Immunofluorescence multiplex combinée à l’analyse d’images spatiales pour l’évaluation clinique et biologique du microenvironnement tumoral
Dans cet article
Résumé
Dans cet article, un protocole d’immunofluorescence multiplex (mIF) manuelle d’amplification du signal tyramide (TSA) combiné à l’analyse d’images et à l’analyse spatiale est décrit. Ce protocole peut être utilisé avec des coupes de paraffine fixées au formol (FFPE) pour la coloration de deux à six antigènes par lame selon le scanner de lames disponible en laboratoire.
Résumé
Le microenvironnement tumoral (TME) est composé d’une pléthore de différents types de cellules, telles que les cellules immunitaires cytotoxiques et les cellules immunomodulatrices. Selon sa composition et les interactions entre les cellules cancéreuses et les cellules péritumorales, le TME peut affecter la progression du cancer. La caractérisation des tumeurs et de leur microenvironnement complexe pourrait améliorer la compréhension des maladies cancéreuses et pourrait aider les scientifiques et les cliniciens à découvrir de nouveaux biomarqueurs.
Nous avons récemment développé plusieurs panels d’immunofluorescence multiplex (mIF) basés sur l’amplification du signal tyramide (TSA) pour la caractérisation du TME dans le cancer colorectal, le carcinome épidermoïde de la tête et du cou, le mélanome et le cancer du poumon. Une fois la coloration et le balayage des panneaux correspondants terminés, les échantillons sont analysés sur un logiciel d’analyse d’images. La position spatiale et la coloration de chaque cellule sont ensuite exportées de ce logiciel de quantification vers R. Nous avons développé des scripts R qui nous permettent non seulement d’analyser la densité de chaque type de cellule dans plusieurs compartiments tumoraux (par exemple, le centre de la tumeur, la marge de la tumeur et le stroma), mais aussi d’effectuer des analyses à distance entre différents types de cellules.
Ce flux de travail particulier ajoute une dimension spatiale à l’analyse de densité classique déjà effectuée régulièrement pour plusieurs marqueurs. L’analyse mIF pourrait permettre aux scientifiques de mieux comprendre l’interaction complexe entre les cellules cancéreuses et le TME et de découvrir de nouveaux biomarqueurs prédictifs de la réponse aux traitements, tels que les inhibiteurs du point de contrôle immunitaire et les thérapies ciblées.
Introduction
Avec le développement de thérapies ciblées et d’inhibiteurs de points de contrôle immunitaires, il est devenu de la plus haute importance de mieux caractériser les interactions entre les cellules cancéreuses et leur microenvironnement tumoral, et c’est actuellement un domaine important de la recherche translationnelle. Le TME est composé d’une pléthore de différents types cellulaires, avec un équilibre de cellules cytotoxiques immunitaires ciblant les cellules cancéreuses et de cellules immunomodulatrices qui pourraient favoriser la croissance tumorale et le caractère invasif 1,2,3,4. La caractérisation de cet environnement complexe pourrait améliorer la compréhension des maladies cancéreuses et pourrait aider les scientifiques et les cliniciens à découvrir de nouveaux biomarqueurs prédictifs et pronostiques afin de mieux sélectionner les patients pour un traitement futur 5,6. Par exemple, Galon et son équipe ont développé l’Immunoscore, qui est une méthode de notation reproductible qui peut être utilisée comme biomarqueur prédictif. L’Immunoscore est calculé en utilisant la densité des lymphocytes T CD3+ et CD8+ dans la marge invasive et au centre de la tumeur 7,8.
Au cours des dernières décennies, des solutions commerciales pour mIF ont été développées, mais celles-ci sont souvent coûteuses et conçues pour des panels spécifiques d’antigènes. Pour surmonter le besoin de panels spécifiques d’antigènes dans la recherche universitaire et translationnelle, nous avons développé une méthode rentable pour effectuer mIF sur des coupes tumorales FFPE, permettant la coloration de deux à six antigènes ajoutés aux noyaux cellulaires contre-coloration sur des échantillons humains et murins.
Une fois que les sections de tissus entiers sont colorées et scannées avec un scanner de lames de fluorescence, les échantillons peuvent être analysés par plusieurs logiciels d’analyse d’images prenant en charge de grands ensembles de données pyramidales. Enfin, les données brutes peuvent être utilisées dans un environnement de calcul statistique et graphique comme le logiciel R (v.4.0.2) afin d’effectuer des analyses de densité et spatiales.
Un protocole optimisé pour la coloration à cinq marqueurs, ainsi que des trucs et astuces pour optimiser les nouveaux panneaux, sont présentés dans ce manuscrit. De plus, les étapes détaillées de l’analyse d’images et les fonctions R utilisées pour l’analyse statistique et spatiale sont expliquées.
Protocole
Tous les échantillons utilisés dans le présent protocole proviennent d’une étude approuvée par les comités d’éthique locaux et autorisée par l’autorité compétente. Tous les participants à l’étude ont fourni un consentement éclairé écrit. L’essai est enregistré auprès de ClinicalTrials.gov (NCT03608046).
1. Immunofluorescence multiplex
- Sectionnement FFPE
- Fixez le tissu dans du paraformaldéhyde à 4% et incorporez le tissu fixe dans de la paraffine.
- Coupez des sections de 5 μm et placez-les sur des lames de microscope adhésif.
- Sécher les lames pendant la nuit à température ambiante (RT).
- Désaffinisation et inhibition des peroxydases endogènes
- Décirer les tissus en immergeant les lames dans du toluène (3x pendant 5 min chacune) et du méthanol (3x pendant 5 min chacune) sous une hotte.
- Inhiber les peroxydases endogènes en immergeant les lames dans du peroxyde d’hydrogène à 3% dilué dans du méthanol pendant 20 min sous une hotte.
- Rincer les lames dans du distillé (d)H2O(1x pendant 3 min).
- Coloration par immunofluorescence multiplex
- Immergez les lames dans un pot de coloration de 300 mL contenant 10 mM de citrate (pH 6) ou d’EDTA (pH 9) complété par du TritonX-100 à 0,1%.
REMARQUE : Le tampon utilisé (pH 6 ou pH 9) dépend de l’antigène coloré (voir le tableau 1). - Placez le pot de coloration avec le couvercle fermé au micro-ondes pendant 3 à 5 minutes à puissance maximale (p. ex., 900 W) jusqu’à ce que le tampon commence à bouillir.
REMARQUE: Le temps optimal pour l’ébullition dépend du micro-ondes et du volume du tampon. Des ajustements peuvent être nécessaires pour trouver le timing parfait. Pour certains antigènes fragiles ou spécimens fragiles et moins adhérents (p. ex. organoïdes et sphéroïdes), l’ébullition au micro-ondes peut être trop dure. Dans ce cas, un autocuiseur peut être utilisé à la place. - Maintenir le tampon à une température proche de l’ébullition en plaçant le pot à colorer fermé au micro-ondes à faible puissance (p. ex. 90 W) pendant 15 minutes.
- Effectuez la dernière étape du chauffage en mettant le micro-ondes à puissance maximale pendant 90 s.
- Retirer le pot du micro-ondes et laisser refroidir le tampon pendant 15 min à TA.
