence des protéines Ku ou 53BP1, sont utilisés comme marqueurs de NHEJ (Figure 1).
Comme les protéines des machines de réparation de l’ADN se recrutent les unes les autres à la rupture et s’assemblent dans des super-complexes, l’ADN-protéine et les interactions protéines-protéines peuvent être déduites en suivant leur localisation individuelle au fil du temps et en analysant la co-localisation des protéines, telle que visualisée par des signaux qui se chevauchent dans la cellule40,41,42. Dans les lignées cellulaires, l’introduction de mutations ponctuelles ou la suppression dans des gènes spécifiques de réparation de l’ADN, soit par l’édition du génome, soit par surexpression de mutants à base de plasmide, permet d’enquêter sur des résidus spécifiques et leur rôle possible dans la reconnaissance des dommages à l’ADN (p. ex., co-localisation avec γH2AX) ou d’assemblage complexe (co-localisation avec une autre ou plusieurs protéines), ainsi que leur impact sur la réparation de l’ADN. Ici, nous utilisons l’immunofluorescence indirecte comme moyen d’étudier la formation et la résolution des DSB en suivant les foyers γH2AX au fil du temps. Nous présentons également un exemple de formation de foyers et d’analyse de co-localisation par un acteur majeur de la réparation de l’ORD : p53 Binding Protein 1 (53BP1)32. Comme mentionné précédemment, 53BP1 est considéré comme central pour le choix de la voie de réparation de l’ADN. Après l’accumulation de 53BP1 et sa co-localisation avec γH2AX fournit des informations précieuses sur la phase de cycle cellulaire, l’accumulation de dommages d’ADN, et la voie utilisée pour réparer les DSB. Le but de l’immunolocalisation indirecte est d’évaluer l’efficacité de la réparation des dommages à l’ADN dans les lignées cellulaires, à la suite de l’IR comme dans cette étude, ou après l’exposition à diverses contraintes dans les cellules, de croisement d’ADN au blocage de la fourche de réplication (une liste d’agents d’endommagement de l’ADN est fournie dans le tableau 1).

Figure 1 : Ruptures de ruptures à double brin d’ADN (DSB) voies de réparation.
La réparation d’ORD implique deux voies principales : la recombinaison homologue (HR, gauche) et l’assemblage non homologue (NHEJ, à droite). Après la pause, les protéines sont activées pour marquer la rupture (γH2AX), participer à la résection finale (MRN), enrober le ssDNA réséqué (pRPA), promouvoir la recombinaison (BRCA1, PALB2, BRCA2, RAD51) ou limiter la résection et promouvoir la SSDNA (pRPA), promouvoir la recombinaison (BRCA1, PALB2, BRCA2, RAD51) ou limiter la résection et promouvoir la resEJ (53BP1). D’autres protéines participent à la réparation des dommages, mais les protéines énumérées sont systématiquement suivies d’immunofluorescence indirecte. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
| Agent d’endommagement d’ADN | Mécanisme d’action | Dose recommandée |
| rayons γ/rayons X | Rayonnement Formation de ruptures à double brin avec certains effets cellulaires incontrôlés | 1-4 Gy |
| 36 Ions Ar | Rayonnement Formation de pauses à double brin | 270 keV/μm |
| α-particules | Rayonnement Formation de pauses à double brin | 116 keV/μm |
| Bleomycine | Inhibiteur de la synthèse de l’ADN | 0,4 à 2 μg/mL |
| Camptothécine | Inhibiteur de la topoisomérase I | 10-200 nM |
| Cisplatine | Agent alcalin (induisant des liens croisés intrastrand) | 0,25 à 2 μM |
| Doxorubicine | Agent intercalaire Inhibiteur de la topoisomerase II | 10-200 nM |
| Etoposide | Inhibiteur de la topoisomerase II | 10 μM |
| Hydroxyurea | Inhibiteur de la synthèse de l’ADN (par ribonucléotide reductase) | 10-200 μM |
| Méthane de méthyle | Agent alcalin | 0,25 à 2 mM |
| Mitomycine C | Agent alcalin | 0,25 à 2 μM |
| Lumière ultraviolette (UV) | Formation de dimers thymidine (générant une distorsion de la chaîne d’ADN) | 50-100 mJ/cm2 |
Tableau 1 : Agents génotoxiques. Exemples d’agents d’ADN nuisibles, leur mécanisme d’action et les dommages induits en fonction de la concentration de travail suggérée.
