
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
הדור של מסומנים מוטאנטים Markerless במודל כחוליות המינים
In This Article
Summary
Introducing multiple genomic alterations into cyanobacteria is an essential tool in the development of strains for industrial and basic research purposes. We describe a system for generating unmarked mutants in the model cyanobacterial species Synechocystis sp. PCC6803 and marked mutants in Synechococcus sp. PCC7002.
Abstract
Cyanobacteria are ecologically important organisms and potential platforms for production of biofuels and useful industrial products. Genetic manipulation of cyanobacteria, especially model organisms such as Synechocystis sp. PCC6803 and Synechococcus sp. PCC7002, is a key tool for both basic and applied research. Generation of unmarked mutants, whereby chromosomal alterations are introduced into a strain via insertion of an antibiotic resistance cassette (a manipulatable fragment of DNA containing one or more genes), followed by subsequent removal of this cassette using a negative selectable marker, is a particularly powerful technique. Unmarked mutants can be repeatedly genetically manipulated, allowing as many alterations to be introduced into a strain as desired. In addition, the absence of genes encoding antibiotic resistance proteins in the mutated strain is desirable, as it avoids the possibility of 'escape' of antibiotic resistant organisms into the environment. However, detailed methods for repeated rounds of genetic manipulation of cyanobacteria are not well described in the scientific literature. Here we provide a comprehensive description of this technique, which we have successfully used to generate mutants with multiple deletions, single point mutations within a gene of interest and insertion of novel gene cassettes.
Introduction
כחוליות מהוות מערכה עתיקה ומגוונת אבולוציונית של חיידקים נמצאים כמעט בכל סביבה טבעית על פני כדור הארץ. בשנת מערכות אקולוגיות ימיות הם במיוחד שופע לשחק תפקיד מפתח במחזורים מזין רבים, והיוו כמחצית קיבוע פחמן 1, רוב קיבוע חנקן 2 ומאות מיליוני טונות של ייצור פחמימנים 3 באוקיינוסים בשנה. כלורופלסטים, אברון האחראי על הפוטוסינתזה אצות איקריוטיים וצמחים, צפויים התפתחו מתוך cyanobacterium כי נבלע על ידי הפונדקאי 4. כחוליות הוכיחו אורגניזמים מודל שימושי לחקר הפוטוסינתזה, אלקטרון התחבורה 5 ו הביוכימיים, שרבים מהם שמורים בצמחים. בשנת כחוליות בנוסף יותר ויותר בשימוש לייצור מזון, דלק ביולוגי 6, חשמל 7 ותעשייתיים תרכובות 8, בשל היי שלהםהמרה יעילה ghly מים ו- CO 2 עד ביומסה באמצעות אנרגיה סולארית 9. מינים רבים יכולים להיות מעובד על קרקע שאינה חקלאית עם חומרים מזינים מינימאליים ומי ים, דבר המצביע על כך כחוליות עלולים להיות מבוגרים בקנה מידה גדולה מבלי להשפיע ייצור חקלאי. מינים מסוימים הם גם מקורות של מוצרים טבעיים, כולל תרכובות נגד פטריות, אנטיבקטריאלי ואנטי סרטן 10,11.
היכולת ליצור מוטציות היא מפתח להבנת פוטוסינתזה כחוליות, ביוכימיה ופיזיולוגיה, וחיוני להתפתחות של זנים למטרות תעשייתיות. רוב המחקרים שפורסמו גנרטור מהונדס גנטי זנים ידי החדרת קלטת עמידות לאנטיביוטיקה לתוך האתר של עניין. זה מגביל את מספר מוטציות יכולות להיות מוחדר זן, כמו שרק כמה קלטות עמידות לאנטיביוטיקה זמינות לשימוש כחוליות. זנים המכילים גנים המקנות מחדש אנטיביוטיsistance לא יכול לשמש לייצור תעשייתי בבריכות פתוחות, אשר צפוי להיות אמצעי היחיד החסכוני לייצר דלק ביולוגי ומוצרים בעלי ערך נמוך אחרים 12. הדור של מוטציות לא מסומנות מתגבר על מגבלות אלה. מוטציות לא מסומנות אינם מכילים דנ"א זר, אלא אם כן נכלל בכוונה, וניתן לטפל מספר פעמים. לכן אפשר ליצור שינויים רבים ב זן כפי רצויות. בנוסף, תופעות קוטב על גנים במורד הזרם של אתר שינוי ניתן למזער, המאפשר שינוי מדויק יותר של האורגניזם 13.
כדי ליצור זנים מוטנטים, פלסמידים התאבדות המכיל שני קטעי דנ"א זהה אזורים בכרומוזום כחוליות איגוף הגן למחיקה (שמכונה 5 'ו 3' אזורי איגוף) בנויים ראשון. שני גנים ואז מוכנס בין אזורי איגוף אלה. אחד מהם מקודד חלבון עמידות לאנטיביוטיקה; השני מקודד SacB, אשר לדרבןuces levansucrase, תרכובת מקנה רגישה סוכרוז. בשלב הראשון של התהליך, מוטציות מסומנות, זנים כלומר המכילים כמה דנ"א זר, נוצרות. הבונה פלסמיד הוא מעורבב עם התאים כחוליות והדנ"א נלקח באופן טבעי על ידי האורגניזם. Transformants נבחר על ידי צמיחה על צלחות אגרו המכילות את אנטיביוטי המתאים ואת הגנוטיפ המוטציה מאומת על ידי PCR. פלסמידים התאבדות לא יכולים לשכפל בתוך הזן של עניין. לכן כל מושבות עמידות לאנטיביוטיקה תגרומנה מאירוע רקומבינציה לפיה הגן של עניין מוכנס לתוך הכרומוזום. כדי ליצור מוטציות לא מסומנות, המוטציה הניכרת הוא מעורב אז עם פלסמיד התאבדות נוסף שהכיל רק 5 'ו 3' אזורי האיגוף. עם זאת, אם החדרת דנ"א זר נדרשה, פלסמיד המורכב של 5 'ו 3' איגוף אזורים עם קסטה המכילה את הגנים של העניין המוכנס בין קטעי דנ"א אלה, ניתן להשתמש. Sele גודלction היא באמצעות צמיחה על צלחות אגר המכיל סוכרוז. כמו סוכרוז הוא קטלני לתאים כאשר מוצר גן sacB מתבטא, התאים היחידים ששורדים הם אלה שבם אירוע רקומבינציה שני התרחש, לפיה גן רגישות סוכרוז, בנוסף גן העמידות לאנטיביוטיקה, כבר recombined מתוך כרומוזום על פלסמיד. כתוצאה מכך של חילופי recombinational, אזורי האיגוף וכל DNA ביניהם מוכנסים לתוך כרומוזום.