- Rincer les lames 3x pendant 5 min chacune dans dH2Oet 1x pendant 5 min dans une solution saline tamponnée contenant 0,1% de Tween 20 (TBS-T).
- Retirez le TBS-T en épongeant les lames sur une serviette en papier
- Placez les lames (à plat) sur un plateau de chambre de coloration ou une boîte de lames de microscope (voir le tableau des matériaux).
- Encerclez le tissu avec un stylo hydrophobe.
- Bloquer les sites de liaison non spécifiques en recouvrant le tissu de 5 % d’albumine sérique bovine (BSA) dissoute dans le TBS-T pendant 30 min.
- Retirez la mémoire tampon bloquante en épongeant les lames sur une serviette en papier.
REMARQUE: Ne rincez pas les lames après l’étape de blocage. - Incuber le tissu pendant 60 min avec l’anticorps primaire (voir tableau 1) dilué dans 1% BSA TBS-T en recouvrant le tissu d’environ 300 μL de la solution.
- Rincer les lames 3x pendant 3 min chacune avec TBS-T.
- Incuber le tissu pendant 40 min avec l’anticorps secondaire poly-HRP (voir tableau 1) en recouvrant le tissu d’environ 300 μL de solution.
- Rincer les lames 3x pendant 3 min avec TBS-T.
- Incuber le tissu pendant 10 min avec un réactif fluorochrome-tyramide (voir tableau 1) dilué 200 fois dans un tampon de borate (0,1 M borate, pH 7,8, 3 M NaCl) complété extemporanément avec 0,003% H2O2 en recouvrant le tissu d’environ 300 μL de la solution.
- Rincer les lames 3x pendant 3 min avec TBS-T.
- Répétez les étapes 1.3.1 à 1.3.16 jusqu’à ce que toute la coloration TSA ait été effectuée.
- Incuber le tissu pendant une nuit à 4 °C avec le dernier anticorps primaire (voir tableau 1) dilué dans du TBS-T BSA à 1%.
REMARQUE: Comme l’incubation dure pendant la nuit, il est important de couvrir le plateau de la chambre de coloration ou la boîte de lame du microscope et d’ajouter dH2O sur une serviette en papier au fond de la boîte (sous les lames) pour s’assurer que les tissus ne sèchent pas pendant l’incubation. - Rincer le mouchoir 3x pendant 5 min chacun avec TBS-T.
- Incuber le tissu pendant 120 min avec l’anticorps secondaire (directement couplé au fluorochrome) dilué 200 fois dans 1% BSA TBS-T.
- Rincer le mouchoir 3x pendant 5 min chacun avec TBS-T.
- Colorer les noyaux en incubant le tissu pendant 5 min dans du bisbenzimide (20 mM) dilué 1 000 fois dans 10% BSA TBS-T.
NOTE: Le bisbenzimide peut être remplacé par DAPI, mais ce dernier est plus toxique et doit être manipulé avec précaution sous une hotte. - Rincer le mouchoir 3x pendant 3 min chacun endH2O.
- Montez les lames à l’aide d’un support de montage à fluorescence et de verres à couverture en borosilicate.
- Immergez les lames dans un pot de coloration de 300 mL contenant 10 mM de citrate (pH 6) ou d’EDTA (pH 9) complété par du TritonX-100 à 0,1%.
2. Numérisation des diapositives
- Numérisez les lames en les numérisant sur un scanner de lames à fluorescence à un grossissement de 20x (les détails du scanner de diapositives sont fournis dans le tableau des matériaux).
REMARQUE : Un balayage représentatif d’un multiplex optimal est illustré à la figure 1.
3. Analyse d’images
- Importez les numérisations dans un logiciel d’analyse d’images (File > Open Image).
- Accédez à l’onglet Classificateurs et sélectionnez le plugin DenseNet AI V2 .
- Entraînez le plugin DenseNet AI V2 à reconnaître les noyaux en entourant environ 500 noyaux dans une image.
- Entraînez l’IA sur plusieurs autres lames du même lot et différents lots de coloration mIF en entourant plusieurs noyaux (50) sur plusieurs lames (environ 10).
REMARQUE: Des instructions détaillées sur la façon d’utiliser le plugin AI peuvent être trouvées dans le manuel du logiciel. L’utilisation de l’IA pour la détection des noyaux est facultative. D’autres méthodes de détection des noyaux sont disponibles en fonction du logiciel d’analyse d’images utilisé. - Enregistrez l’IA entraînée (actions du classificateur > sauvegarde).
- Accédez à l’onglet Annotations et créez une annotation pour chaque région d’intérêt (ROI), telle que le centre de la tumeur et la marge de la tumeur, à l’aide de l’outil d’annotation du stylet.
- Si nécessaire, supprimez les régions avec des plis et les régions qui apparaissent floues à l’aide de l’outil d’annotation d’exclusion.
REMARQUE: La coloration à l’hématoxyline-éosine d’une section adjacente à celle utilisée pour la mIF peut être effectuée avant la coloration mIF pour s’assurer que les cellules tumorales sont présentes dans l’échantillon et pour aider les anatomopathologistes à déterminer les ROI. - Accédez à l’onglet Analyse et sélectionnez l’algorithme HighPlex FL (Actions de paramètres > charge > HighPlex FL).
- Sélectionnez l’onglet Sélection du colorant , puis sélectionnez le colorant qui vous intéresse.
- Dans l’onglet Détection nucléaire, accédez à Type de segmentation nucléaire et sélectionnez Personnalisation de l’IA.
- Dans Nuclear Segmentation Classifier, sélectionnez l’IA enregistrée à l’étape 3.5.
- Dans l’onglet Détection de la membrane et du cytoplasme, choisissez le rayon maximal du cytoplasme (dans cette étude, 1,5 a été utilisé) et le nombre de colorants membranaires.
- Pour chaque colorant, sélectionnez le seuil positif du noyau, le seuil positif du cytoplasme et le seuil positif de la membrane.
REMARQUE : Le seuil est différent pour chaque coloration et doit être ajusté pour chaque lot de lames et chaque antigène coloré. L’utilisation de l’outil Paramètres d’affichage (Paramètres d’affichage > d’affichage) peut vous aider à sélectionner un seuil adéquat en utilisant la valeur d’intensité à la fin du pic d’intensité (à droite). - Pour chaque colorant, sélectionnez le pourcentage de noyau, de membrane et de cytoplasme des valeurs d’exhaustivité.
REMARQUE : Ce paramètre est important pour éviter la détection de faux positifs lorsque deux cellules avec des colorations différentes sont proches l’une de l’autre (Figure 2). - Enregistrez l’algorithme (Actions de paramètres > Enregistrer).
- Analysez les ROI (Analyze > Annotation Layer).
- Accédez à l’onglet Résultats et sélectionnez toutes les données dans Données d’objet (ctrl + A).
- Exportez les données dans .csv format (clic droit > Exporter > données d’objet . CSV).
Remarque : Ce tableau contient la position (Xmin, Xmax; Ymin, Ymax) et la positivité de chaque marqueur de chaque cellule analysée.
4. Bioinformatique utilisant R
REMARQUE: Un script R fournissant plus de détails sur les étapes suivantes est disponible sur GitHub (benidovskaya / Ring: Pipeline pour l’analyse des colorations d’immunofluorescence multiplexe. [github.com])
- À l’aide du tableau exporté, définissez d’abord les différents types de cellules en fonction des colorations de colocalisation. Par exemple, définissez les lymphocytes T cytotoxiques par des lymphocytes CD3+/CD8+ doublement positifs.