Protocole
1. Culture cellulaire HeLa
- Prétraiter les couvercles ronds en verre de 1 M HCl à 50 °C pendant 16-18 heures. Rincer avec ddH2O et conserver dans 100% EtOH.
- Préparer le milieu de culture cellulaire : ajouter 10 % (v/v) FBS au milieu DMEM.
- Cultiver 4,0 x 104 cellules/puits dans une plaque stérile de 12 puits avec une couverture en verre rond de 18 mm à 37 °C et 5 % de CO2 dans un incubateur humidifié. Développer les cellules à 80% de confluence (environ 24 heures).
2. Traitement cellulaire par rayonnement (IR)
- Pour induire la formation de ruptures à double brin, exposez les cellules à 4 Gy γ-irradiation (contrôle : Pas d’irradiation, t=0). Dans la cellule Gamma -40, cela correspond à 4,54 min, à 0,815 Gy par min.
- Incuber les cellules à 37 °C et 5 % de CO2 dans un incubateur humidifié pour la durée appropriée (points de temps choisis ici t=1, 2, 4, 16 h).
3. Extraction nucléaire et fixation cellulaire
- Préparer des solutions de stock : 0,2 M PIPES (pH 6,8), 5 M NaCl, 2 M de saccharose, 1 M MgCl2, 0,1 M EGTA (pH 8,0).
- Préparer un tampon d’extraction nucléaire (NEB): dissoudre 10 mM PIPES (pH 6.8), 100 mM NaCl, 300 mM saccharose, 3 mM MgCl2, 1 mM EGTA (pH 8.0) et 0,5% (v/v) Triton X-100 en ddH2O. Mix jusqu’à dissolution complète.
- Préparer 4% (v/v) paraformaldéhyde (PFA): dissoudre 10 mL de 16% PFA solution aqueuse dans 30 mL PBS. Mélanger jusqu’à dissolution complète.
- Au moment opportun (t=0, 1, 2, 4, 16 h), laver les cellules deux fois avec 1 mL de PBS. Retirez complètement PBS.