השתמשנו בהצלחה בשיטות אלה כדי ליצור מוטציות כרומוזומליות מרובות באותו הזן של Synechocystis sp. PCC6803 (להלן המכונה Synechocystis) 13,14, להציג מוטציות נקודה אחת לתוך גן של עניין 13 ו לביטוי של קלטות גן. בעוד הדור של knockouts המסומנת הודגם לפני עבודתנו Synechocystis 15,16, שיטה מפורטת, שנעזרמצגת ויזואלית של השלבים הקריטיים, הוא לא זמין לציבור. גם יש לנו ליישם אותה השיטה לייצור של knockouts ניכרה cyanobacterium מודל אחר, sp Synechococcus. PCC7002 (להלן המכונה Synechococcus). פרוטוקול זה מספק שיטה ברורה, פשוטה ליצירת מוטציות ובפרוטוקול מהיר על תיקוף ואחסון זנים אלה.
Protocol
1. הכנה של התקשורת בתרבות
- כן בינוני BG11 פי Castenholz 1988 17.
- הכינו מלאי של פתרונות BG11 100x, יסודות קורט ומלאי ברזל (טבלה 1).
- הכינו פתרונות נפרדים ממלאי פוספט, מניית Na 2 CO 3, N - [טריס (hydroxymethyl) מתיל] חומצה -2-aminoethanesulfonic (TES) חיץ NaHCO 3 (טבלה 1).
- חיטוי פוספט Na 2 CO 3 מניות. מסנן לעקר TES חיץ NaHCO 3 עם 0.2 מיקרומטר מסננים.
- הכן BG11 ידי שילוב 976 מ"ל מים, 10 מ"ל של BG11 100x, 1 מ"ל של יסודות קורט 1 מ"ל של המניה ברזל ו החיטוי הפתרון. לאחר פתרון זה מתקרר לטמפרטורת החדר, להוסיף 1 מ"ל של המניה פוספט, 1 מ"ל של המניה Na 2 CO 3 ו -10 מ"ל של NaHCO 3.
- לקבלת בינוני BG11 מוצק, להוסיף 15 גרם של אגר ו -700 מ"ל מים לאחד flask. אל הבקבוק השני, להוסיף 3 גרם של Na 2 S 2 O 3, 226 מ"ל מים, 10 מ"ל של BG11 100x, 1 מ"ל של יסודות קורט 1 מ"ל של המניה ברזל. חיטוי שני הפתרונים. לאחר פתרונות אלה התקררו לטמפרטורת החדר, לשלב אותם ולהוסיף 1 מ"ל של המניה פוספט, 1 מ"ל של המניה Na 2 CO 3, 10 מ"ל של חיץ TES, ו -10 מ"ל של NaHCO 3.
הערה: פתרונות מוכנים בנפרד, כדי למנוע משקעים של מלחים מסוימים.
- לבחירה על סוכרוז, להכין 50% (w / v) פתרון סוכרוז. סנן לעקר את הפתרון עם 0.2 מיקרומטר מסננים ומוסיפים BG11 (100 מ"ל של סוכרוז 50% ל -900 מ"ל של BG11) לייצר BG11 / 5% הצלחות סוכרוז.
הערה: אל תוסיף NaHCO 3 עד BG11 / 5% צלחות אגר סוכרוז. להוסיף Na 2 CO 3 כרגיל. - עבור culturing של Synechococcus להוסיף 10 מ"ל של 1 M 4- (2-hydroxyethyl) piperazine-1-ethanesulfonic חומצה, N - (2-hydroxyethyl) piperazine- ע"נ42; - (2-ethanesulfonic חומצה) (HEPES) ו 1 מ"ל של ויטמין B 12 (טבלה 1) עד 1 ליטר של המדיום BG11.
הערה: טרנספורמציה של זנים בתרבית תקשורת BG11 הזמין המסחרי היא משמעותית פחות יעיל מאשר מתכוני תקשורת BG11 המתואר כאן ולכן אינו מומלצת.
צמיחת 2. כחוליות זנים
- תרבות זני צלוחיות חרוטי 100 מיליליטר עם נפח של עד 50 מ"ל ולנער ב 120 סל"ד. חותם צלחות BG11 עם Parafilm לנקב שלושה חורים קטנים בצד של הצלחת לאפשר חילוף גזים. דגירת כל הזנים על 30 מעלות צלזיוס מתחת נורות פלורסנט בתוך photobioreactor בעצימות אור בין 20-40 μmol פוטונים מ -2 שניות -1.
- השימוש הטוב ביותר בטכניקות סטריליות. טיפול בכל הזנים כחוליות במנדף זרימה למינרית.
הערה: זה חשוב במיוחד כאשר זנים מתורבתים עם סוכרוז המכיל התקשורת, אשר ניתן בקלות contaminגוונים.
3. דור של בונה פלסמיד
- ערכות עיצוב של פריימרים, לרבות באתרי אנזים הגבלה הנדרש, באמצעות תוכנת עיצוב פריימר כגון Primer3 (http://frodo.wi.mit.edu/primer3/), כדי להגביר שני ~ 1 kb אזורים 5 'ו 3' של גן של עניין. התייעצו עם רצף הגנום של מינים כחוליות דרך Cyanobase (http://genome.kazusa.or.jp/cyanobase). ראה טבלה 2 לכל פריימרים משמשים כאן. בעת תכנון פריימרים בחשבון את הגורמים הבאים:
- ודא כי אזורים מוגברים כוללים 5 'ו 3' אזורים של הגן כי יהיה מוטציה, למשל איור 1.