- Ensuite, reconstruisez une image simplifiée de la diapositive sur un graphique à points à l’aide des coordonnées exportées du logiciel d’analyse d’images et de ggplot2 (Graphique 3). À l’aide de ces données, plusieurs types d’analyses peuvent être effectués :
- Analyse de densité
Remarque : L’analyse la plus simple est une analyse de densité.- Effectuer une analyse de densité pour tous les types de cellules en utilisant la lame entière pour les biopsies ou une zone spécifique du tissu. Par exemple, calculer la densité des lymphocytes T CD3+ et CD8+ au centre de la tumeur et la marge de la tumeur (Figure 4A-C).
- Pour calculer ces densités, utilisez un logiciel d’analyse d’images pour produire une base de données spécifique par échantillon avec le phénotype et les coordonnées de chaque cellule. Grâce à une fonction de regroupement (k plus proche voisin) sur R, créez un objet polygonal en utilisant les bordures de la biopsie étudiée et calculez la densité des types de cellules d’intérêt à l’intérieur de celui-ci.
REMARQUE: Cela permet de comparer les densités de différents types de cellules entre différentes conditions (telles que différents points temporels, types de traitement, types de tissus et réponse au traitement) et localisations (centre de la tumeur, marge de la tumeur, fibrose du stroma et zone de nécrose) en fonction de l’hypothèse biologique. En raison de la grande proximité entre les cellules cancéreuses, les cellules péritumorales et les cellules infiltrant la tumeur, le logiciel d’analyse d’images peut détecter les cellules doublement positives en tant que cellules immunitaires et cancéreuses en même temps. Dans ce cas, il faut corriger bio-informatique ce problème en mentionnant ce que sont ces cellules doublement positives. Dans ce cas, les cellules CD3 + CD8 + cytokératine + ont été marquées comme cellules cytotoxiques parce que la positivité de la cytokératine était due aux cellules tumorales entourant les lymphocytes infiltrants.
- Cartes thermiques
- En utilisant la densité de chaque type de cellule à partir de différents panels et en appliquant une normalisation (p. ex., centrage de mise à l’échelle), dessinez des cartes thermiques (Figure 5) représentant l’abondance des cellules dans la population d’échantillons.
- En utilisant le regroupement hiérarchique non supervisé basé sur la densité cellulaire, regrouper les patients ayant des compositions similaires de TME et corréler ces clusters avec des paramètres cliniques tels que la réponse au traitement et la survie.
REMARQUE: Les cartes thermiques et la clusterisation peuvent être facilement effectuées avec le package R ComplexHeatmap9.
- Distribution cellulaire spatiale
- Calculer bio-informatique les distances entre les cellules (p. ex., cellules immunitaires et tumorales; Figure 6A, B) basée sur les coordonnées de cellule fournies par l’analyse d’image. Utilisez les distances médiane et moyenne entre les types de cellules d’intérêt pour comparer la proximité cellulaire dans tous les échantillons d’une cohorte.
- Fonctions descriptives spatiales
- Utilisez la fonction G-cross du plus proche voisin croisé, disponible via le package R spatstat10, pour déterminer la probabilité pour une cellule d’intérêt, X (par exemple, une cellule tumorale), rencontrant la cellule la plus proche, Y (par exemple, une cellule T), dans un certain rayon autour de la cellule X.
- Calculer l’aire sous la courbe empirique pour obtenir une valeur numérique représentant l’infiltration tumorale des lymphocytes T CD3+ autour des cellules tumorales11 (Figure 6C). Utilisez d’autres fonctions descriptives spatiales telles que la fonction F ou la fonction J12.
- Analyse des immunoscores
- Calculer l’Immunoscore (I), développé par l’équipe de Galon7,8, en utilisant la densité des lymphocytes T CD3+ et CD8+ au centre de la tumeur et la marge invasive de la tumeur.
REMARQUE: Le score varie de I0 à I4. Une faible densité de lymphocytes T CD3+ et CD8+ au centre et dans la marge de la tumeur est associée à un score I0, tandis qu’une densité élevée de lymphocytes T CD3+ et CD8+ dans les deux régions est associée à un score I4. Récemment, l’impact pronostique de l’Immunocore a été validé dans une étude avec des échantillons de 2 681 patients atteints d’un cancer du côlon de stade I-III provenant de 14 centres dans 13 pays7. Cependant, pour être calculé, Immunoscore nécessite un échantillon réséqué chirurgicalement contenant à la fois le centre et la marge de la tumeur. Pour les biopsies, qui manquent généralement de marge, un Immunoscore adapté à la biopsie a été récemment mis au point13. - Pour calculer l’immunoscore adapté à la biopsie, convertissez la valeur de la densité des lymphocytes T CD3+ et CD8+ en centile, puis utilisez le percentile moyen des lymphocytes T CD3+ et CD8+ pour classer l’une des trois catégories suivantes (c.-à-d. faible, intermédiaire et élevé)13.
- Calculer l’Immunoscore (I), développé par l’équipe de Galon7,8, en utilisant la densité des lymphocytes T CD3+ et CD8+ au centre de la tumeur et la marge invasive de la tumeur.