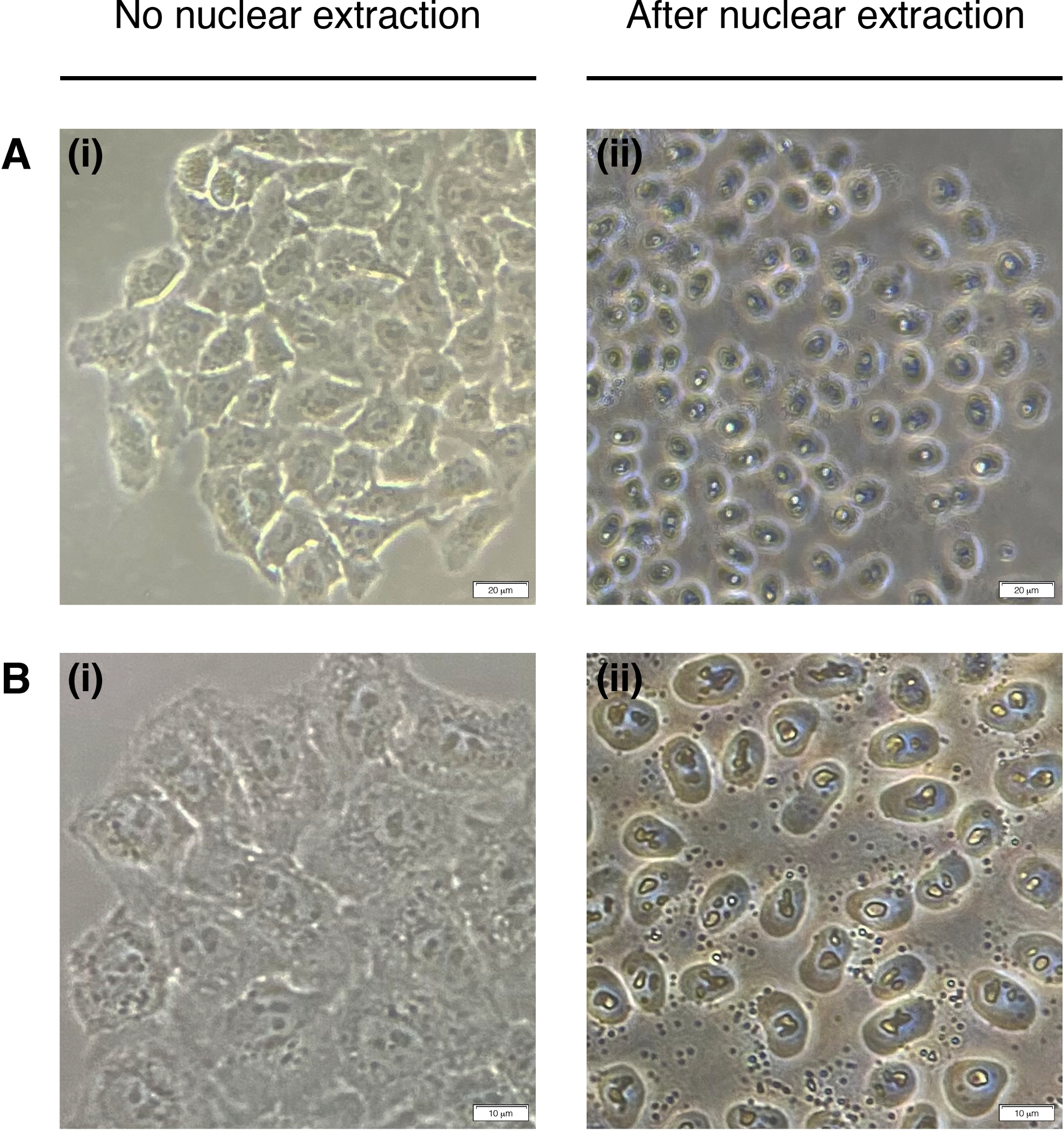
- Ajouter 200 μL d’ONÉ à chaque puits pour l’extraction des noyaux cellulaires (le cytoplasme est dégradé, il ne reste que le noyau) (figure 2). Incuber pendant 2 minutes à température ambiante et retirer complètement.
REMARQUE : Ne dépassez pas 2 minutes.

Figure 2 : Extraction nucléaire.
Images représentatives des cellules avant (à gauche) et post (droite) extraction nucléaire. Le cytoplasme doit être digéré, mais la structure nucléaire doit rester intacte après l’extraction (à droite). (A) grossissement 20x; barre d’échelle = 20 μm et (B) 40x grossissement; barre d’échelle = 10 μm. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
- Laver les cellules avec 1 mL de PBS. Retirez complètement PBS. Ajoutez PBS soigneusement, les cellules sont très fragiles à cette étape.
- Ajouter 200 μL de 4 % (v/v) PFA à chaque puits pour la fixation cellulaire. Incuber pendant 10 minutes à 4 °C. Retirez complètement la PFA.
- Ajouter 1 mL de PBS.
REMARQUE : Les cellules peuvent être stockées dans pbs à 4 °C.
4. Coloration d’immunofluorescence
- Préparer la solution de blocage : dissoudre 5 % de BSA (w/v) dans PBS et ajouter 0,3 % (v/v) Triton X-100. Mélanger jusqu’à dissolution complète.
- Préparer la tampon de dilution : dissoudre 1 % de BSA (w/v) dans pbs et ajouter 0,3 % (v/v) Triton X-100. Mélanger jusqu’à dissolution complète.
- Pour le blocage, ajoutez 200 μL de solution de blocage à chaque puits. Incuber pendant 2 heures à température ambiante ou 16-18 heures à 4 °C.