- אין מוטציה אזורים גניים להימנע מוטציה מכוונת של antisense ו RNAs ללא קידוד. עבור דור של מוטנטים ב Synechocystis, עיין ברשימת האתרים תחילת תעתיק מתועד Mitschke et al., 2011 18, על מנת למנוע מוטציה של antisenseאו RNAs קידוד שאינם.
- בעת בחירת אזורי איגוף אינם כוללים את מסגרת קריאה פתוחה השלמה של גנים סמוכים כמו ביטוי של גנים אלה coli Escherichia עלול להפריע שיבוט.
- להגביר מוצרים על ידי PCR באמצעות DNA פולימרז איכות גבוהה פי הוראות היצרן.
הערה: מניסיוננו אנזים זה מייצר כמה שגיאות.- גדר 50 μl PCR תגובות המכילות חיץ HF ואו 0, 1.5 או 3 μl של DMSO. השתמש 100 ננוגרם של הדנ"א הגנומי לכל תגובה. השתמש בתכנית מורכב צעד denaturation ראשוני של 98 מעלות צלזיוס למשך 30 שניות, 35 סיבובים של 98 מעלות צלזיוס למשך 10 שניות, 67 ° C למשך 30 שניות, 72 ° C למשך 30 שניות, ואחריו צעד ארכה סופי של 72 מעלות צלזיוס למשך 5 דקות. זה בדרך כלל נותן מוצרים עקביים.
- ודא מוצרי PCR דגימות מתעכלים עם אנזימי endonuclease עבור הגודל הנכון באמצעות ג'ל אלקטרופורזה. הפעל 1% (w / v) agarose ג'ל המכיל 0.02%(V / v) ברומיד ethidium עבור 45 דקות ב 100 V.
זהירות: ברומיד Ethidium הוא mutagen פוטנציאלי צריך להיות מטופלים עם הגנה מתאימה. - לטהר מוצרי PCR באמצעות ערכת טיהור DNA על פי הוראות היצרן. כמו כן להשתמש ערכה זו לטיהור שברה פלסמיד, כוללים חתיכות לחתוך ג'ל agarose. Elute DNA מטוהרים 14 μl של מים.
- לקבלת שלבים שיבוט, דגירה תערובות התגובה endonuclease הגבלה ב 37 מעלות צלזיוס למשך> 1 hr בנפח כולל של 30 μl על פי הוראות היצרן.
- לקבלת שלבי קשירה, קטעי DNA ולקשור בטמפרטורת חדר למשך> 1 hr בנפח כולל של 20 μl, המכילה 5 μl של פלסמיד מתעכל מטוהר, 12 μl של כנס מתעכל מטוהר, 2 μl של חיץ 1 μl של אנזים.
- כן Escherichia coli DH5α תאי transformant פי השיטה הבאה.
- לגדול E. לילה coli תרבות ב 10 מ"ל לוריא Bertani (LB) בתקשורת.
- לחסן 400 מ"ל LB בבקבוק חרוטי 1 L המכיל 6 מ"ל 1 M MgCl 2 (טבלה 1) עם 1 מ"ל של תרבות לילה.
- לגדול התרבות ב 37 מעלות צלזיוס ב 220 סל"ד במשך כ -4 שעות או עד OD 600nm מגיע 0.4-0.6.
- מניחים את התאים על הקרח במשך שעה 1.
- צנטריפוגה ב 2800 XG במשך 10 דקות עד גלולת תאים ב 4 ° C..
- הסר supernatant ו resuspend ב 160 מ"ל פתרון (טבלה 1) ו דגירה על קרח למשך 20 דקות.
- צנטריפוגה ב 2800 XG במשך 10 דקות עד גלולת תאים ב 4 ° C..
- הסר supernatant ו resuspend ב 4 מ"ל פתרון A + גליצרול (טבלה 1).
- כן 50 aliquots μl, להקפיא בנוזל N 2, ולאחסן ב -80 מעלות צלזיוס.
- מערבבי 5 μl של תערובת קשירה עם 50 μl של תאי מוסמכות דגירה במשך שעה 1 על קרח.
- מחממים לזעזע את התאים ב 42 מעלות צלזיוס למשך 90 שניות, folloד על ידי דגירה על קרח במשך 2 דקות.
- להוסיף 950 μl של תקשורת LB (טבלה 1) ו לדגור על 37 מעלות צלזיוס במשך שעה 1.
- Aliquot 50 ו -200 μl על צלחות עם אנטיביוטי מתאים, או אמפיצילין (100 מיקרוגרם / מ"ל) ו / או kanamycin (30 מיקרוגרם / מ"ל).
זהירות: שניהם kanamycin ו אמפיצילין רעילים צריכים להיות מטופלים עם הגנה מתאימה. - פיק דגירה מושבות אחת ב 2 מ"ל התקשורת LB מחוסן עם אנטיביוטי מתאים.
- לטהר את כל פלסמידים באמצעות ערכת טיהור פלסמיד miniprep פי הוראות היצרן.
- צור פלסמידים, בדוגמה הספציפית הזו לאחר שחיסל את הגנים cpcC1C2, על פי השלבים הבאים.
- להגביר את אזור איגוף '1,012 נ"ב 5 (שבר שמאלה) באמצעות פריימרים cpcC1C2leftfor ו cpcC1C2leftrev (ראה שלב 3.2, טבלה 2). הסר כמות קטנה של תגובת PCR ולאשר אםמוצר גודל נכון כבר מוגבר באמצעות ג'ל אלקטרופורזה (שלב 3.3). תקציר שבר זה pUC19 עם Xba אני Bam HI (שלב 3.5).
- לטהר הוא ההכנות (שלב 3.4), ולקשור (שלב 3.6), להפוך (שלב 3.7) ובשל ארבעה 2 מיליליטר LB תרבויות נוזליות עם אמפיצילין (100 מיקרוגרם / מיליליטר) ממושבות נפרדות עבור טיהור פלסמיד באמצעות minipreps (שלב 3.8).
- בדוק החדרת את הפיסה לתוך pUC19 באמצעות Xba I / Bam HI עיכול ג'ל אלקטרופורזה (שלב 3.3). להקות של 2,660 נ"ב 1,012 נ"ב מצביעות מבוא נכון של הכנס לתוך פלסמיד.