- Analyse des points chauds
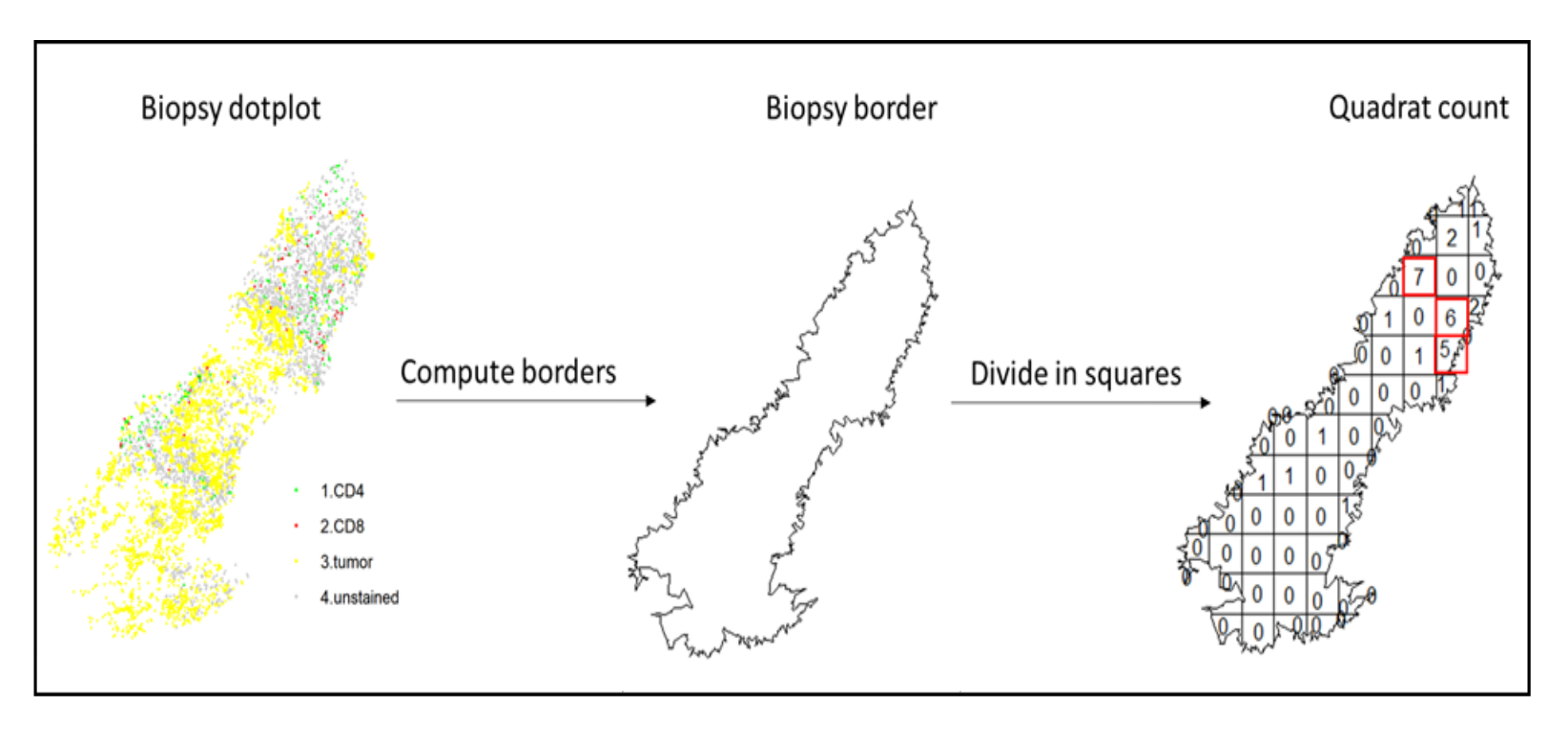
- Utilisez l’analyse des points chauds, en utilisant la fonction quadratcount (spatstat)10, pour comparer les densités de différents types de cellules dans la zone la plus infiltrée du tissu. Par exemple, il est possible de calculer un score de type « Immunoscore » en utilisant la valeur de la densité des lymphocytes T CD3 et CD8 des carrés les plus infiltrés du tissu (Figure 7). Appliquez cette méthode pour l’analyse de tout type de cellule avec une distribution non homogène à travers le tissu.
- Analyse de densité
Résultats Représentatifs
En suivant ce protocole, plusieurs paramètres doivent être étudiés pour s’assurer que le tissu est correctement coloré. Tout d’abord, la coloration TSA doit afficher une bonne plage dynamique lors de l’utilisation de temps d’exposition faibles (généralement de 2 à 100 ms) pendant le processus de numérisation. Un temps d’exposition faible implique que l’amplification a été effectuée correctement lors de la réaction avec HRP. Pour les antigènes colorés avec l’anticorps secondaire directement couplé au fluorochrome, le temps d’exposition pourrait être beaucoup plus long, ce qui pourrait entraîner un photoblanchiment (une diminution de l’intensité du signal due à un long temps d’exposition). Deuxièmement, il est important de vérifier que chaque coloration affiche un rapport signal/bruit élevé. Un signal de fond élevé avec un signal antigénique faible peut indiquer que l’anticorps primaire n’est pas assez spécifique, que les peroxydases endogènes n’ont pas été inactivées correctement ou qu’une étape du protocole n’a pas été effectuée correctement. Troisièmement, en fonction du scanner de diapositives et des jeux de filtres utilisés pour la numérisation, il est possible de voir des chevauchements entre deux couleurs (par exemple, AF555, AF594 et AF647). Le choix des bons jeux de filtres sur le scanner et la bonne dilution des anticorps primaires sont cruciaux pour éviter d’éventuelles détections croisées. Le contrôle de la qualité consiste à détecter des cellules colorées uniques pour chaque marqueur sur le fichier numérisé. Enfin, il est également important d’ajouter un contrôle positif et négatif pour chaque lot de coloration. Pour les cellules immunitaires, l’amygdale est un bon contrôle positif. Un résultat représentatif de la coloration optimale est présenté à la figure 1.

Figure 1 : Cancer du rectum localement avancé coloré par immunofluorescence multiplexe. Abréviations :-1 = protéine de mort cellulaire programmée 1; -L1 = ligand de mort programmé 1; ROR-γ = récepteur gamma orphelin lié au RAR; CD3 = groupe de différenciation 3; hPanCK = pan-cytokératine humaine. Chaque coloration antigénique est scannée en niveaux de gris, et les couleurs présentées dans la figure sont des pseudo-couleurs. Faible grossissement de la barre d’échelle: 200 μm; Barre d’échelle Grossissement élevé : 100 μm. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Détection des noyaux et des colorations d’un cancer du rectum localement avancé à l’aide d’un logiciel d’analyse d’images. Sans le paramètre de pourcentage d’exhaustivité correctement défini, le logiciel détecte deux cellules CD8+ (cercle vert) parce qu’elles sont proches l’une de l’autre, mais une seule cellule est colorée. L’utilisation d’une exhaustivité de 70 % permet d’éviter cette détection de faux positifs. Vert = hPanCK; Jaune = CD3; Orange = CD8. Barre d’échelle : 100 μm Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3 : Analyse d’images et reconstitution par diagramme de points R d’une métastase du cancer colorectal du foie. Sur la coloration multiplex (à gauche), la pancytokératine humaine est en jaune, CD3 en vert, CD8 en bleu clair et IDO en orange. Sur le diagramme à points (à droite), les cellules pancytokératine+ humaines sont en jaune, les cellules CD3+CD8− en vert, les cellules CD3+CD8+ en bleu et les cellules IDO+ en orange. Veuillez cliquer ici pour voir une version agrandie de cette figure.
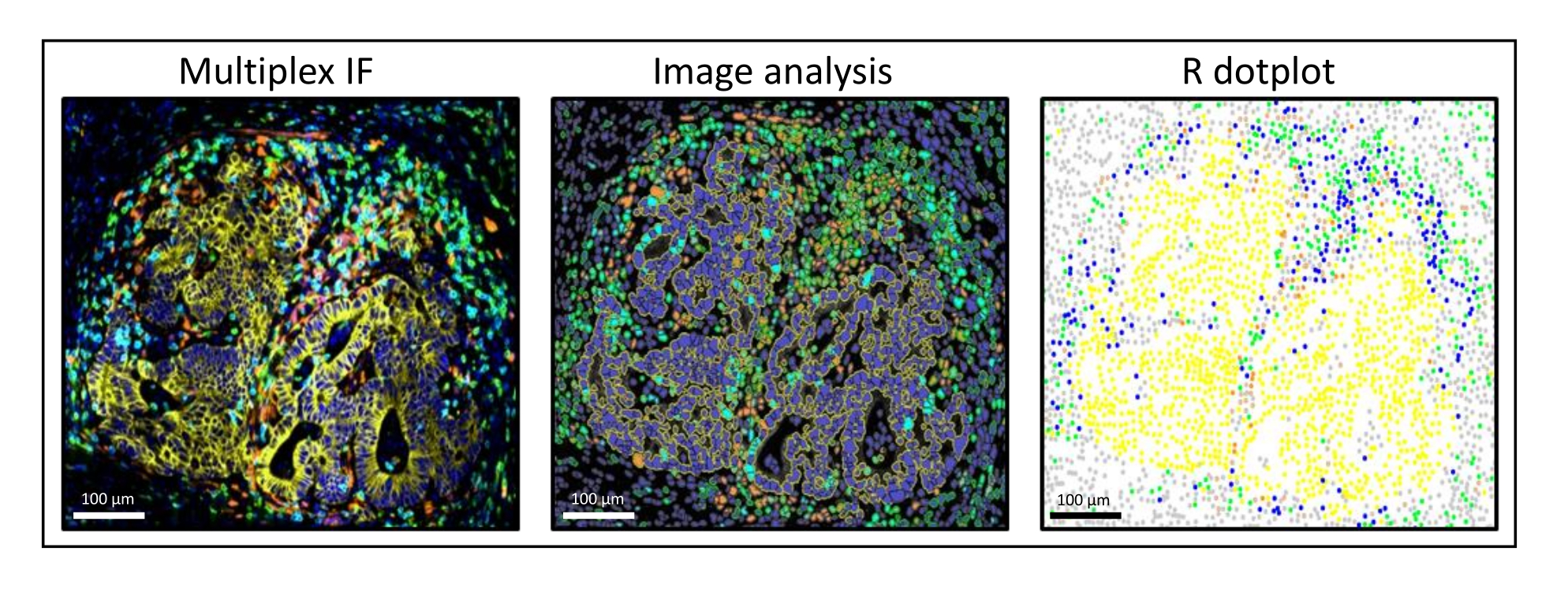{kind=link}

Figure 4 : Analyse d’une section chirurgicale d’un HNSCC. (A) Section chirurgicale d’un HNSCC. Les cellules cancéreuses sont visibles en vert. Les cellules péri-tumorales sont visualisées autour des îlots tumoraux (CD3 en jaune et CD8 en violet). (B) Le centre de la tumeur (en jaune avec une bordure noire) est calculé bio-informatique par l’algorithme k-plus proche-voisin basé sur la distance entre les îlots tumoraux d’une seule zone. Autour de cette zone, une marge invasive (jaune clair avec une bordure grise) est calculée sur une base arbitraire de 500 μm. (C) Les lymphocytes T invasifs sont mis en évidence par des points noirs au centre de la tumeur et des points gris dans la marge invasive. Les autres lymphocytes T sont surlignés par des points vert clair. Barre d’échelle: 1 mm. Veuillez cliquer ici pour voir une version agrandie de cette figure.
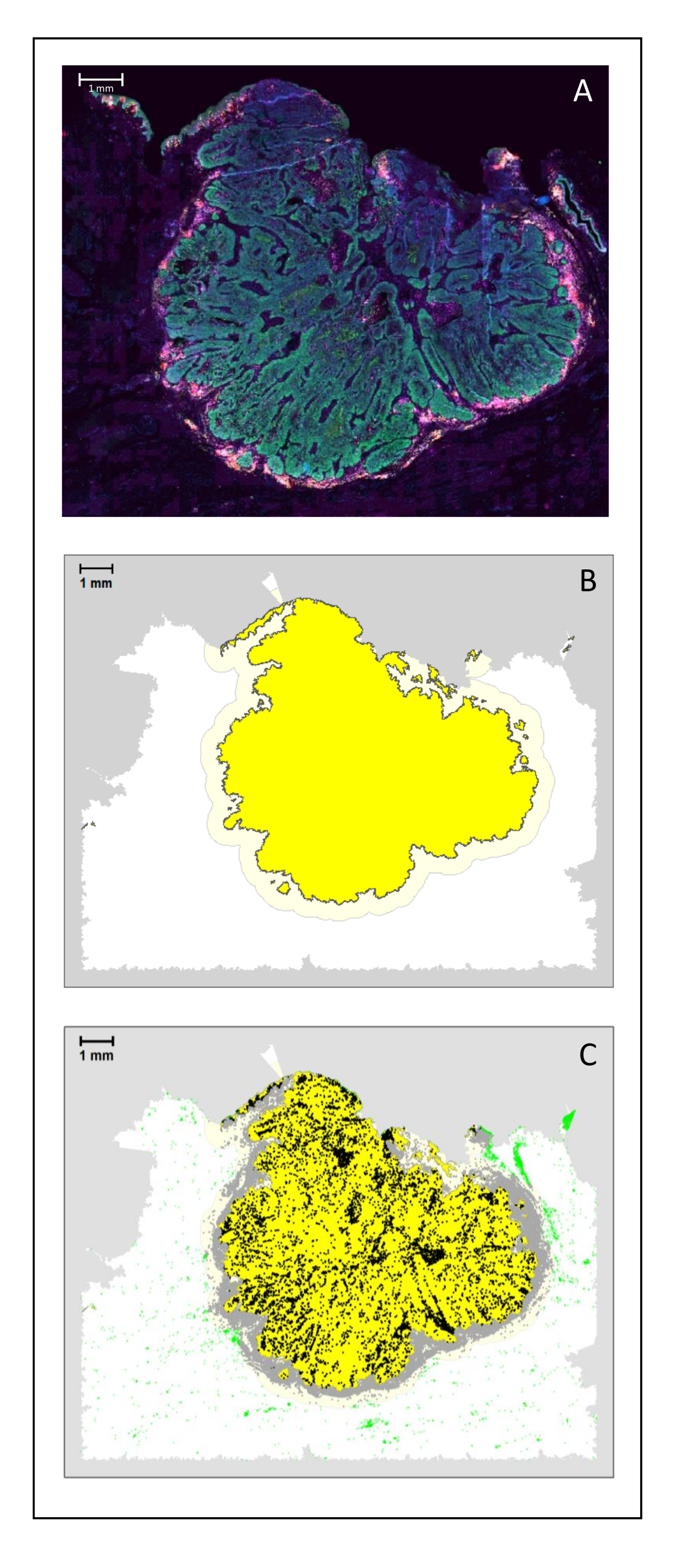{kind=link}

Figure 5 : Carte thermique de la densité de différents types cellulaires de biopsies du cancer du rectum localement avancées. La carte thermique a été dessinée à l’aide d’un regroupement non supervisé des densités de différents types de cellules à partir de différents panneaux multiplex avec le package ComplexHeatmap. La mise à l’échelle et le centrage ont été utilisés pour la normalisation. Veuillez cliquer ici pour voir une version agrandie de cette figure.
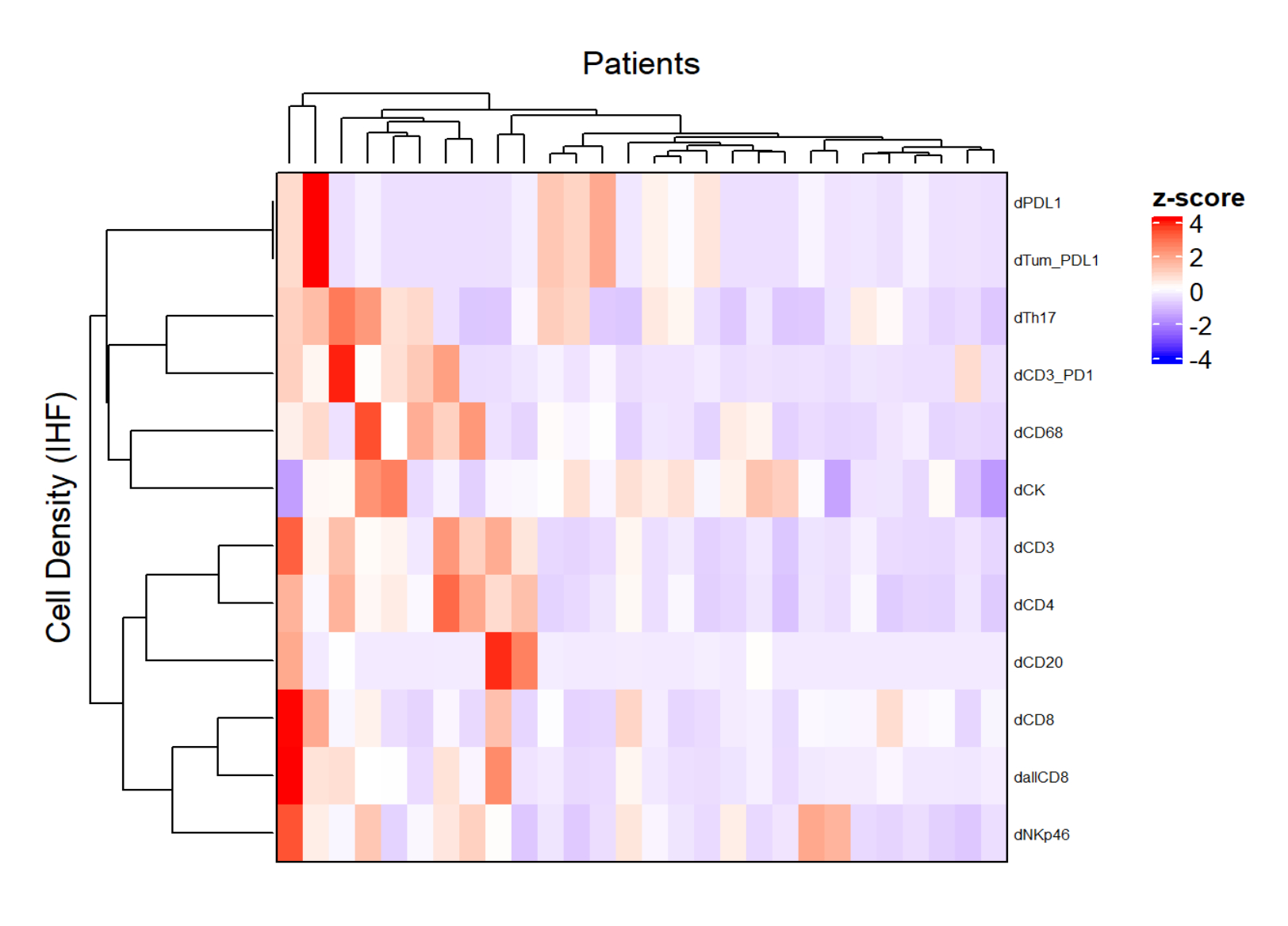{kind=link}

Figure 6 : Distances des cellules CD4+ et CD8+ à chaque cellule IDO+ ou cellule tumorale. Les cellules pancytokératines+ humaines sont en jaune, les cellules CD3+CD8− en vert, les cellules CD3+CD8+ en bleu et les cellules IDO+ en orange. (A) La distance la plus proche entre les cellules tumorales et chaque lymphocytes T CD8+. (B) Diagrammes à barres des distances entre les cellules IDO+ et chaque lymphocyte T CD8+ (bleu) ou T CD4+ (vert). (C) Exemple d’échantillon analysé par la fonction G-cross. L’axe des y montre la probabilité qu’une cellule tumorale rencontre un lymphocyte CD3+ dans un rayon allant de 0 à 200 μm autour de la cellule tumorale. Trois courbes sont représentées; la courbe théorique est en pointillé vert (distribution de Poisson), la courbe empirique corrigée avec correction de km est en noir et la courbe empirique corrigée avec correction de bordure est en pointillé rouge. Veuillez cliquer ici pour voir une version agrandie de cette figure.
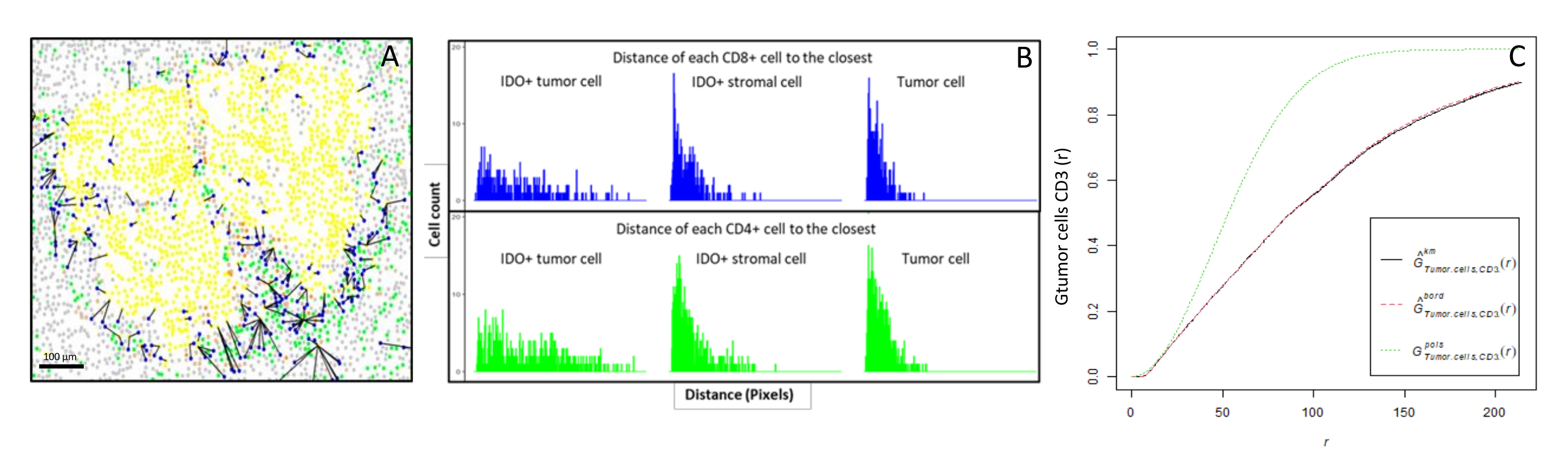{kind=link}

Figure 7 : Illustration d’un quadratcount. Le calcul des frontières et le comptage des quadrats ont été effectués à l’aide du progiciel spatstats. Les carrés les plus infiltrés (hotspots) peuvent être utilisés pour les statistiques en aval. CD4 est en vert, CD8 est en rouge et les cellules tumorales sont en jaune. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

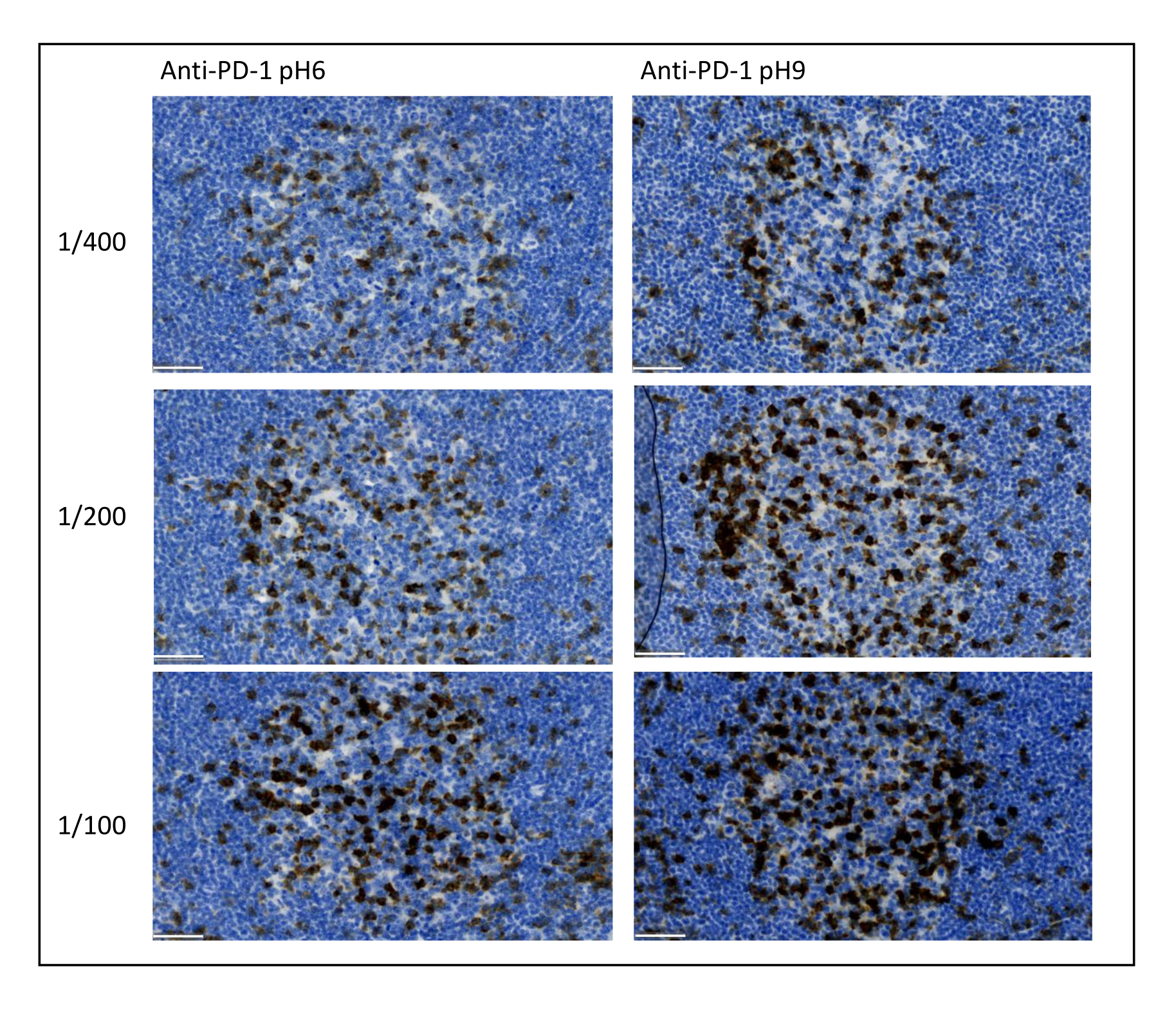
Figure 8 : Dilution des anticorps et optimisation de l’extraction des antigènes. Détection chromogénique de-1 à l’aide de trois dilutions différentes et de deux solutions différentes de récupération d’antigènes de l’anticorps primaire (Citrate pH 6 et EDTA pH 9). Barre d’échelle: 50 μm. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
| Anticorps primaire | Dilution | Récupération d’antigènes | Anticorps secondaire | Fluorochrome | Position |
| -1 | 1/100 | EDTA (pH 9) | Anti-lapin | AF647 | 1 |
| -L1 | 1/1000 | EDTA (pH 9) | Anti-lapin | AF488 | 2 |
| ROR-γ | 1/200 | EDTA (pH 9) | Anti-souris | ATT0-425 | 3 |
| CD3 | 1/100 | Citrate (pH 6) | Anti-lapin | AF555 | 4 |
| hPanCK | 1/50 | Citrate (pH 6) | Anti-souris couplé avec AF750 | 5 | |
Tableau 1 : Exemple d’un panneau multiplex optimisé. Abréviations :-1 = protéine de mort cellulaire programmée 1; -L1 = ligand de mort programmé 1; ROR-γ = récepteur gamma orphelin lié au RAR; CD3 = groupe de différenciation 3; hPanCK = pancytokératine humaine; AF = AlexaFluor; EDTA = acide éthylènediaminetétraacétique. CD3 est utilisé pour détecter les lymphocytes T; -1 est utilisé pour détecter les lymphocytes épuisés; ROR-γ est utilisé pour détecter Th-17; et hPanCK est utilisé pour détecter les cellules tumorales. La colonne position indique l’ordre dans lequel le multiplex séquentiel doit être exécuté.
Discussion
Les paramètres les plus importants à prendre en considération pour optimiser la coloration multiplex sont la dilution, la spécificité et le prélèvement d’antigène utilisé pour chaque anticorps primaire. Avant de commencer un protocole multiplex, la dilution optimale de chaque anticorps primaire et la récupération optimale des épitopes (pH 6 ou pH 9) doivent être testées par coloration chromogène (DAB). Nous conseillons de tester trois dilutions pour chaque tampon de récupération d’antigène : la dilution qui est habituellement spécifiée par la marque commercialisant l’anticorps, la même dilution divisée deux fois et la même dilution multipliée par deux (Figure 8). Le choix de la bonne dilution est une étape très importante pour vérifier la spécificité des anticorps et optimiser le rapport signal sur bruit (SNR) de la coloration. Après avoir choisi la bonne dilution dans le DAB, la même dilution doit être testée pour chaque anticorps primaire à l’aide de TSA uniplex. Une fois que le tampon de dilution et de récupération d’épitopes est sélectionné pour chaque coloration antigénique, il est également important de configurer correctement la séquence du multiplexe; Plus précisément, certains antigènes sont mieux colorés à la première position et d’autres à la dernière position. Nous conseillons de tester le marquage multiplex en utilisant toutes les permutations d’ordre possibles pour choisir quelle coloration antigénique doit venir en premier, en second, etc. C’est aussi une étape très importante car certains antigènes fragiles peuvent être dégradés après plusieurs cycles de récupération d’épitopes, et certains antigènes sont mieux colorés après plusieurs cycles de récupération d’épitopes. Par exemple, le SNR est toujours plus élevé en dernière position pour le CD3 et en première position pour la coloration-1. De plus, la coloration de plusieurs antigènes co-localisés peut être entravée par un effet parapluie (la saturation des sites réactifs au tyramide). Cela peut être atténué en diminuant la concentration de tyramide. Lorsque l’expression d’un antigène est conditionnée par l’expression d’un autre (CD8 uniquement présent sur les lymphocytes T exprimant CD3), nous conseillons de colorer l’antigène avec l’expression la plus large (CD3 dans ce cas) après l’autre. Enfin, choisir le bon fluorochrome pour chaque coloration antigénique en fonction des spécificités du scanner est également une étape importante pour éviter les détections croisées.
Les principaux avantages de cette technique sont l’amplification et le rapport signal sur bruit obtenu. Cependant, cette technique est limitée par le fait que la coloration est séquentielle et que les fluorochromes sont liés de manière covalente au tissu. Néanmoins, après avoir effectué toutes les tournées d’amplification du signal tyramide, il est également possible d’ajouter une dernière coloration avec un anticorps secondaire directement couplé à un fluorochrome (pas de TSA). Dans certains panneaux, nous avons utilisé cette méthode pour ajouter des taches dans le canal 750. Cela était nécessaire car aucun tyramide-AF750 n’était disponible dans le commerce à cette époque. Il est à noter que le temps d’exposition (pendant l’analyse) de l’antigène coloré avec AF750 sera beaucoup plus long que pour les autres antigènes colorés avec TSA. Dans ce cas, nous conseillons de colorer une protéine fortement exprimée telle que la cytokératine ou d’augmenter la concentration de l’anticorps primaire. Ce faisant, il est possible de colorer un maximum de cinq à six antigènes par lame dans un lot en fonction du scanner à fluorescence.
En revanche, plusieurs techniques disponibles dans le commerce utilisent la coloration en série avec plusieurs cycles de coloration, de balayage et de décapage ou de photoblanchiment pour améliorer le nombre d’antigènes pouvant être colorés sur une seule section de tissu. Cependant, ces techniques sont souvent longues, coûteuses, n’ont pas d’amplification du signal, nécessitent des étapes de calcul avancées pour fusionner correctement les scans série et, selon notre expérience, peuvent induire des lésions tissulaires irréversibles en raison des nombreuses étapes de la procédure. Néanmoins, il a été rapporté que jusqu’à 30 antigènes pouvaient être colorés sur un seul tissu en utilisant cette méthode14.
En conclusion, notre méthode est une technique d’immunohistofluorescence robuste, reproductible, facile à utiliser et rentable qui peut être utilisée dans n’importe quel laboratoire possédant un scanner de lames de fluorescence. Tout anticorps primaire commercialisé adapté à l’IHC peut être utilisé, et les panels ne sont pas spécifiques à des kits commerciaux. L’analyse d’image peut être effectuée sur plusieurs programmes différents, y compris des programmes open-source tels que QuPath et R. Cependant, nous pensons que cette méthode pourrait même être améliorée à l’avenir pour les grands panneaux d’antigènes, permettant d’effectuer une coloration / balayage en série de la même lame avec différents panneaux d’antigènes et avec l’avantage de l’amplification du signal.
Déclarations de divulgation
Les auteurs n’ont aucun conflit d’intérêts à déclarer.
Remerciements
Les auteurs tiennent à remercier la Dre Derouane F pour son aide et son soutien. Nicolas Huyghe est chercheur soutenu par une bourse du Fonds national de la recherche scientifique (Télévie/FNRS 7460918F).
matériels
| Name | Company | Catalog Number | Comments |
| anti-CD3 primary antibody | Abcam | ab16669 | rabbit monocolonal |
| anti-CD8 primary antibody | DAKO | M710301 | mouse monoclonal |
| anti-hPanCK primary antibody | DAKO | M3515 | mouse monoclonal |
| anti-PD-1 primary antibody | Cell Signalling | D4W2J | rabbit monocolonal |
| anti-PD-L1 primary antibody | Cell Signalling | 13684 | rabbit monocolonal |
| anti-RORC primary antibody | Sigma | MABF81 | mouse monoclonal |
| ATTO-425 | ATTOtec | ||
| Axioscan Z1 | Zeiss | Light source: Colibri 7 (385, 430, 475, 555, 590, 630, 735 nm) Filtersets: Excitation 379/34 – beam splitter 409 – emission 440/40; Excitation 438/24 – beam splitter 458 – emission 483/32; Excitation 490/20 – beam splitter 505 – emission 525/20; Excitation 546/10 – beam splitter 556 – emission 572/23; Excitation 592/21 – beam splitter 610 – emission 630/30; Excitation 635/18 – beam splitter 652 – emission 680/42; Excitation 735/40 – beam splitter QBS 405 + 493 + 611 + 762 - emission QBP 425/30 + 524/51 + 634/38 + 785/38; Objective: Plan-Apochromat 20x/0.8; Camera : Orca Flash 4.0 V3 | |
| Borosilicate Cover Glass | VWR | 631-0146 | |
| Envision+ anti-mouse | DAKO | K4001 | |
| Envision+ anti-rabbit | DAKO | K4003 | |
| Fluorescence mounting medium | DAKO | S3023 | |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 750 | ThermoFischer | A-21037 | |
| HALO software | Indicalabs | ||
| Hoescht | Sigma | 14533 | |
| Superfrost plus microscope slides | Fisherscientific/Epredia | 10149870 | |
| Tyramide-AF488 | ThermoFischer | B40953 | |
| Tyramide-AF555 | ThermoFischer | B04955 | |
| Tyramide-AF647 | ThermoFischer | B04958 |
Références
- Ge, P., et al. Profiles of immune cell infiltration and immune-related genes in the tumor microenvironment of colorectal cancer. Biomedicine & Pharmacotherapy. 118, 109228 (2019).
- Fridman, W. H. The immune microenvironment as a guide for cancer therapies. Oncoimmunology. 1 (3), 261-262 (2012).
- Fridman, W. H., Pages, F., Sautes-Fridman, C., Galon, J. The immune contexture in human tumours: Impact on clinical outcome. in Nature Reviews. Cancer. 12 (4), 298-306 (2012).
- Hanahan, D., Weinberg, R. A. Hallmarks of cancer: The next generation. Cell. 144 (5), 646-674 (2011).
- Calu, V., et al. Key biomarkers within the colorectal cancer related inflammatory microenvironment. Scientific Reports. 11 (1), 7940 (2021).
- Havel, J. J., Chowell, D., Chan, T. A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nature Reviews. Cancer. 19 (3), 133-150 (2019).
- Pages, F., et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 391 (10135), 2128-2139 (2018).
- Mlecnik, B., et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 44 (3), 698-711 (2016).
- Gu, Z., Eils, R., Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 32 (18), 2847-2849 (2016).
- Baddeley, A., Rubak, E., Turner, R. . Spatial Point Patterns: Methodology and Applications with R. , (2022).
- Barua, S., et al. Spatial interaction of tumor cells and regulatory T cells correlates with survival in non-small cell lung cancer. Lung Cancer. 117, 73-79 (2018).
- Parra, E. R. Methods to determine and analyze the cellular spatial distribution extracted from multiplex immunofluorescence data to understand the tumor microenvironment. Frontiers in Molecular Biosciences. 8, 668340 (2021).
- El Sissy, C., et al. A diagnostic biopsy-adapted immunoscore predicts response to neoadjuvant treatment and selects patients with rectal cancer eligible for a watch-and-wait strategy. Clinical Cancer Research. 26 (19), 5198-5207 (2020).
- Bolognesi, M. M., et al. Multiplex staining by sequential immunostaining and antibody removal on routine tissue sections. The Journal of Histochemistry and Cytochemistry. 65 (8), 431-444 (2017).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.