REMARQUE : Si l’anticorps de chèvre est utilisé, ajoutez 5 % de sérum de chèvre à la solution de blocage. - Diluer l’anticorps primaire dans le tampon de dilution (1:500; voir tableau 2 pour la liste des anticorps) et vortex jusqu’à ce qu’il soit bien mélangé.
REMARQUE : Si l’anticorps de chèvre est utilisé, ajoutez 1% de sérum de chèvre au tampon de dilution. - Dans une boîte d’humidité/incubation, adhérez à un morceau de parafilm. Ajouter 10 μL d’anticorps primaires (en une seule goutte). Aligner un bord du couvercle avec la goutte et baisser lentement sur le parafilm, pour que le liquide se répande tout au long (éviter les bulles si possible). Incuber pendant 2 heures à température ambiante.
- Laver les couvercles trois fois dans PBS pendant 1 minute.
- Diluer l’anticorps secondaire dans le tampon de dilution (concentration finale : 2 μg/mL) et vortex jusqu’à ce qu’il soit bien mélangé.
- Appliquer 10 μL d’anticorps secondaires tels que décrits pour les anticorps primaires. Incuber pendant 2 heures à température ambiante.
REMARQUE : Protégez-vous de la lumière.
| Anticorps | Société | Référence | Source |
| 53BP1 | Signalisation cellulaire | 4937 | Lapin |
| Anti-Mouse IgG H&L (Alexa Fluor 647) | Abcam | ab150103 | Âne |
| Anti-phospho-Histone H2A. X (Ser139), clone JBW301 | Millipore | 05-636 | Souris |
| Anti-Rabbit IgG H&L (Alexa Fluor 488) | Abcam | ab150081 | Chèvre |
Tableau 2 : Anticorps utilisés. Liste des anticorps utilisés dans cette étude.
- Laver les couvercles trois fois dans PBS pendant 1 minute.
- Laver les couvercles avec H2O pendant 1 minute.
- Contrestain DE L’ADN avec DAPI : appliquer 10 μL de 300 nM DAPI (comme décrit pour les anticorps), incuber pendant 30 minutes à température ambiante, puis monter sur une lame de verre avec un milieu de montage à base de glycérol. Sinon, ajoutez une goutte (10 μL) de support de montage antifade commercial contenant du DAPI sur une diapositive et appliquez un couvercle. Appuyez doucement sur le couvercle et retirez l’excès de liquide autour de lui avec une serviette en papier.
- Sceller les couvercles avec du vernis à ongles transparent et les laisser sécher pendant 20 minutes.
- Stockez les diapositives à 4 °C.
5. Acquisition d’images
- Placez une goutte d’huile d’immersion sur l’objectif 60x. Utilisez DAPI pour localiser les noyaux à travers un morceau d’œil.
- Pour l’acquisition d’images XYZ, logiciel d’acquisition ouverte et certains paramètres : Type de scanner : Galvano ; Mode scanner : Aller-retour; Taille de l’image: 512×512; Mode PMT: VBF; Moyenne pmt : cadre (4 fois); Analyse séquentielle PMT : ligne.
- Sélectionnez le colorant et les détecteurs :
Canal (CH1), Colorant (DAPI), Détecteur (SD1)
Canal (CH2), Colorant (Alexa Fluor 488), Détecteur (HSD3)
Canal (CH3), Colorant (Alexa Fluor 647), Détecteur (HSD4) - Sélectionnez ON dans « Z ».
- Ajustez l’image en direct. Appuyez sur le bouton En direct de la fenêtre En direct.
- Ajuster la mise au point et définir l’intensité laser (%), la sensibilité (HV), le gain et le décalage sur la fenêtre d’outils « PMT ».
- Ajuster l’intensité laser (%): pour la luminosité et le blanchiment. Plus l’intensité laser est élevée, plus le signal est fort, mais le spécimen se photoblera.
- Ajuster la sensibilité (HV): niveau de bruit. Plus le HV est élevé, plus le signal est fort, mais l’image sera bruyante si elle est trop haute.
REMARQUE : Maintenez toujours la tension constante. - Ajuster le décalage : niveau d’arrière-plan.