- להגביר את אזור איגוף '1,016 נ"ב 3 (שבר מימין) באמצעות פריימרים cpcC1C2rightfor ו cpcC1C2rightrev (ראה שלב 3.2, טבלה 2). הסר כמות קטנה של תגובת PCR ולאשר אם המוצר הגודל הנכון כבר מוגבר באמצעות ג'ל אלקטרופורזה (שלב 3.3). תקציר שבר זה pUC19 עם Sac I ו- Eco RI (stEP 3.5).
- לטהר הוא ההכנות (שלב 3.4), ולקשור (שלב 3.6), להפוך (שלב 3.7) ובשל ארבעה 2 מיליליטר LB תרבויות נוזליות עם אמפיצילין (100 מיקרוגרם / מיליליטר) ממושבות נפרדות עבור טיהור פלסמיד באמצעות minipreps (שלב 3.8).
- בדוק החדרת את הפיסה לתוך pUC19 באמצעות Sac I / העיכול Eco RI (שלב 3.5) ו ג'ל אלקטרופורזה (שלב 3.3). להקות של 2,660 נ"ב 1,016 נ"ב מצביעות מבוא נכון של הכנס לתוך פלסמיד.
הערה: Xba I / Bam HI אתרי שיבוט של 5 'באזור Sac I / Eco RI עבור שיבוט של 3' אזור pUC19 משמשת בכל מקום אפשרי. אם הדבר אפשרי, תמיד כוללים אתר בם HI על פריימר הפוכה במשך 5 'באזור או פריימר קדימה עבור 3' באזור על מנת להבטיח כי לאחר מכן השלבים שיבוט קלים יותר לביצוע. - רצף השני מוסיף לקבוע אם הרצף נכון באמצעות פריימרים פורשו הכניסה לאתר, למשל M13 קדימה לאחור M13 (טבלה 2). הרצף חייב להיות נכון כדי להבטיח שאין טעויות מוכנסות אזורי איגוף.
- והבלו שבר שמאלה מן pUC19 באמצעות Xba I / Bam HI העיכול. לעכל את pUC19 + שבר תקין עם Xba I / Bam HI (שלב 3.5).
- לטהר את שבר 1,012 נ"ב שמאלה 3,676 נ"ב pUC19 + שבר כבר מן ג'ל agarose (שלב 3.3) באמצעות כריתה של ה- DNA באמצעות להב סכין המנתחים.
- לטהר הוא ההכנות (שלב 3.4), ולקשור (שלב 3.6), להפוך (שלב 3.7) ובשל ארבעה 2 מיליליטר LB תרבויות נוזליות עם אמפיצילין (100 מיקרוגרם / מיליליטר) ממושבות נפרדות עבור טיהור פלסמיד באמצעות minipreps (שלב 3.8).
- בדוק החדרת את הפיסה לתוך pUC19 + שבר תקין באמצעות Xba I / Bam HI העיכול (שלב 3.5) ו ג'ל אלקטרופורזה (שלב 3.3). להקות של 3,676 נ"ב 1,012 נ"ב מצביעים הכנסה נכונה של להכניס לתוך פלסמיד (קוראים לזה B פלסמיד).
- ובלו קלטת npt1 / sacB מ -19 pUM24cm באמצעות עיכול בם HI. תקציר פלסמיד B עם Bam HI (שלב 3.5).
הערה: הקלטת npt1 / sacB לא חייב להיות מטוהרים מן ג'ל agarose מאז pUM24cm המקודד חלבון המקנה עמידות chloramphenicol. לכן אם מושבות גדלות על LB / אמפיצילין / צלחות אגרו kanamycin השילוב האפשרי היחיד שיוביל מושבות עמידות הוא שילוב של הקלטת npt1 / sacB לתוך B. פלסמיד - לטהר הכנות הן (שלב 3.4), ולקשור (שלב 3.6), להפוך (שלב 3.7) ובשל ארבעה 2 מיליליטר LB תרבויות נוזליות עם אמפיצילין (100 מיקרוגרם / מיליליטר) ו kanamycin (30 מיקרוגרם / מיליליטר) ממושבות נפרדות עבור טיהור פלסמיד באמצעות minipreps (שלב 3.8).
- בדוק להכנסה של הקלטת npt1 / sacB לתוך B פלסמיד באמצעות עיכול בם HI (שלב 3.5) ו ג'ל אלקטרופורזה (שלב 3.3). להקות של 4,688 נ"ב 3,894 נ"ב מצביעות הכנסה נכונה של הדואר להכניס לתוך פלסמיד (קוראים לזה פלסמיד).
- לחלופין, סוף להקהות את npt1 / sacB קלטת השיבוט לתוך אתר endonuclease ההגבלה שונה שבין קרעי השמאל וימין ב פלסמיד npt1 / קלטת sacB חייבים להיות משובט בין השמאל ושבר תקינים.
הערה: אם ביטוי של קלטת זרים נדרש אז זה צריך להיות מוכנס שבין הקרעים על ימין ועל שמאל של B. פלסמיד פלסמיד זה משמש לאחר מכן בשלבים בנוקאאוט המסומנים.
דור 4. מסומן מוטאנטים Synechocystis ו Synechococcus
- הגדרת תרבות טריה ידי ייחס לולאה מלאה של תאים לתוך 30-50 מיליליטר של מדיום BG11. לגדל את התרבות במשך 2-3 ימים כדי 750nm OD = 0.2 כדי 0.6.
הערה: בדרך כלל מושבות בודדות הם קטנות מדי לשימוש עבור חיסון וחשיפה של תאים בודדים אפילו לרמות נמוכות של אור תגרומנה photoinhibition ובחירהעבור מוטנטים עמידים אור. - צנטריפוגה 1-2 מ"ל של התרבות ב 2,300 XG במשך 5 דקות וזורקים supernatant. אל צנטריפוגות כל התרבויות כחוליות ב> 2,300 XG מכיוון שהדבר עלול לגרום נזק לתאים. שטוף את הכדור פעם אחת עם מדיום BG11.
הערה: אל resuspend התאים על ידי vortexing מכיוון שהדבר עלול לגרום לאובדן של פילי שחיוניים ספיגת DNA. Resuspend התאים על ידי pipetting עדין. - הוסף בינוני BG11 לנפח סופי של 100 μl. העברת התאים צינור מסביב לתחתית 14 מ"ל.
- הוסף 1 מיקרוגרם של פלסמיד לתאים ומערבבים ידי הקשה עדינה. הוסף <10 μl של פלסמיד.
הערה: רצוי הפלסמיד צריך להיות בריכוז של> 100 ng / μl אך ריכוזים נמוכים מזה הם מספיקים טרנספורמציה מוצלחת. - הנח צינורות למטה אופקי בחממה. דגירת תרבויות במשך 4-6 שעות.
הערה: תאים יכולים להיות מעורבים בקצרה על ידי קשה בכל שעה 1-2 אבל זה לא הכרחי. ניתן למקם דגימותרועד חממה אף זו אינה משפרת יעילות משמעותית. - מורחים aliquots של תערובת DNA תרבית תאים / פלסמיד על צלחות אגר BG11 ללא אנטיביוטיקה. בדרך כלל 20 μl ו -80 aliquots μl פרושים על צלחות נפרדות.
- ~ 24 שעות לאחר מכן, להוסיף 2.5-3 מ"ל של 0.6% פתרון אגר במים המכילים kanamycin (מחיר 20 מ"ל: 0.12 גר 'אגר, 100 μl של 100 מ"ג / מ"ל kanamycin) לצלחת אגר. מגניבי פתרון זה ~ 42 ° C, ולהוסיף את שולי הצלחת אגרה. הטה את הצלחת לכן הפתרון מהווה גם 'אגר למעלה שכבה על פני השטח.
- דגירת צלחות אגרו לתקופה נוספת של זמן. מושבות צריכות להיות גלויות לאחר כ 7 ימים.
הערה: צלחות אגרו יכולות להיערם 3 גבוהים באינקובטור. בדרך כלל מאות מושבות מתקבלים לכל שינוי. - מושבות בודדים Streak על BG11 + kanamycin (30 מיקרוגרם / מ"ל) צלחות אגר. מחלק את הצלחת אגרה לתוך 6 סקטורים ולהשתמש קיסם בקצה קהה לשעוט החוצההמושבות על כל מגזר פרט. קבלת מושבות אחת אינה צמיחה חשובה, רק של transformants.
- אשר בנוקאאוט בסימן PCR באמצעות פולימראז תקי DNA על פי הוראות היצרן. הוסף 2 μl של MgCl 2 (25 מ"מ) לכל תגובה.
- הסרת חלק קטן של תאים ולהעביר לתוך צינור המכיל 50 מים μl ו ~ 20 425-600 חרוזי זכוכית מיקרומטר. Shake ב ויברטור במשך 5 דקות ב ~ 2,000 סל"ד. צנטריפוגה ב 15,700 XG במשך 5 דקות ולהשתמש 5 μl של supernatant לכל 50 μl תגובת PCR.
הערה: אל resuspend הפתרון. פסולת התא צריכה להישאר בתחתית של התחתית.
- הסרת חלק קטן של תאים ולהעביר לתוך צינור המכיל 50 מים μl ו ~ 20 425-600 חרוזי זכוכית מיקרומטר. Shake ב ויברטור במשך 5 דקות ב ~ 2,000 סל"ד. צנטריפוגה ב 15,700 XG במשך 5 דקות ולהשתמש 5 μl של supernatant לכל 50 μl תגובת PCR.
- אמת מוטנטים
- פריימרים עיצוב אשר span באזור בנוקאאוט באמצעות תוכנת עיצוב פריימר (כגון Primer3). פריימרים עיצוב החל מ ~ 200 נ"ב משני צדי באזור בנוקאאוט.
הערה: Primers לאימות מוטצית cpcC1C2 מתואר בטבלה 2ו מכונים cpcC1C2for ו cpcC1C2rev. - להגביר מוצרים באמצעות תכנית מורכבת צעד denaturation ראשוני של 95 מעלות צלזיוס למשך 2 דקות, 35 סיבובים של 95 מעלות צלזיוס במשך 1 דקות, 60 מעלות צלזיוס במשך 1 דקות, 72 מעלות צלזיוס במשך 1 דקות לכל קילו של רצף, ואחריו צעד ארכה סופי של 72 מעלות צלזיוס למשך 5 דקות. כלול שליטה wild-type. זה בדרך כלל נותן מוצרים עקביים.
- ודא הגנוטיפ באמצעות ג'ל אלקטרופורזה. Transformants בנוקאאוט המסומן יציג להקה של ~ 4 kb (0.2 kb משני השמאל ושהברים תקינים בתוספת קלטת npt1 / sacB) והיעדר להקת wild-type (איור 2).
הערה: במקרים מסוימים להקת kb ~ 4 לא הוא ציינה המוטציה הניכרת בשל גודלו של מוצר PCR זה. עם זאת, אם להקה המתאימה לגודל הצפוי של wild-type לא הוא ציין אז בדרך כלל זן זה הוא בנוקאאוט ניכר.
- פריימרים עיצוב אשר span באזור בנוקאאוט באמצעות תוכנת עיצוב פריימר (כגון Primer3). פריימרים עיצוב החל מ ~ 200 נ"ב משני צדי באזור בנוקאאוט.
- אם להקת wild-type עדיין קיימת אז מחדש פס המתח עלBG11 + kanamycin טריים (30 מיקרוגרם / מ"ל) צלחת אגר וחזור על PCR. חזור על התהליך מחדש פסים עד המוטציה היא פרדה כך שאף להקת wild-type הוא ציינה את תגובת PCR.
הערה: הגדלת כמות kanamycin לריכוז של 50 מיקרוגרם / מ"ל, ואז 100 מיקרוגרם / מ"ל לפעמים הכרחי על מנת להפריד מוטציה ניכרת במלואה. - אם הזן מציג פרופיל מוטציה מסומן באמצעות PCR, אז מחדש פס על BG11 + kanamycin טרי (30 מיקרוגרם / מיליליטר) צלחת אגרה. השתמש זן זה על מנת ליצור את בנוקאאוט המסומנת.