- Sélectionnez Démarrer/Fin (15 tranches) pour les piles Z.
- Ajuster la mise au point et définir l’intensité laser (%), la sensibilité (HV), le gain et le décalage sur la fenêtre d’outils « PMT ».
- Démarrer l’acquisition.
- Sélectionnez le dossier pour enregistrer des images. Appuyez sur le bouton Démarrer LSM pour commencer à acquérir l’image. Appuyez sur le bouton Série Done pour compléter l’acquisition d’image.
6. Analyse des données
- Pour l’analyse d’image, ouvrez le logiciel d’analyse.
- Appuyez sur la fenêtre de l’outil Batch, sélectionnez les images à analyser et sélectionnez le dossier de sortie.
- Appuyez sur la fenêtre de l’outil Analyse et sélectionnez Projection (affichez la projection d’intensité maximale à partir de 15 tranches).
- Sous Paramètre Entrée/Sortie, sélectionnez le lot créé.
- Appuyez sur Processus pour que les images soient traitées.
- Exporter des images sous forme de fichiers .tiff.
- Pour la quantification des foyers nucléaires, ouvrez CellProfiler.
- Ouvrez le pipeline de quantification γH2AX et 53BP1 foyers (voir Informations supplémentaires).
- Graphiques à l’aide d’un logiciel de table.
- Pour l’analyse de co-localisation, ouvrez CellProfiler.
- Ouvrez le pipeline colocalisation (voir Informations supplémentaires). Un fichier de feuille de calcul sera créé et enregistré à l’emplacement préféré. Toutefois, les graphiques eux-mêmes ne seront pas automatiquement enregistrés. Il est suggéré de prendre un instantané des fenêtres pour garder pour enregistrer les résultats.
- Graphiques à l’aide d’un logiciel de table.
Résultats
Le jour 2, ou 24 h après les cellules d’ensemencement sur les couvercles, les cellules ont subi une division et sont confluentes à 80%. Des baisses ou des mutations spécifiques dans les protéines de réparation de l’ADN peuvent augmenter le temps de doublement, ou sensibiliser les cellules au traitement génotoxique, et la densité d’ensemencement ainsi que les doses de traitement devraient être ajustées en conséquence. Pour déterminer les meilleures conditions de travail, le moment de la réponse aux domma...
Discussion
L’analyse du moment et de l’efficacité de la réparation des dommages à l’ADN par microscopie s’est avérée essentielle pour comprendre le fonctionnement de la machinerie de réparation de l’ADN, dans les cellules normales et dans les pathologies humaines telles que le cancer.
Le développement d’anticorps spécifiques qui permettent la détection de protéines activées dans leur version phosphorylé (tels que γH2AX, pRPA, pRAD50 et autres10,...
Déclarations de divulgation
Les auteurs n’ont rien à révéler.
Remerciements
Ce travail a été soutenu par une subvention de la San Antonio Area Foundation. Le Mays Cancer Center est soutenu par NCI cancer center support core grant P30 CA054174. Nous tenons à remercier Stephen Holloway pour son aide à l’approvisionnement des réactifs, et le laboratoire Sung pour fournir l’espace et la capacité de microscopie.