הערה: הפרוטוקול יכול לשמש כדי ליצור מוטציות המסומנות רק קלטת עמידות לאנטיביוטיקה כלומר על ידי החלפת קלטת npt1 / sacB רק עם קלטת npt1 מ pUC18K 20 בין השמאל ושבר תקינים..
5. דור של מוטאנטים מסומנים Synechocystis
- הגדרת תרבות טריה של בנוקאאוט בסימן ייחס לולאה מלאה גאמות לתוך 30-50 מ"ל של מדיום BG11. לגדל את התרבות במשך 2-3 ימים כדי 750nm OD = 0.2 כדי 0.6.
- צנטריפוגה 10 מ"ל של התרבות ב 2,300 XG במשך 5 דקות וזורקים supernatant. שטפי פעם עם מדיום BG11.
הערה: אל resuspend התאים על ידי vortexing מכיוון שהדבר עלול לגרום לאובדן של פילי שחיוניים ספיגת DNA. Resuspend התאים על ידי pipetting עדין. - להוסיף BG11 לנפח סופי של 200 μl. העברת התאים צינור מסביב לתחתית 14 מ"ל.
- הוסף 1 מיקרוגרם של ה- DNA פלסמיד B לתאים ומערבבים ידי הקשה עדינה.
- דגירת הדגימות במשך 4-6 שעות. הנח צינורות למטה אופקי.
הערה: תאים יכולים להיות מעורבים בקצרה על ידי קשה בכל שעה 1-2 אבל זה לא הכרחי. דוגמאות ניתן להציב חממה רועדת אם כי זה לא לשפר את היעילות. - הוסף 1.8 מ"ל של מדיום BG11 דגירה דגימות עבור סכום כולל של 4 ימים עם רעד. זהו זמן מספיק כדי לאפשר רקומבינציה להתרחש עותקי כרומוזומליות המרובים.
- aliquots פלייט של התערובת טרנספורמציה על צלחות אגר סוכרוז BG11 / 5%. פלייט 50 μl, 10 μl ו 1 μl לכל צלחת אגר. אם מדשאת מושבה מופיעה על כל צלחות אגרו אלה לדלל את התמיסה נוספת aliquot על צלחות טריות. מושבות צריכות להיות גלויות לאחר כ 7 ימים.
- תיקון 30-50 מושבות בודדות על BG11 + kanamycin (30 מיקרוגרם / מיליליטר) אגרו צלחות ראשונות BG11 / 5% צלחות אגרו סוכרוז שני, באמצעות קיסם בקצה קהה. כל חיידקים הגדלים על צלחות סוכרוז BG11 / 5% אך לא BG11 + צלחות kanamycin הן knockouts המסומן פוטנציאלי. חיידקים הגדלים על הצלחות הם צפויים להיות סוכרוז עמיד עקב מוטציה בגן sacB.
- ודא knockouts מסומנת באמצעות אותו פריימרים והשיטה כמו שמש לבדוק את knockouts ניכרה. למשל cpcC1C2for ו cpcC1C2rev (טבלה 2) לאימות בנוקאאוט המסומנת cpcC1C2. בנוקאאוט מסומנת תציג להקה על t המתאים ג'ל agaroseo גודל wild-type מינוס באזור שנמחק (איור 2).
- אם הזן מציג פרופיל מוטציה מסומן באמצעות PCR (שלב 4.11.2) ו ג'ל אלקטרופורזה (איור 2), ואז שוב פס על צלחת אגר BG11 טרי ללא אנטיביוטיקה.
6. אחסון לטווח ארוך של זנים
- הגדרת תרבות טריה של המתח על ידי ייחס לולאה מלאה של תאים לתוך 30-50 מיליליטר של מדיום BG11. לגדל את התרבות במשך 3-4 ימים כדי 750nm OD = 0.4 ל -0.7.
- שטפו תאים פעם עם BG11 ו resuspend ב ~ 2 מ"ל של BG11.
- להוסיף 0.8 מ"ל של תאים מרוכזים כדי צינור אחד. לאחר מכן מוסיפים 0.2 מ"ל של 80% גליצרול מעוקרים הסינון.
- אופציונאלי: להוסיף 0.93 מיליליטר של תאים מרוכזים צינור נוסף. להוסיף 0.07 מ"ל של DMSO כדי הצינור הזה.
זהירות: DMSO הוא רעיל צריך להיות מטופלים עם הגנה מתאימה. - אחסן את שני הצינורות ב -80 מעלות צלזיוס. כדי להחיות זנים להסיר את הצינור לגרד כמה תאים עם שן בוטהלאסוף לצלחת אגרה ללא אנטיביוטיקה. Streak החוצה כרגיל באמצעות לולאה סטרילי.