matériels
| Name | Company | Catalog Number | Comments |
| 16% (v/v) paraformaldehyde (PFA) aqueous solution | Electron Microscopy Sciences | 15710 | Microscopy quality of the PFA ensures best images. If using "home-made PFA", filter prior to use. |
| Bovine serum albumin (BSA) | Sigma-Aldrich | A3059 | Heat-shock fraction is recommended, to avoid precipitation/background. |
| Coverglass #1, 18 mm round (coverslips) | Neuvitro | NC0308920 | Coverslips need to be cleaned and sterilized prior using, with HCl or ethanol. |
| DMEM, High Glucose [(+) 4.5 g/L D-Glucose, (+) L-Glutamine, (-) Sodium Pyruvate] | Gibco | 11965118 | Adjust the growing media to the needs of cell line used. |
| DPBS, no calcium, no magnesium | Gibco | 14190144 | PBS for tissue culture. |
| Ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) | Research Products International | E57060 | Nuclear extraction buffer. |
| Fetal Bovine Serum (FBS) | Life Technologies | 104370028 | The quality of FBS can be assessed by testing gH2AX foci formation. If traces of genotoxic endotoxin are present in the batch, gH2AX will be positive in the absence of stress. |
| Magnesium chloride (MgCl2) | Research Products International | M24000 | Nuclear extraction buffer. |
| Piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) | Research Products International | P40150 | Nuclear extraction buffer. |
| SlowFade Diamond Antifade Mountant with DAPI | Invitrogen | S36973 | 300 nM DAPI with VECTASHIELD Antifade Mounting Medium can be used instead. |
| Sodium chloride (NaCl) | Research Products International | S23020 | Nuclear extraction buffer. |
| Sucrose | Research Products International | S24060 | Nuclear extraction buffer. |
| Superfrost Plus Microscope Slides | Fisherbrand | 1255015 | Polysine Slides can be used instead. |
| TC-Treated Multiple Well Plates, size 12 wells | Costar | 3513 | Seeding on coverslips is done in 12-wells plate. |
| Triton X-100 | AmericanBio | AB02025 | Nuclear extraction buffer. |
| TrypLE Express Enzyme (1X), No Phenol Red | Gibco | 12604021 | Trypsin-EDTA can be used instead. |
| Trypsin-EDTA (0.5%), No Phenol Red | Gibco | 15400054 | TrypLE can be used instead. |
Références
- Prakash, R., Zhang, Y., Feng, W., Jasin, M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harbor Perspectives in Biology. 7 (4), 016600 (2015).
- Jalan, M., Olsen, K. S., Powell, S. N. Emerging Roles of RAD52 in Genome Maintenance. Cancers (Basel). 11 (7), (2019).
- Oh, J., Symington, L. S. Role of the Mre11 Complex in Preserving Genome Integrity. Genes (Basel). 9 (12), (2018).
- Uziel, T., et al. Requirement of the MRN complex for ATM activation by DNA damage. The EMBO Journal. 22 (20), 5612-5621 (2003).
- Lee, J. H., Paull, T. T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 308 (5721), 551-554 (2005).
- Rogakou, E. P., Pilch, D. R., Orr, A. H., Ivanova, V. S., Bonner, W. M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. Journal of Biological Chemistry. 273, 5858-5868 (1998).
- Kinner, A., Wu, W., Staudt, C., Iliakis, G. γ-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Research. 36 (17), 5678-5694 (2008).
- Martin, O. A., Pilch, D. R., Redon, C., Bonner, W. M. Histone H2AX in DNA damage repair. Cancer Biology & Therapy. 2 (3), 233-235 (2003).
- Rogakou, E. P., Boon, C., Redon, C., Bonner, W. M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. Journal of Cell Biology. 146 (5), 905-916 (1999).
- Stucki, M., et al. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 123 (7), 1213-1226 (2005).
- Lou, Z., et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Molecular Cell. 21 (2), 187-200 (2006).
- Chapman, J. R., Jackson, S. P. Phospho-dependent interaction between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Reports. 9 (8), 795-801 (2008).
- Melander, F., et al. Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. Journal of Cell Biology. 181 (2), 213-226 (2008).
- Branzei, D., Foiani, M. Regulation of DNA repair throughout the cell cycle. Nature Review. Molecular Cell Biology. 9 (4), 297-308 (2008).
- Chiruvella, K. K., Liang, Z., Wilson, T. E. Repair of double-strand breaks by end joining. Cold Spring Harbor Perspectives in Biology. 5 (5), 012757 (2013).
- Mehta, A., Haber, J. E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harbor Perspectives in Biology. 6 (9), 016428 (2014).
- Symington, L. S., Gautier, J. Double-strand break end resection and repair pathway choice. Annual Review of Genetics. 45, 247-271 (2011).
- Huertas, P. DNA resection in eukaryotes: deciding how to fix the break. Nature Structural & Molecular Biology. 17 (1), 11-16 (2010).
- Nimonkar, A. V., et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes & Development. 25 (4), 350-362 (2011).
- Garcia, V., Phelps, S. E. L., Gray, S., Neale, M. J. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature. 479 (7372), 241-244 (2011).
- Sturzenegger, A., et al. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. Journal of Biological Chemistry. 289 (39), 27314-27326 (2014).
- Daley, J. M., Niu, H., Miller, A. S., Sung, P. Biochemical mechanism of DSB end resection and its regulation. DNA Repair. 32, 66-74 (2015).
- Sartori, A. A., et al. Human CtIP promotes DNA end resection. Nature. 450 (7169), 509-514 (2007).
- Chen, L., Nievera, C. J., Lee, A. Y. L., Wu, X. Cell cycle-dependent complex formation of BRCA1-CtIP-MRN is important for DNA double-strand break repair. Journal of Biological Chemistry. 283, 7713-7720 (2008).
- Yun, M. H., Hiom, K. CtIP-BRCA1 modulates the choice of DNA double-strand break repair pathway throughout the cell cycle. Nature. 459 (7245), 460-463 (2009).
- Sung, P., Klein, H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nature Review. Molecular Cell Biology. 7, 739-750 (2006).
- San Filippo, J., Sung, P., Klein, H. Mechanisms of eukaryotic homologous recombination. Annual Review of Biochemistry. 77, 229-257 (2008).
- Jasin, M., Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harbor Perspectives in Biology. 5 (11), 012740 (2013).
- Dynan, W. S., Yoo, S. Interaction of Ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Research. 26 (7), 1551-1559 (1998).
- Lieber, M. R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annual Review of Biochemistry. 79, 181-211 (2010).
- Cejka, P. DNA end resection: nucleases team up with the right partners to initiate homologous recombination. Journal of Biological Chemistry. 290 (38), 22931-22938 (2015).
- Mirman, Z., de Lange, T. 53BP1: a DSB escort. Genes & Development. 34, 7-23 (2020).
- Cao, L., et al. A selective requirement for 53BP1 in the biological response to genomic instability induces by BRCA1 deficiency. Molecular Cell. 35 (4), 534-541 (2009).
- Zimmermann, M., de Lange, T. 53BP1: Pro choice in DNA repair. Trends in Cell Biology. 24 (2), 108-117 (2014).
- Mavragani, I. V., Nikitaki, Z., Kalospyros, S. A., Georgakilas, A. G. Ionizing Radiation and Complex DNA Damage: From Prediction to Detection Challenges and Biological Significance. Cancers (Basel). 11 (11), (2019).
- Nikitaki, Z., et al. Measurement of complex DNA damage induction and repair in human cellular systems after exposure to ionizing radiations of varying linear energy transfer (LET). Free Radical Research. 50, 64-78 (2016).
- Redon, C., et al. Histone H2A variants H2AX and H2AZ. Current Opinion in Genetics & Development. 12 (2), 162-169 (2002).
- Fernandez-Capetillo, O., Lee, A., Nussenzweig, M., Nussenzweig, A. H2AX: the histone guardian of the genome. DNA Repair. 3 (8-9), 959-967 (2004).
- Paull, T. T., et al. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Current Biology. 10 (15), 886-895 (2000).
- Sy, S. M. H., Huen, M. S. Y., Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proceedings of the National Academy of Sciences. 106 (17), 7155-7160 (2009).
- Buisson, R., Masson, J. Y. PALB2 self-interaction controls homologous recombination. Nucleic Acids Research. 40 (20), 10312-10323 (2012).
- Belotserkovskaya, R., et al. PALB2 chromatin recruitment restores homologous recombination in BRCA1-deficient cells depleted of 53BP1. Nature Communications. 11 (1), 819 (2020).
- Betts, J. A., et al. Long noncoding RNAs CUPID1 and CUPID2 mediate breast cancer risk at 11q13 by modulating the response to DNA damage. American Journal of Human Genetics. 101 (2), 255-266 (2017).
- Dray, E., et al. Molecular basis for enhancement of the meiotic DMC1 recombinase by RAD51 associated protein 1 (RAD51AP1). Proceedings of the National Academy of Sciences. 108 (9), 3560-3565 (2011).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.