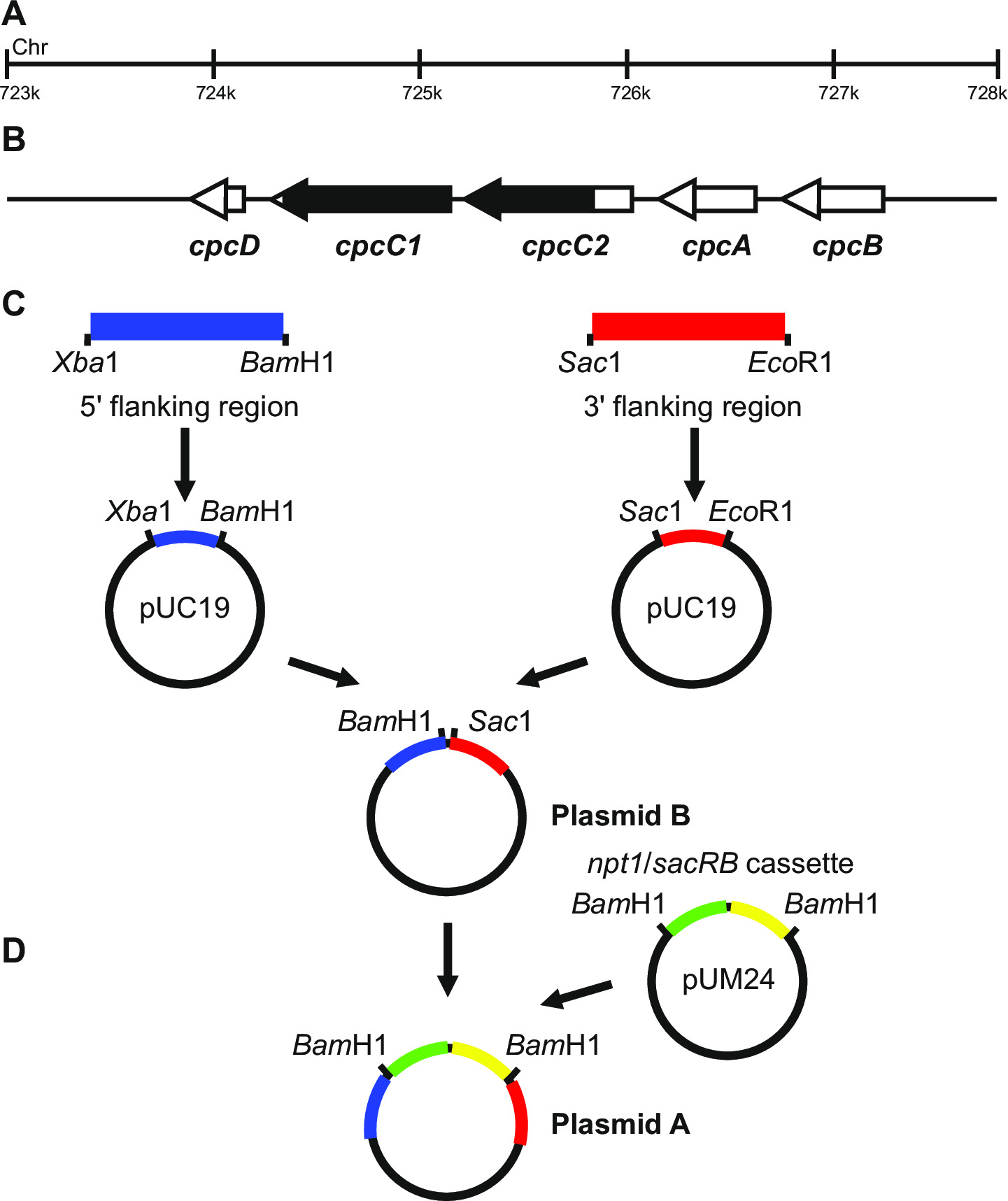
איור 1: הבנייה פלסמיד עבור הדור של knockouts מסומנים ולא מסומנים, למשל cpcC1 ו cpcC2 ב Synechocystis (א) אזור של הגנום Synechocystis שם (B) cpcC1 ו cpcC2 וגנים סמוכים נמצאים.. מודגש בשחור הוא האזור של הגנום למחיקה של המוטציה. (ג) אתרים של הגנום אשר מוגבר על ידי PCR. ה 'באזור האיגוף (מסומן בכחול) ו 3' 5 באזור איגוף (המסומן באדום) הם מוגברים עם אתרי endonuclease הגבלה עבור שיבוט לתוך pUC19. 5 '(או 3') איגוף באזור הוא נכרת מתוך pUC19 ונוסף pUC19 + 3 '(או 59;) איגוף פלסמיד באזור לייצר ב פלסמיד (ד) קלטת npt1 / sacB מ pUM24 הוא נכרת באמצעות עיכול בם HI והחדיר בין 5 'ו 3' איגוף אזורים ליצור פלסמיד א אנא לחץ כאן כדי להציג גדול גרסה של נתון זה.
{kind=link}
תוצאות
עיצוב פלסמיד הוא קריטי עבור דור מוצלח של שני מוטנטים מסומנים ולא מסומנים. איור 1 נותן דוגמא של פלסמיד ו- B השתמשו כדי ליצור מוטציה מחיקה בגני Synechocystis cpcC1 ו cpcC2 13. בכל מקרה 5 'ו 3' אזורי האיגוף כ 900-1,000 נ"ב. אזורי איגוף מופ?...
Discussion
השלבים הקריטיים ביותר בדור של מוטציות לא מסומנות הם: 1) עיצוב פלסמיד זהיר כדי להבטיח את אזור המיקוד רק משתנה; 2) להבטיח כי דגימות להישאר axenic, במיוחד כאשר בתרבית על סוכרוז; 3) ציפוי טרנספורמציה תאים עבור דור מוטציה מסומן בתחילה על צלחות אגרו BG11 חסרי אנטיביוטיקה, ואחריו תו?...
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgements
אנו מודים טראסט לחינוך סביבתי שירותים האגודה, ביולוגיה סינתטית בקרן קיימברידג SynBio ומשרד צדק חברתי והעצמה, ממשלת הודו, עבור תמיכה כספית.
Materials
| Name | Company | Catalog Number | Comments |
| NaNO3 | Sigma | S5506 | |
| MgSO4.7H2O | Sigma | 230391 | |
| CaCl2 | Sigma | C1016 | |
| citric acid | Sigma | C0759 | |
| Na2EDTA | Fisher | EDT002 | |
| H3BO3 | Sigma | 339067 | |
| MnCl2.4H2O | Sigma | M3634 | |
| ZnSO4.7H2O | Sigma | Z4750 | |
| Na2MoO4.2H2O | Sigma | 331058 | |
| CuSO4.5H2O | Sigma | 209198 | |
| Co(NO3)2.6H2O | Sigma | 239267 | |
| Ferric ammonium citrate | Sigma | F5879 | |
| K2HPO4 | Sigma | P3786 | |
| Na2CO3 | Fisher | SODC001 | |
| TES | Sigma | T1375 | |
| NaHCO3 | Fisher | SODH001 | |
| HEPES | Sigma | H3375 | |
| cyanocobalamin | Sigma | 47869 | |
| Na2S2O3 | Sigma | 72049 | |
| Bacto agar | BD | 214010 | |
| Sucrose | Fisher | SUC001 | |
| Petri dish 90 mm triple vented | Greiner | 633185 | |
| 0.2 µm filters | Sartorius | 16534 | |
| 100 ml conical flasks | Pyrex | CON004 | |
| Parafilm M 100 mm x 38 m | Bemis | FIL003 | |
| Phusion high fidelity DNA polymerase | Phusion | F-530 | |
| Agarose | Melford | MB1200 | |
| DNA purification kit | MoBio | 12100-300 | |
| Restriction endonucleases | NEB | ||
| T4 ligase | Thermo Scientific | EL0011 | |
| Luria Bertani broth | Invitrogen | 12795-027 | |
| MES | Sigma | M8250 | |
| Kanamycin sulfate | Sigma | 60615 | |
| Ampicillin | Sigma | A9518 | |
| GeneJET plasmid miniprep kit | Thermo Scientific | K0503 | |
| 14 ml round-bottom tube | BD falcon | 352059 | |
| GoTaq G2 Flexi DNA polymerase | Promega | M7805 | |
| 425-600 µm glass beads | Sigma | G8772 | |
| Glycerol | Sigma | G5516 | |
| DMSO | Sigma | D8418 | |
| Fluorescent bulbs | Gro-Lux | 69 | |
| HT multitron photobioreactor | Infors |
References
- Zwirglmaier, K., et al. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol. 10, 147-161 (2008).
- Galloway, J. N., et al. Nitrogen cycles: past, present, and future. Biogeochemistry. 70, 153-226 (2004).
- Lea-Smith, D. J., et al. Contribution of cyanobacterial alkane production to the ocean hydrocarbon cycle. Proc Natl Acad Sci U S A. , (2015).
- Howe, C. J., Barbrook, A. C., Nisbet, R. E. R., Lockhart, P. J., Larkum, A. W. D. The origin of plastids. Philos Trans R Soc Lond B Biol Sci. 363, 2675-2685 (2008).
- Lea-Smith, D. J., Bombelli, P., Vasudevan, R., Howe, C. J. Photosynthetic, respiratory and extracellular electron transport pathways in cyanobacteria. Biochim Biophys Acta. , (2015).
- McCormick, A. J., et al. Hydrogen production through oxygenic photosynthesis using the cyanobacterium Synechocystis sp PCC 6803 in a bio-photoelectrolysis cell (BPE) system. Energy Environ. Sci. 6, 2682-2690 (2013).
- Bradley, R. W., Bombelli, P., Lea-Smith, D. J., Howe, C. J. Terminal oxidase mutants of the cyanobacterium Synechocystis sp. PCC 6803 show increased electrogenic activity in biological photo-voltaic systems. Phys Chem Chem Phys. 15, 13611-13618 (2013).
- Ducat, D. C., Way, J. C., Silver, P. A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 29, 95-103 (2011).
- Dismukes, G. C., Carrieri, D., Bennette, N., Ananyev, G. M., Posewitz, M. C. Aquatic phototrophs: efficient alternatives to land-based crops for biofuels. Curr Opin Biotechnol. 19, 235-240 (2008).
- Tan, L. T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 68, 954-979 (2007).
- Volk, R. B., Furkert, F. H. Antialgal, antibacterial and antifungal activity of two metabolites produced and excreted by cyanobacteria during growth. Microbiol Res. 161, 180-186 (2006).
- Scott, S. A., et al. Biodiesel from algae: challenges and prospects. Curr Opin Biotechnol. 21, 277-286 (2010).
- Lea-Smith, D. J., et al. Phycobilisome-deficient strains of Synechocystis sp. PCC 6803 have reduced size and require carbon-limiting conditions to exhibit enhanced productivity. Plant Physiol. 165, 705-714 (2014).
- Lea-Smith, D. J., et al. Thylakoid terminal oxidases are essential for the cyanobacterium Synechocystis sp. PCC 6803 to survive rapidly changing light intensities. Plant Physiol. 162, 484-495 (2013).
- Liu, X., Sheng, J., Curtiss, R. Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci U S A. 108, 6899-6904 (2011).
- Xu, H., Vavilin, D., Funk, C., Vermaas, W. Multiple deletions of small cab-like proteins in the cyanobacterium Synechocystis sp PCC 6803 - Consequences for pigment biosynthesis and accumulation. J Biol Chem. 279, 27971-27979 (2004).
- Castenholz, R. W. Culturing methods for Cyanobacteria. Method Enzymol. 167, 68-93 (1988).
- Mitschke, J., et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp PCC6803. Proc Natl Acad Sci U S A. 108, 2124-2129 (2011).
- Ried, J. L., Collmer, A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 57, 239-246 (1987).
- Vieira, J., Messing, J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 19, 259-268 (1982).
- Vermaas, W. F. J., Williams, J. G. K., Rutherford, A. W., Mathis, P., Arntzen, C. J. Genetically Engineered Mutant of the Cyanobacterium Synechocystis 6803 Lacks the Photosystem-Ii Chlorophyll-Binding Protein Cp-47. Proc Natl Acad Sci U S A. 83, 9474-9477 (1986).
- Westphal, S., Heins, L., Soll, J., Vothknecht, U. C. Vipp1 deletion mutant of Synechocystis: A connection between bacterial phage shock and thylakoid biogenesis?. Proc Natl Acad Sci U S A. 98, 4243-4248 (2001).
- Zhang, S. Y., Shen, G. Z., Li, Z. K., Golbeck, J. H., Bryant, D. A. Vipp1 Is Essential for the Biogenesis of Photosystem I but Not Thylakoid Membranes in Synechococcus sp PCC 7002. J Biol Chem. 289, 15904-15914 (2014).
- Taroncher-Oldenberg, G., Nishina, K., Stephanopoulos, G. Identification and analysis of the polyhydroxyalkanoate-specific beta-ketothiolase and acetoacetyl coenzyme A reductase genes in the cyanobacterium Synechocystis sp strain PCC6803. Appl Environ Microbiol. 66, 4440-4448 (2000).
- Hein, S., Tran, H., Steinbuchel, A. Synechocystis sp. PCC6803 possesses a two-component polyhydroxyalkanoic acid synthase similar to that of anoxygenic purple sulfur bacteria. Arch Microbiol. 170, 162-170 (1998).
- Ng, A. H., Berla, B. M., Pakrasi, H. B. Fine tuning of photoautotrophic protein production by combining promoters and neutral sites in Synechocystis 6803, a cyanobacterium. Appl Environ Microbiol. , (2015).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved