
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
RNA פולימראז קשור DNA לתכנות תמלול במבחנה וחישוב מולקולרי
In This Article
Summary
אנו מתארים את ההנדסה של פולימראז T7 RNA חדשני הקשור לדנ"א כדי לווסת את תגובות התמלול במבחנה. אנו דנים בצעדים לסינתזה ואפיון חלבונים, מאמתים רגולציה של תמלול הוכחת הרעיון, ודנים ביישומיה במחשוב מולקולרי, אבחון ועיבוד מידע מולקולרי.
Abstract
ננוטכנולוגיה של DNA מאפשרת הרכבה עצמית ניתנת לתכנות של חומצות גרעין לצורות ודינמיקה שנקבעו על ידי המשתמש עבור יישומים מגוונים. עבודה זו מדגימה כי מושגים מננוטכנולוגיה DNA ניתן להשתמש כדי לתכנת את הפעילות אנזימטית של פולימראז T7 RNA נגזר phage (RNAP) ולבנות רשתות רגולטוריות גנים סינתטיים מדרגיים. ראשית, T7 RNAP הקשור לאוליגונוקלאוטיד מתוכנן באמצעות ביטוי של RNAP מתויג N-סופני SNAP וצימוד כימי לאחר מכן של תג SNAP עם אוליגונוקלאוטיד בנצילגואנין (BG). לאחר מכן, עקירת גדיל חומצת גרעין משמשת לתכנות שעתוק פולימראז לפי דרישה. בנוסף, מכלולי חומצת גרעין עזר יכולים לשמש "גורמי שעתוק מלאכותיים" כדי לווסת את האינטראקציות בין RNAP T7 מתוכנת DNA עם תבניות ה- DNA שלה. מנגנון רגולציה זה של תמלול במבחנה יכול ליישם מגוון התנהגויות מעגלים כגון לוגיקה דיגיטלית, משוב, מדורג ומולטיפלקסינג. המורכבות של ארכיטקטורה רגולטורית זו של הגנים מאפשרת הפשטה, סטנדרטיזציה ושינוי קנה מידה של עיצוב. תכונות אלה יאפשרו יצירת סוג של מכשירים גנטיים במבחנה עבור יישומים כגון חישה ביולוגית, זיהוי מחלות ואחסון נתונים.
Introduction
מחשוב דנ"א משתמש בערכה של אוליגונוקלאוטידים מעוצבים כמדיום לחישוב. אוליגונוקלאוטידים אלה מתוכנתים עם רצפים להרכבה דינמית על פי לוגיקה שצוינה על-ידי המשתמש ולהגיב לכניסות ספציפיות של חומצת גרעין. במחקרי הוכחת הרעיון, הפלט של החישוב מורכב בדרך כלל מסט של אוליגונוקלאוטידים שכותרתם פלואורסצנטית שניתן לזהות באמצעות אלקטרופורזה ג'ל או קוראי לוחות פלואורסצנטיות. במהלך 30 השנים האחרונות, מעגלים חישוביים DNA מורכבים יותר ויותר הודגמו, כגון מפלי לוגיקה דיגיטלית שונים, רשתותתגובהכימית, ורשתות עצביות 1,2,3. כדי לסייע בהכנת מעגלי DNA אלה, מודלים מתמטיים שימשו לחיזוי הפונקציונליות של מעגלי גנים סינתטיים4,5, וכלים חישוביים פותחו עבור עיצוב רצף DNA אורתוגונל6,7,8,9,10 . בהשוואה למחשבים מבוססי סיליקון, היתרונות של מחשבי DNA כוללים את יכולתם להתממשק ישירות עם ביומולקולים, לפעול בפתרון בהיעדר ספק כוח, כמו גם את הקומפקטיות והיציבות הכוללות שלהם. עם הופעתו של רצף הדור הבא, העלות של סינתזה של מחשבי DNA כבר יורדת בשני העשורים האחרונים בקצב מהר יותר מאשר חוק מור11. יישומים של מחשבים מבוססי DNA כאלה מתחילים כעת לצוץ, כגון לאבחון מחלות12,13, להפעלת ביופיסיקה מולקולרית14, וכפלטפורמות אחסון נתונים15.

איור 1:מנגנון של תזוזת גדיל DNA בתיווך דריסת רגל. דריסת הבוהן, δ, היא רצף חופשי ולא מאוגד על דופלקס חלקי. כאשר תחום משלים (δ*) מוצג על גדיל שני, הדומיין δ החופשי משמש דריסת רגל להכלאה, ומאפשר לשאר הגדיל (ɑ*) לעקור לאט את המתחרה שלו באמצעות תגובה הפיכה רוכסת / בלתי מתפשרת המכונה העברת גדילים. ככל שאורך δ גדל, ה- ΔG לתגובה הקדמית פוחת, והעקירה מתרחשת ביתר קלות. אנא לחץ כאן כדי להציג גירסה גדולה יותר של איור זה.
{kind=link}
עד כה, רוב מחשבי הדנ"א משתמשים במוטיב מבוסס היטב בתחום הננוטכנולוגיה הדינמית של הדנ"א המכונה עקירת גדיל DNA בתיווך בוהן (TMDSD, איור 1)16. מוטיב זה מורכב דופלקס DNA כפול תקוע חלקית (dsDNA) המציגים overhangs קצרים "דריסת רגל" (כלומר, 7- עד 10 נוקלאוטידים (nt)). גדילי "קלט" חומצת גרעין יכולים לקיים אינטראקציה עם הדופלקסים החלקיים דרך דריסת המכהן. זה מוביל לעקירתו של אחד הגדילים מהדופלקס החלקי, והגדיל המשוחרר הזה יכול לשמש כקלט לדופלקסים חלקיים במורד הזרם. לכן, TMDSD מאפשרת סילוק את אותות ועיבוד מידע. באופן עקרוני, מוטיבים אורתוגונליים של TMDSD יכולים לפעול באופן עצמאי בפתרון, ומאפשרים עיבוד מידע מקביל. היו מספר וריאציות על תגובת TMDSD, כגון החלפת גדיל DNA בתיווך דריסת רגל (TMDSE)17, דריסות "ללא דליפות" עם תחומים ארוכים כפולים18, דריסות רצף לא תואמות19, ו "אחיזת יד", עקירת גדילים בתיווך20. עקרונות עיצוב חדשניים אלה מאפשרים אנרגטיות ודינמיקה של TMDSD מכוונות יותר לשיפור ביצועי מחשוב ה- DNA.
מעגלי גנים סינתטיים, כגון מעגלי גנים תמלול, מסוגלים גם לחשב21,22,23. מעגלים אלה מוסדרים על ידי גורמי שעתוק חלבונים, המפעילים או מדחיקים שעתוק של גן על ידי קשירה לאלמנטים ספציפיים של DNA רגולטורי. בהשוואה למעגלים מבוססי דנ"א, למעגלי התמלול יש מספר יתרונות. ראשית, לתעתיק אנזימטי יש שיעור תחלופה גבוה בהרבה ממעגלי ה- DNA הקטליטיים הקיימים, ובכך יוצר יותר עותקים של פלט לכל עותק יחיד של קלט ומספק אמצעי יעיל יותר להגברת אותות. בנוסף, מעגלי שעתוק יכולים לייצר מולקולות פונקציונליות שונות, כגון aptamers או קידוד RNA שליח (mRNA) עבור חלבונים טיפוליים, כמו יציאות חישוב, אשר ניתן לנצל עבור יישומים שונים. עם זאת, מגבלה מרכזית של מעגלי התמלול הנוכחיים היא חוסר המדרגיות שלהם. הסיבה לכך היא שיש קבוצה מוגבלת מאוד של גורמי שעתוק מבוססי חלבון אורתוגונל, ועיצוב דה נובו של גורמי שעתוק חלבונים חדשים נשאר מאתגר מבחינה טכנית וגוזל זמן רב.

איור 2: הפשטה ומנגנון של קומפלקס פולימראז "לקשור" ו"כלוב". (A ו- B) קשירה אוליגונוקלאוטיד מסומנת באופן אנזימטי לפולימראז T7 באמצעות תגובת SNAP-tag. כלוב המורכב ממקדם T7 "מזויף" עם תוחם משלים קשירה מאפשר לו להתיידד לקשירה ולחסום פעילות שעתוק. (C)כאשר המפעיל (a*b*) קיים, הוא נקשר לאיכות הבוהן על הרצועה אוליגונוקלאוטיד (ab) ומזיז את אזור b * של הכלוב, ומאפשר שעתוק להתרחש. נתון זה שונה מצ'ו ושי27. קיצורים: RNAP = RNA פולימראז. אנא לחץ כאן כדי להציג גירסה גדולה יותר של איור זה.
{kind=link}
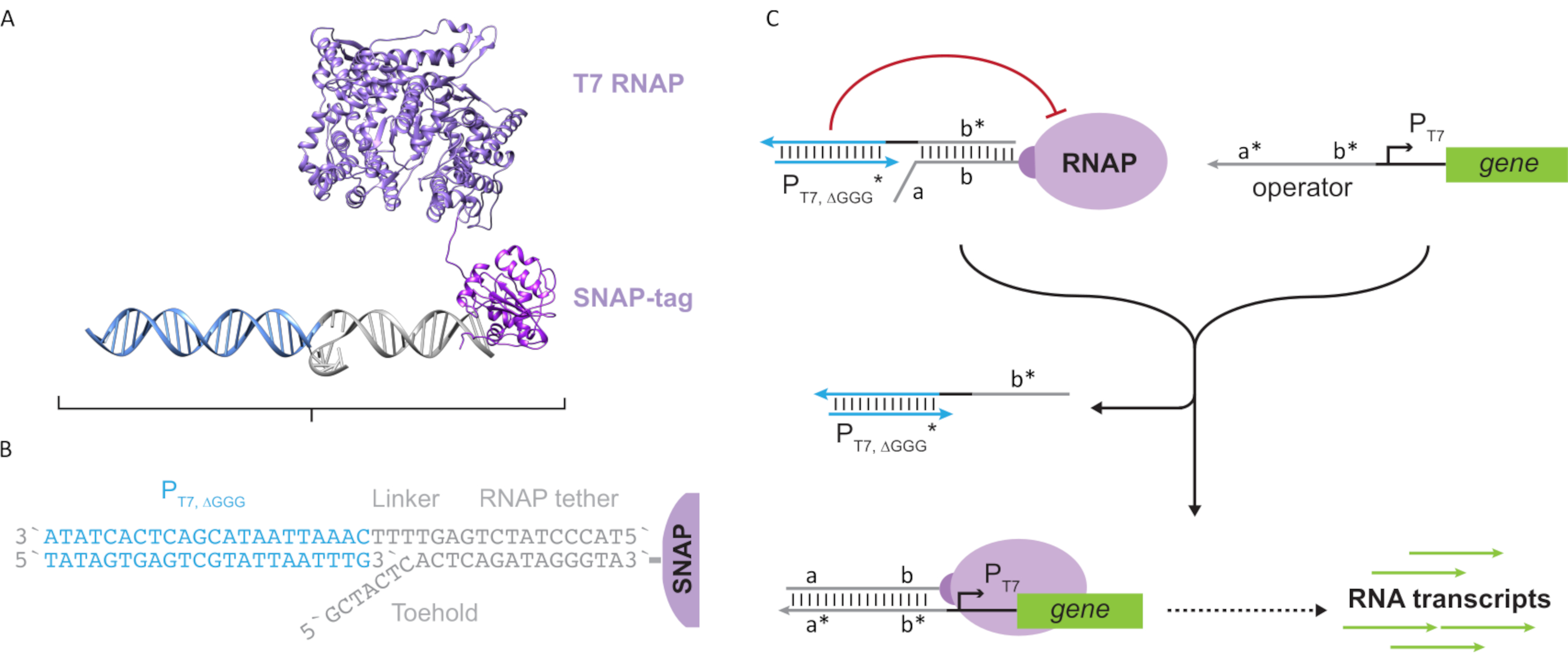
מאמר זה מציג אבן בניין חדשנית למחשוב מולקולרי המשלבת את הפונקציות של מעגלי שעתוק עם מדרגיות של מעגלים מבוססי DNA. אבן בניין זו היא T7 RNAP המחוברת באופן קוולנטי עם רצועת DNA חד-גדילית(איור 2A). כדי לסנתז את ה-T7 RNAP הזה, הפולימראז הותך לתגSNAP-24 של N-terminal והתבטא מחדש באשריצ'יה קולי. תג SNAP הגיב אז עם אוליגונוקלאוטיד פונקציונלי עם מצע BG. הרצועה אוליגונוקלאוטיד מאפשרת מיקום של אורחים מולקולריים בסמיכות לפולימראז באמצעות הכלאת DNA. אורח אחד כזה היה חוסם תמלול תחרותי המכונה "כלוב", המורכב ודופלקס DNA "מזויף" של מקדם T7 ללא גנים במורד הזרם(איור 2B). כאשר הוא מאוגד לרנ"פ באמצעות קשירת האוליגונוקלאוטיד שלו, הכלוב מעכב פעילות פולימראז על ידי אי-התאמה של תבניות DNA אחרות עבור כריכת RNAP, ועיבוד ה-RNAP במצב "כבוי"(איור 2C).
כדי להפעיל את הפולימראז למצב "ON", תוכננו תבניות DNA T7 עם תחומי "אופרטור" חד-גדיליים במעלה הזרם של מקדם T7 של הגן. תחום המפעיל (כלומר, תחום a*b* איור 2C) יכול להיות מתוכנן לעקור את הכלוב מה- RNAP באמצעות TMDSD ולמקם את ה- RNAP הקרוב למקדם T7 של הגן, ובכך ליזום שעתוק. לחלופין, תבניות DNA תוכננו גם כאשר רצף המפעיל היה משלים לגדילי חומצת גרעין עזר המכונים "גורמי שעתוק מלאכותיים" (כלומר, גדילי TFA ו- TFB באיור 3A). כאשר שני גדילים מוצגים לתוך התגובה, הם יתאספו באתר המפעיל, יצירת תחום פסאודו-רציף חדש a *b*. תחום זה יכול לאחר מכן לעקור את הכלוב באמצעות TMDSD כדי ליזום תמלול (איור 3B). גדילים אלה יכולים להיות מסופקים או אקסוגני או מיוצר.

איור 3: תכנות סלקטיבי של פעילות פולימראז באמצעות מפעיל מתג בעל שלושה רכיבים. (A)כאשר קיימים גורמי התמלול (TFA ו- TFB), הם נקשרים לתחום המפעיל במעלה הזרם של המקדם, ויוצרים רצף פסאודו חד-גדילי (a*b*) המסוגל לעקור את הכלוב באמצעות תזוזת דנ"א מתווכת. (B)תחום a*b* זה יכול לעקור את הכלוב באמצעות TMDSD כדי ליזום תמלול. נתון זה שונה מצ'ו ושי27. קיצורים: TF = גורם שעתוק; RNAP = RNA פולימראז; TMDSD = תזוזת גדיל DNA בתיווך דריסת רגל. אנא לחץ כאן כדי להציג גירסה גדולה יותר של איור זה.
{kind=link}
השימוש בגורמי שעתוק מבוססי חומצת גרעין עבור ויסות תמלול במבחנה מאפשר יישום מדרגי של התנהגויות מעגל מתוחכמות כגון לוגיקה דיגיטלית, משוב, ומדדי אותות. לדוגמה, ניתן לבנות מפלי שער לוגיים על ידי תכנון רצפי חומצות גרעין כך שהתעתיקים מגן במעלה הזרם מפעילים גן במורד הזרם. יישום אחד המנצל את המדורגים והמכפילים המסוגלים בטכנולוגיה מוצעת זו הוא פיתוח מעגלי מחשוב מולקולריים מתוחכמים יותר לאבחון נייד ועיבוד נתונים מולקולריים. בנוסף, שילוב יכולות המחשוב המולקולרי וסינתזת דה נובו RNA יכול לאפשר יישומים חדשים. לדוגמה, ניתן לתכנן מעגל מולקולרי כדי לזהות אחד או שילוב של RNAs המוגדרים על-ידי המשתמש כתשומות ופלט של RNAs טיפוליים או mRNAs המקודדים פפטידים פונקציונליים או חלבונים עבור יישומים רפואיים נקודתיים.
Protocol
1. הכנת מאגר
הערה: הכנת מאגר טיהור חלבון יכולה להתרחש בכל יום; כאן, זה נעשה לפני תחילת הניסויים.
- הכן מאגר תמוגה/שיווי משקל המכיל 50 מ"מ טריס (הידרוקסימתיל)אמינומטן (טריס), 300 מ"מ נתרן כלורי (NaCl), 5% גליצריל ו-5 מ"מ β-מרצפטואתנול (BME), pH 8. הוסף 1.5 מ"ל של 1M טריס, 1.8 מ"ל של 5M NaCl, 1.5 מ"ל של גליצלול, 25.2 מ"ל של מים דהויונים (ddH2O) לתוך צינור צנטריפוגה 50 מ"ל, ולהוסיף 10.5 μL של 14.2 M BME רק לפני השימוש.
הערה: טריס יכול לגרום רעילות חריפה; לכן, להימנע לנשום את האבק שלה, ולהימנע ממגע עור ועיניים. BME רעיל ויש להשתמש בו רק במכסה אדים. חשוב להוסיף BME אחרון, רק לפני resuspension ותסיסה תא. ראה טבלה 1 עבור נוסחת מאגר תמה. - הכן חוצץ כביסה (pH 8) המכיל 50 מ"מ טריס, 800 מ"מ NaCl, 5% גליצריל, 5 mM BME ו 20 mM imidazole. הוסף 1.5 מ"ל של 1 M טריס, 4.8 מ"ל של 5 M NaCl, 1.5 מ"ל של גליצרל, ו 22.2 מ"ל של ddH2O לתוך צינור צנטריפוגה 50 מ"ל. רק לפני השימוש, להוסיף 7 μL של 14.2 M BME ו 200 μL של 2 M imidazole כדי 20 מ"ל של הפתרון לעיל.
הערה: כדי למנוע רעילות חריפה עקב imidazole, להשתמש בציוד מגן אישי. חשוב להוסיף BME ו imidazole האחרון, רק לפני שטיפת החלבון מהעמוד. ראה טבלה 2 לקבלת נוסחת מאגר כביסה. - הכן חוצץ אלוטיון (pH8) המכיל 50 מ"מ טריס, 800 מ"מ NaCl, 5% גליצריל, 5 mM BME ו 200 mM imidazole. הוסף 0.5 מ"ל של 1 M טריס, 1.6 מ"ל של 5 M NaCl, 0.5 מ"ל של גליצרל, ו 6.4 מ"ל של ddH2O לצינור צנטריפוגה 15 מ"ל. רק לפני השימוש, להוסיף 3.5 μL של 14.2 M BME ו 1 מ"ל של 2 M imidazole כדי 10 מ"ל של הפתרון לעיל.
הערה: חשוב להוסיף BME ו imidazole האחרון, רק לפני שאילוט החלבון מתוך העמודה. ראה טבלה 3 עבור נוסחת מאגר אלוטיון. - הכן 2x חוצץ אחסון (להיות מעורב 1:1 עם גליצרל) המכיל 100 mM טריס, 200 mM NaCl, 40 mM BME, ו 2 mM אתילנדימינטראטראקטי (EDTA), 0.2% של פעילי שטח לא יוניים (ראה טבלת החומרים). הכן 50 מ"ל של מאגר האחסון על ידי הוספת 5 מ"ל של 1 M טריס, 2 מ"ל של 5 M NaCl, 42.56 מ"ל של ddH2O, 200 μL של 0.5 M EDTA, 100 μL של פעילי שטח לא יוניים לצינור צנטריפוגה 50 מ"ל. מערבבים עד שהפתרון הומוגני, מסננים את מאגר האחסון דרך מסנן מזרק של 0.2 מיקרומטר ומוסיפים 140.8 μL של BME לפתרון לעיל לפני השימוש.
הערה: כדי למנוע רעילות חריפה עקב EDTA, להימנע לנשום את האבק שלה, ולהימנע ממגע עור ועיניים. חשוב להוסיף BME אחרון ולערבב את כל חיץ האחסון 1:1 עם גליצריל, רגע לפני אחסון החלבון המטוהר. ראה טבלה 4 לקבלת נוסחת מאגר אחסון.
2. צמיחת תרבות הלילה: יום 1
- הכן 1,000x קנאמיצין מלאי על ידי המסת 500 מ"ג של kanamycin ב 10 מ"ל של ddH2O.
הערה: השתמש בציוד מגן אישי כדי למנוע רעילות חריפה עקב kanamycin. - הוסף 20 μL של 1,000x קנאמיצין מלאי ל 20 מ"ל של ציר ליסוגניה. באמצעות טיפ פיפטה סטרילי, לתקוע BL21 E. coli גליצרל המרה ולאחר מכן לחסן את התרבות על ידי החדרת הקצה לתוך מרק מדיה הצמיחה.

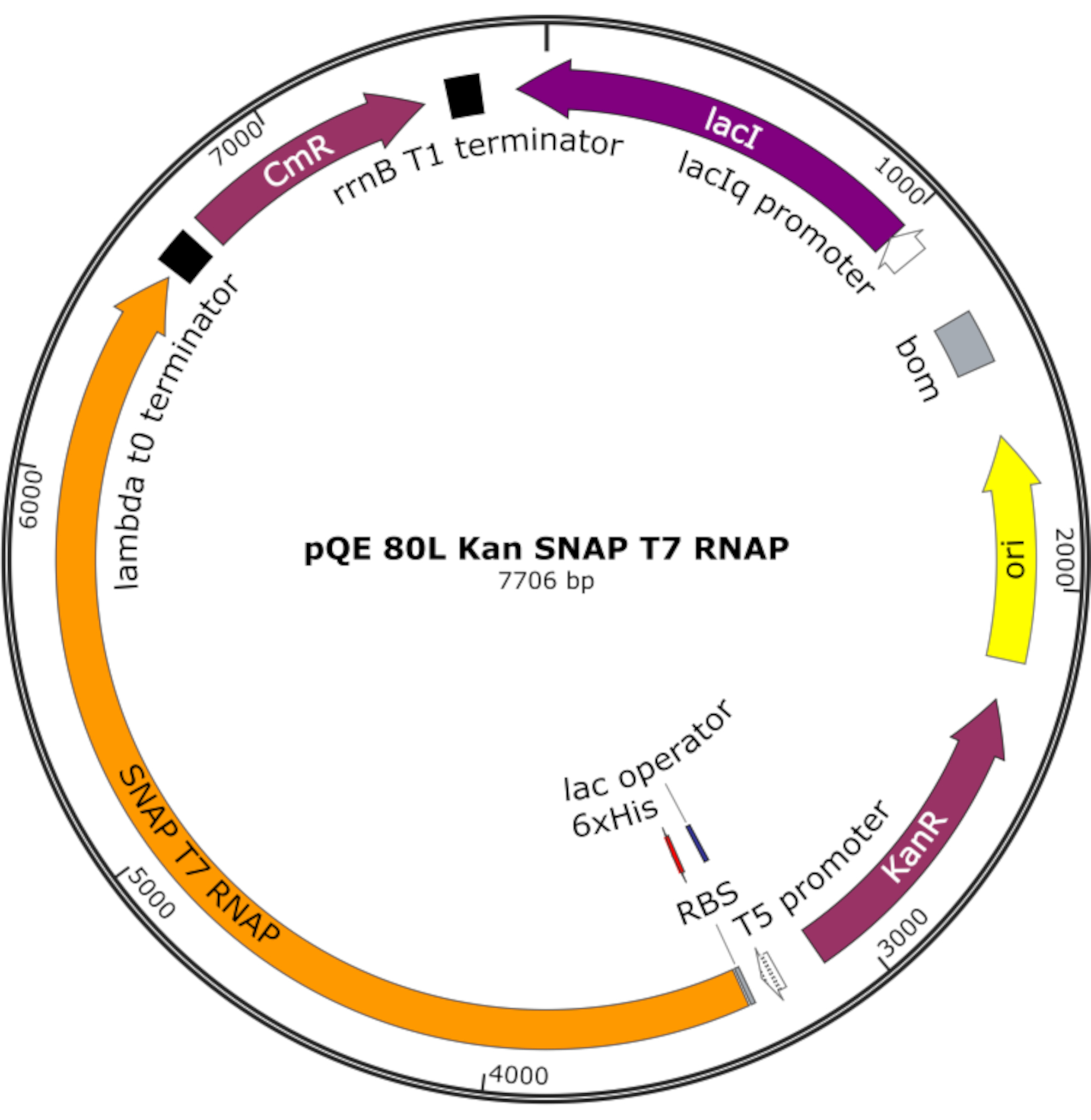
איור 4: מפת פלסמיד עבור SNAP T7 RNAP. הפלסמיד מקודד T7 RNAP המכיל תג היסטידין N-terminal (6x שלו) ו- SNAP-tag תחום (SNAP T7 RNAP) תחת מדכא לאק (lacI) על עמוד שדרה pQE-80L. תכונות אחרות כוללות עמידות קנאמיצין (KanR) ו גנים עמידות כלורמפניקול (CmR). קיצור: RNAP = RNA פולימראז. אנא לחץ כאן כדי להציג גירסה גדולה יותר של איור זה.
{kind=link}
הערה: הפלסמיד מקודד T7 RNAP המכיל תג היסטידין N-terminal ותחום SNAP-תג (SNAP T7 RNAP), כמו גם גן התנגדות קנאמיצין תחת עמוד שדרה pQE-80L (איור 4)25.
- שוב, להוסיף 20 μL של 1,000x קנאמיצין מלאי בקבוקון תרבות נפרד המכיל 20 מ"ל של ציר ליזוגניים, ולדגירה אותו כמו שליטה.
- לדגור על שתי הדגימות (ממדרגות 2.2 ו 2.3) לילה עבור 12-18 שעות ב 37 °C (50 °F), תוך סיבוב ב 10 × גרם.
3. צמיחת תאים ואינדוקציה: יום 2
- לחסן 400 מ"ל של ציר ליזוזני המכיל 400 μL של מלאי kanamycin עם 4 מ"ל של תרבות הצמיחה הלילה מן שלב 2.4. לדגור על בקבוקיות התרבות ב 37 °C (50 °F), תוך סיבוב ב 10 × גרם.
- לאחר התרבות הגיעה צפיפות אופטית (OD) ב 600 ננומטר של ~ 0.5, להוציא 1 מ"ל של מדגם מבקבוק הצמיחה כמו שליטה. אחסן את דגימת הבקרה ב- 4 °C (7°F).
- לגרום לתאים עם איזופרופיל β-D-1-thiogalactopyranoside (IPTG) על ידי הוספת 40 μL של 1M IPTG לכל 100 מ"ל של תרבות כדי להשיג ריכוז סופי של 0.4 mM IPTG. לדגור את המדגם במשך 3 שעות ב 37 °C (50 °F), מסתובב ב 10 × גרם, ולאחר מכן לסובב את התרבות המושרה ב 8,000 × גרם במשך 10 דקות כדי כדורי התאים. הסר את supernatant, ולאחסן את הכדור ב -20 °C (70 °F) עד לשימוש נוסף.
הערה: כדי למנוע רעילות חריפה עקב IPTG, להימנע לנשום את האבק שלה, ולהימנע ממגע עור ועיניים. במידת הצורך, אתה יכול להשהות את הניסוי כאן ולהמשיך למחרת.
4. תמוגת תאים, טיהור חלבונים: יום 3
- יש להזרים מחדש את גלולה התא המאוחסנת עם 10 מ"ל של חיץ תמוז על הקרח, ולהסתובב בעדינות כדי להבטיח את הכדור כולו הוא resuspended. לאחר מכן, פיפטה 1 מ"ל של מדגם לתוך עשרה צינורות 1.5 מ"ל שנשמרים על קרח.
- Sonicate כל מדגם על הגדרת משרעת של "1", פעמו עבור 2 s עם מחזור חובה 50% על פני תקופה של 30 s. לפני ואחרי כל דגימה, לנקות את קצה sonication עם 70% אתנול ו ddH2O. שמור את כל הדגימות על קרח במהלך ואחרי sonication.
הערה: יש להרחיק 70% אתנול מחום ולהבות פתוחות. - שיווי משקל חומצה nitrilotriacetic טעון ניקל (Ni-NTA) עמוד ספין טיהור לטמפרטורת עבודה של 4 °C (50 °F). מקם/אחסן את העמודה ב-4 °C (7°F) והמשיך להחזיק בקרח במהלך השימוש.
- צנטריפוגות עשר 1 דגימות מ"ל ב 15,000 × גרם במשך 20 דקות ב 4 °C (55 °F). בזהירות pipette את supernatant המכיל את RNAP רקומביננטי מבלי להפריע לכדור. במידת הצורך, השתמש במאגר שיווי משקל נוסף כדי לכוונן את עוצמת הקול הכוללת ל- ≥ 6 מ"ל.
- הסר בעדינות את הכרטיסיה התחתונה מעמודת הסיבוב של Ni-NTA כדי לאפשר זרימה דרך העמודה. מניחים את העמוד בצינור צנטריפוגה, ושומרים אותו על קרח.
הערה: השתמש בצינור צנטריפוגה 50 מ"ל עם עמודות ספין Ni-NTA 3 מ"ל. - צנטריפוגה את העמודה ב 700 × g ו 4 °C (5 °F) במשך 2 דקות כדי להסיר את מאגר האחסון. שפל את העמודה על-ידי הוספת 6 מ"ל של מאגר שיווי משקל לעמודה. אפשר למאגר להיכנס באופן מלא למיטת השרף.
- הסר את מאגר השקיבות מהעמודה על ידי צנטריפוגה ב 700 × גרם ו 4 °C (7 °F) במשך 2 דקות. לפני הוספת תמצית התא המוכנה לעמודה, מקם תקע תחתון בעמודה כדי להימנע מאובדן מוצר כלשהו. לאחר מכן, מוסיפים את תמצית התא לעמודה, ומערבבים על מערבל שייקר מסלולי במשך 30 דקות ב 4 °C (70 °F).
- הסר את התקע התחתון מהעמודה והצב את העמודה בצינור צנטריפוגה של 50 מ"ל המסומן בתווית Flow דרך. צנטריפוגות העמוד ב 700 × g במשך 2 דקות כדי לאסוף את הזרימה דרך.
- הוסף 6 מ"ל של מאגר כביסה לעמוד כדי לשטוף את השרף. צנטריפוגה את העמודה ב 700 × גרם במשך 2 דקות כדי לאסוף את השבר בצינור צנטריפוגה חדש שכותרתו לשטוף 1. חזור על שלב זה פעמיים נוספות עבור סך של 3 שברים נפרדים, ולאסוף את השברים בצינורות צנטריפוגות נפרדים (לשטוף 2 לשטוף 3).
- הוסיפו 3 מ"ל של חוצץ אלוטיון כדי לחמוק מהחלבונים המתויגים שלו מהשרף. צנטריפוגה את העמודה ב 700 × g במשך 2 דקות כדי לאסוף שבר בצינור צנטריפוגה חדש שכותרתו eluate 1. חזור על שלב זה פעמיים נוספות עבור סך של 3 שברים נפרדים, ולאסוף את השברים לתוך צינורות צנטריפוגות נפרדים (eluate 2 ו eluate 3).
- מערבבים את האלואטים ומבצעים התפלה להסרת מלחים מתמיסת החלבון.
- Pipette 15 mL של 0.05 % w/v פוליסורבט 20 על פני יחידת מסנן צנטריפוגלית של 100 kDa. צנטריפוגה ב 4,000 × g במשך 40 דקות ולהשליך את הזרימה דרך.
- השתמש במסנן המצופה כדי לרכז את eluates 1, 2, ו 3 (9 מ"ל של סך של חלבון eluate + 6 מ"ל של חוצץ אחסון) כדי ~ 1,500 μL. צנטריפוגה המסנן ב 3,220 × גרם במשך 20 דקות, בעדינות pipette לשטוף את הממברנה כדי למנוע משקעים.
- לדלל את המדגם ל 15 מ"ל עם מאגר אחסון. בצע חילופי מאגרים באמצעות מאגר אחסון 1:1,000 על-ידי חזרה על שלב 4.11.2 פעמיים נוספות.
- לכמת את החלבון המטוהר על ידי מדידת ספיגת השבר ב 280 ננומטר. רוקן את ספקטרופוטומטר עם מאגר אחסון (מאגר אחסון 2x ב 4 °C (4 °C). מערבבים בעדינות את המדגם של eluates המשולב ולמדוד את ספיגתו.
הערה: בצע שלוש קריאות נפרדות בדילול של 1x, פי 10 ו- 50x של דגימת החלבון כדי לכמת את החלבון בממוצע. לדלל דוגמאות במאגר האחסון. - התאם את דגימות החלבון ל-100 מיקרומטר באמצעות חיץ אחסון פי 2. לדלל את המדגם המותאם 1:1 לפי נפח עם 100% גליצריל. יש לאחסן את תמיסה חלבון וכתוצאה מכך ב -80 °C (70 °F).
5. ניתוח אלקטרופורזה של ג'ל סודיל דודסיל סולפט-פוליאקרילמיד (SDS-PAGE) של מוצר חלבון: יום 3
- הפעל ג'ל SDS-PAGE לניתוח חלבונים. מערבבים 9 μL של המדגם עם 3 μL של 4x ליתיום דודסיל סולפט (LDS) חלבון טעינת צבע. מחממים את הדגימות ב 95 °C (55 °F) במשך 10 דקות.
- טען את הדגימות על הגדרת ג'ל ביס-טריס SDS-PAGE של 4-12%. טען את סולם החלבון היטב 1, ולאחר מכן עם דגימות (משמאל לימין): זרימה דרך, לשטוף 1, לשטוף 2, לשטוף 3, elution 1, אלוטיון 2, elution 3, ואת elution מותפל הכולל.
הערה: טבלה 5 מכילה טבלת טעינה לדוגמה עבור ג'ל SDS-PAGE. - הפעל את דגימות ג'ל טעון 2-(N- morpholino) חומצה אתנולפונית (MES) חוצץ במשך 35 דקות ב 200 V. לשטוף את הג'ל במגש נקי שלוש פעמים במשך 10 דקות כל אחד באמצעות 200 מ"ל של ddH2O, עם עצבנות עדינה כדי להסיר כל SDS מטריצת הג'ל.
הערה: ללבוש ציוד מגן אישי כדי למנוע רעילות חריפה עקב MES. - מכתימים את הג'ל בכחול קומאסי ב-20 מ"ל, ודגר את הג'ל למשך הלילה בטמפרטורת החדר בתסיסה עדינה. דה-להכתים את הג'ל פעמיים במשך 1 שעה כל אחד עם 200 מ"ל של ddH2O עם עצבנות עדינה על שייקר מסלולית.
הערה: שטיפת הג'ל לתקופה ארוכה יותר או החלפת מים לעתים קרובות תשפר את הרגישות. בנוסף, הצבת רקמת ניגוב עדינה מקופלת במיכל לספיגת צבע עודף תאיץ את תהליך ההכתמה.
6. אימות פונקציונלי של SNAP T7 RNAP באמצעות תמלול במבחנה
הערה: פרוטוקול זה משתמש בתבנית DNA, המקודדת עבור אפטאמר RNA ברוקולי פלואורסצנטי ומאפשרת שימוש בפלואורסצנטיות כדי לפקח על הקינטיקה של שעתוק על קורא לוחות פלואורסצנטיות.
- הגדר שלוש תגובות תמלול במבחנה (IVT) כדי להשוות את הפעילות של SNAP T7 RNAP עם RNAP T7 מסוג פראי (WT) ממקור מסחרי ובקרת מאגר בלבד. כוונן את עוצמת הקול של כל תגובה ל- 20 μL.
- הכן את תגובת SNAP T7 RNAP IVT על ידי ערבוב 2 μL של מאגר תמלול 10x, 0.4 μL של 25 mM ribonucleoside טריפוספט (rNTP) תערובת, 5 μL של 500 nM תבנית DNA, 2 μL של 500 nM SNAP T7 RNAP, ו 10.6 μL של ddH2O.
- הכן את תגובת WT RNAP IVT על ידי ערבוב 2 μL של מאגר שעתוק 10x, 0.4 μL של 25 mM rNTP לערבב, 5 μL של תבנית DNA 500 nM, 2 μL של WT T7 RNAP, ו 10.6 μL של ddH2O.
- הכן את תגובת IVT המאגר בלבד על ידי ערבוב 2 μL של מאגר שעתוק 10x, 0.4 μL של 25 mM rNTP לערבב, 5 μL של תבנית DNA 500 nM, ו 12.6 μL של ddH2O.
הערה: הוסף את הרנ"א אחרון, תוך שמירה על הדגימות על קרח עד הצגתו. טבלה 6, טבלה 7 וטבלה 8 מכילות את נוסחאות התגובה של IVT.
- נטר את קינטיקה שעתוק על קורא צלחת פלואורסצנטית במשך 2 שעות במרווחים של 2 דקות ב 37 °C (57 °F) באמצעות אורך גל עירור של 470 ננומטר ואורך גל פליטה של 512 ננומטר.
7. הכנת אוליגונוקלאוטידים ב-BG מותאמים: יום 1
- להמיס את oligonucleotide עם שינוי 3'-אמין ב ddH2O לריכוז סופי של 1 mM. תייג S1זה.
- לערבב 25 μL של 1 M נתרן ביקרבונט (NaHCO3), 284 μL של 100% דימתיל סולפוקסיד (DMSO), 125 μL של S1 (מניית אוליגונוקלאוטיד), ו 66 μL של 50 mM של BG-N-הידרוקסיסוקינימיד (NHS) אסתר (BG-GLA-NHS) מדולל עם DMSO, כוונן את הנפח ל 500 μL, ודגורה לילה בטמפרטורת החדר ב 100 × גרם.
הערה: יש להרחיק את DMSO מחום ומלהבה מכיוון שהוא נוזל דליק. טבלה 9 מכילה את נוסחת התגובה עבור ההטיות BG לאוליגונוקלאוטיד.
- לערבב 25 μL של 1 M נתרן ביקרבונט (NaHCO3), 284 μL של 100% דימתיל סולפוקסיד (DMSO), 125 μL של S1 (מניית אוליגונוקלאוטיד), ו 66 μL של 50 mM של BG-N-הידרוקסיסוקינימיד (NHS) אסתר (BG-GLA-NHS) מדולל עם DMSO, כוונן את הנפח ל 500 μL, ודגורה לילה בטמפרטורת החדר ב 100 × גרם.
8. משקעים אתנול/אצטון של הצטיידות BG-אוליגונוקלאוטיד: יום 2
- צנטריפוגה המוצר של שלב 7.1.1. ב 13,000 × g במשך 5 דקות. בזהירות להעביר את supernatant לצינור טרי ולהשליך כל BG מזורץ. לפצל את התגובה לשני aliquots שווה μL כדי למנוע גלישה, ולבצע את השלבים הבאים על שני aliquots.
- הוסף 1/10th של נפח של 3 M נתרן אצטט (25 μL), ואחריו 2.5x את הנפח ב 100% אתנול (625 μL). דגירה ב -80 °C (50 °F) למשך שעה.
הערה: השתמש בציוד מגן אישי בעת טיפול הן נתרן אצטט (עלול לגרום לגירוי בעיניים, בעור, במערכת העיכול ובמערכת הנשימה) ואתנול (דליק מאוד, גורם לגירוי במגע). במידת הצורך, השהו את הניסוי כאן והמשיכו למחרת. - מניחים את הצינורות בצנטריפוגה ומסמנים את הקצה החיצוני. צנטריפוגות הצינורות ב 17,000 × גרם במשך 30 דקות ב 4 °C (50 °F).
הערה: גלולה אוליגונוקלאוטיד תופיע בקצה המסומן של הצינור. - מבלי להפריע לכדור, השלך את הסופר-טבעי. למעלה עם 750 μL של 70% אתנול צונן, ולסובב ב 17,000 × גרם במשך 10 דקות ב 4 °C (7 °F).
- מבלי להפריע לכדור, השלך את הסופר-טבעי. למעלה עם 750 μL של 100% אצטון, ולסובב ב 17,000 × גרם במשך 10 דקות ב 4 °C (7 °F).
הערה: השתמש בציוד מגן אישי בעת טיפול אצטון כפי שהוא דליק מאוד וגורם לגירוי במגע. - עם מכסה הצינור פתוח, אוויר יבש במשך 5 דקות כדי להסיר כל אצטון עודף באמצעות אידוי. להמיס מחדש את האולגונוקלאוטיד ב- 250 μL של חיץ 1x Tris-EDTA (TE) כדי לייצר פתרון ~850 μM BG-oligonucleotide.
- חזור על שלבים 8.2 עד 8.6 והתמוסס מחדש ב- 70 μL של מאגר TE 1x. תייג S2זה.
9. ניקוי BG-אוליגונוקלאוטיד באמצעות כרומטוגרפיה של סינון ג'ל
- להשעות את המטריצה על ידי היפוך נמרץ של העמודות מספר פעמים; הסר את המכסה העליון והצמד את הקצה התחתון של העמודה. מניחים את העמוד בצינור צנטריפוגה של 1.5 מ"ל, וצנטריפוגה של הצינור ב-1,000 × גרם למשך דקה אחת בטמפרטורת החדר. השלך את צינור המאגר והאיסוף הנבה.
הערה: חשוב למנוע היווצרות ואקום. השתמש בעמודות מוכנות באופן מיידי. - מניחים את העמודים הארוזים בצינורות צנטריפוגות נקיים של 1.5 מ"ל. הוסף 300 μL של מאגר TE 1x למרכז מיטת העמודה וצנטריפוגה ב- 1,000 × גרם למשך 2 דקות כדי להחליף את פתרון המאגר. שוב, להשליך את חוצץ eluted וצינור האיסוף.
- מקם את העמודים שהוחלפו במאגר בצינורות צנטריפוגות נקיים בגודל 1.5 מ"ל. יש למרוח עד 75 מיקרו-אל של דגימה על מרכז המיטה. ספין ב 1,000 × גרם במשך 4 דקות.
הערה: אל תפריע למיטה או תיגע בצידי העמודה; הנקודה הגבוהה ביותר של מדיית הג'ל צריכה להצביע לכיוון הרוטור החיצוני. - לאסוף את eluate מצינור האיסוף, כפי שהוא מכיל את חומצת הגרעין מטוהר. כדי לכמת את המדגם, למדוד את ספיגתה ב 260 ננומטר; תייג S3 זה.
הערה: שים לב לאורך הנתיב המשמש במדידה, וחשב את הריכוז באמצעות חוק באר-למברט.
10. ניתוח דף דנטורינג של התקהלות BG-אוליגונוקלאוטיד
- יצוק 18% טריס-בוראט-EDTA (TBE)-אוריאה דף ג'ל. להמיס 4.8 גרם של UREA, 4.5 מ"ל של 40% אקרילאמיד (19:1), ו 1 מ"ל של 10x TBE ב 2.8 מ"ל של ddH2O; מוסיפים 5 μL טטרמתילאתילנדיאמין (TEMED) ומערבבים היטב. חזור עם 100 μL של 10% אמוניום אסולפט (APS). יוצקים את התמיסה לתוך קלטת ג'ל ריקה ומאפשרים פילמור במשך 40 דקות.
הערה: השתמש בציוד מגן אישי מתאים בעת טיפול אוריאה (גורם לגירוי בעיניים ובעור), אקרילאמיד (רעיל ומסרטן) ו TEMED (רעיל, דליק, מאכל). טבלה 10 מכילה את נוסחת התגובה עבור ג'ל פוליאקרילמיד TBE-UREA 18%. - מיקרוגל 500 מ"ל של חוצץ TBE (0.5x) במשך 2 דקות ו 30 s או עד ~ 70 °C (70 °F) ויוצקים לתוך מנגנון ג'ל. הכן פורממיד (denaturing) טוען צבע המכיל 95% פורממיד + 1 mM EDTA ו ברומופנול כחול. מערבבים את צבע הטעינה עם כל דגימה, וטוען את התערובת על ג'ל polyacrylamide.
הערה: השתמש בציוד מגן אישי מתאים בעת טיפול פורממיד מכיוון שהוא מסרטן. טבלה 11 מכילה טבלת טעינת ג'ל לדוגמה. - הפעל את הג'ל ב 270 V במשך 35 דקות, או עד חזית הצבע נודד עד הסוף. מניחים את הג'ל בקופסת ג'ל וכתים בצבע ציאנין לחומצות גרעין למשך 15 דקות בטמפרטורת החדר לפני ההדמיה.
הערה: השתמש בציוד מגן אישי מתאים בעת טיפול בצבע ציאנין כפי שהוא דליק.
11. הטיות של אוליגונוקלאוטיד ל- SNAP T7 RNAP וניתוח דף
- הכן את הריאגנטים עבור צימוד בקנה מידה אנליטי של BG-oligonucleotide ל SNAP T7 RNAP: להפוך 9 דילול של DNA חד גדילי (ssDNA) אוליגו עם ddH2O כדי ליצור יחסי אוליגו: RNAP הנעים בין 5:1 ל 1:5. לדלל את מלאי החלבון ל 50 מיקרומטר.
הערה: ניתן למצוא יחסי דוגמה בטבלה 12; יחסים אלה מחושבים באמצעות ריכוז RNAP של 50 מיקרומטר. - עבור כל דילול של אוליגו ssDNA, לעשות 10 μL של תערובת התגובה המכילה 2 μL של חיץ SNAP, 4 μL של BG-אוליגונוקלאוטיד, ו 4 μL של SNAP T7 RNAP.
הערה: טבלה 13 מכילה נוסחאות תגובה לתגובת תיוג SNAP..- הכן שתי דוגמאות בקרה נוספות: 1) פקד RNAP על-ידי החלפת BG-אוליגונוקלאוטיד ב- ddH2O; 2) בקרת DNA על ידי החלפת SNAP T7 RNAP עם ddH2O (לריכוז האוליגונוקלאוטיד הנמוך ביותר של SNAP T7 RNAP). לדגור על כל הדגימות בטמפרטורת החדר במשך שעה אחד, ולשמור על קרח עד הצורך.
- הגדר 11 תגובות 10 μL על ידי הוספת 2 μL של כל מדגם ל 4 μL של חיץ SNAP ו 2 μL של צבע טעינת חלבון, וחום ב 70 °C (70 °F) במשך 10 דקות. העמיסו 2 מיקרו-אל של כל דגימה על ג'ל החלבון ביס-טריס 4-12%, ובצעו אלקטרופורזה של ג'ל על קרח ב-200 V למשך 35 דקות.
הערה: טבלה 14 מכילה נוסחאות תגובה עבור דגימות טעינת הג'ל.- לשטוף SDS באמצעות החלפת מים 3x על שייקר, כל לשטוף נמשך 10 דקות כל אחד. כתם עם צבע ציאנין לחומצות גרעין במשך 15 דקות לפני ההדמיה. מכתים את הג'ל שוב באמצעות 20 מ"ל של כתם כחול קומאסי למשך שעה. דה-כתם עם ddH2O עבור 1 שעה (או לילה) לפני הדמיה.
הערה: בג'ל, אחת התגובות תייצר את הפולימראז הקשור ביותר יחד עם הכמות הנמוכה ביותר של עודף חינם BG-oligonucleotide; זהו היחס האופטימלי.
- לשטוף SDS באמצעות החלפת מים 3x על שייקר, כל לשטוף נמשך 10 דקות כל אחד. כתם עם צבע ציאנין לחומצות גרעין במשך 15 דקות לפני ההדמיה. מכתים את הג'ל שוב באמצעות 20 מ"ל של כתם כחול קומאסי למשך שעה. דה-כתם עם ddH2O עבור 1 שעה (או לילה) לפני הדמיה.
- הכן ריאגנטים עבור צימוד בקנה מידה הכנה BG-אוליגונוקלאוטיד ל SNAP T7 RNAP. בצע את תגובת הצימוד עם היחס האופטימלי שנמצא בסולם האנליטי.
הערה: צמצם את החשיפה לחלבון לטמפרטורת החדר על-ידי הנחת החלבון על הקרח כאשר אינו בשימוש.
12. טיהור של SNAP-T7 קשור לאוליגונוקלאוטיד באמצעות עמודות חילופי יונים
- בצע את הוראות היצרן עבור הגדרת צינור אם הוא סוטה מההוראות המפורטות כאן. הכן חוצץ טיהור עם pH גבוה יותר מהנקודה האיזואלקטרית של החלבון.
הערה: עבור חלבון לדוגמה בפרוטוקול זה, חוצץ טיהור של 10 mM נתרן פוספט חוצץ (pH 7) שימש.- הכן 1,000 μL של חוצץ אלוטיון המכיל ריכוזים סופיים של 50 mM טריס ו 0.5 M NaCl. לערבב 50 μL של 1 M טריס, 100 μL של 5 M NaCl, ו 850 μL של ddH2O.
הערה: טבלה 15 מכילה את נוסחת התגובה עבור מאגר ההתחמקות.
- הכן 1,000 μL של חוצץ אלוטיון המכיל ריכוזים סופיים של 50 mM טריס ו 0.5 M NaCl. לערבב 50 μL של 1 M טריס, 100 μL של 5 M NaCl, ו 850 μL של ddH2O.
- מניחים עמוד בצינור צנטריפוגה 2 מ"ל, ולשטוף עם חוצץ טיהור ב 2,000 × גרם במשך 15 דקות, או עד כל החיץ כבר eluted. זרוק את המאגר הנבה.
- לדלל כל מדגם עם מאגר טיהור ב 3:1 מאגר טיהור:יחס מדגם, ולטעון את המדגם לתוך עמודה 400 μL בכל פעם. ספין ב 2,000 × g במשך 10 דקות, או עד כל החיץ כבר eluted. אסוף את הזרימה דרך וסמן אותה כזרימה דרך.
- הוסף 400 μL של מאגר טיהור למרכז העמודה. ספין ב 2,000 × g במשך 15 דקות, או עד כל החיץ כבר eluted. לאסוף את הזרימה דרך ולתייג אותו כמו לשטוף 1. חזור פעמיים נוספות לכביסה 2 ולשטוף 3.
- הוסף 50 μL של מאגר elution למרכז העמודה. ספין ב 2,000 × g במשך 5 דקות, או עד כל החיץ כבר eluted. לאסוף את הזרימה דרך ולתייג אותו כמו eluate 1. חזור פעמיים נוספות עבור eluate 2 ואלט 3.
- הבריכה אלווה 1, 2 ו -3 (תווית זה eluate הכולל), משאיר חלק קטן של כל eluate עבור הג'ל, ולמדוד ספיגה ב 260 ננומטר (A260) ו 280 ננומטר (A280). לאחר המדידה, להוסיף גליצריל ביחס של 1:1 ולאחסן ב -20 °C (70 °F) עד לשימוש נוסף.
- השתמש ביחידת סינון צנטריפוגלית (0.5 מ"ל; 30 kDa) כדי להחליף מאגר הכולל eluate עם מאגר אחסון 2x (~ 1:100) (תווית מוצרזה). מדוד A260/280 שוב. הוסף גליצריל ביחס של 1:1 ואחסן ב--20 °C (70 °F) עד לשימוש נוסף.
- טענו כל אלוט: זרימה דרך, לשטוף 1-3, אלוט הכולל, ומוצר בג'ל 4-12% Bis-Tris SDS-PAGE, יחד עם סולם חלבונים. הפעל ב 200 V במשך 35 דקות, או עד חזית הצבע נודד עד הסוף.
13. הדגמה של שליטה לפי דרישה בפעילות RNA פולימראז קשורה
- הכן 5x חישול המכיל 25 mM טריס, 5 mM EDTA, ו 25 mM מגנזיום כלוריד (MgCl2). לערבב 2.4 μL של כל תבנית (1 μM) עם 5 μL של חישול חוצף ו 14.2 μL של ddH2O כדי ליצור 25 μL של 1 μM dsDNA כלוב. לדגור על פתרון זה ב 75 °C (75 °F) במשך 2 דקות. באופן דומה, חין גדילי תחושה ואנטיסנס של האמרגן ותבנית DNA aptamer ירוק מלאכי. הכן פתרון 1mM של אוקסלט ירוק מלאכיט.
הערה: טבלה 16 מכילה את נוסחת התגובה עבור חישול 5x, טבלה 17 מכילה את נוסחת התגובה לחישול שתי תבניות ssDNA. - לדגור על SNAP T7 RNAP קשור עם כלוב dsDNA ביחס טוחנת 1:5 בטמפרטורת החדר במשך 15 דקות לריכוז סופי של RNAP 500 ננומטר. יש לשמור על קרח עד שיידרש.
- מחממים את קורא הלוחות ל-37 מעלות צלזיוס. הגדרת שלוש תגובות IVT 25 μL על קרח
- הגדר תגובה המכילה את SNAP T7RNAP בכלוב עם גורמי שעתוק חומצת גרעין. לערבב 2.5 μL של 10x IVT חוצץ, 1 μL של 25 mM rNTP לערבב, 1 μL של 1 mM ירוק מלאכיט, 2.5 μL של תערובת כלוב RNAP, 2.5 μL כל אחד 1 μM גורם שעתוק A ו B oligonucleotide גדילים, ו 3 μL של 1 mM malachite ירוק תבנית aptamer ב 10 μL של ddH2O.
- הגדר תגובה המכילה את SNAP T7RNAP בכלוב ללא גורמי שעתוק חומצת גרעין. לערבב 2.5 μL של 10x IVT חוצץ, 1 μL של 25 mM rNTP לערבב, 1 μL של 1 mM ירוק מלאכי, 2.5 μL של תערובת כלוב RNAP, ו 3 μL של 1 mM malachite ירוק תבנית aptamer ב 15 μL של ddH2O.
- הגדר תגובה המכילה מאגר בלבד. לערבב 2.5 μL של 10x IVT חוצץ, 1 μL של 25 mM rNTP לערבב, 1 μL של 1 mM ירוק מלכיט, ו 3 μL של 1 mM malachite ירוק תבנית aptamer ב 17.5 μL של ddH2O.
הערה: טבלה 18 מכילה הפניה כללית לתגובות התמלול במבחנה.
- מעבירים כל תגובה לצלחת 384-well. צג תמלול של aptamer ירוק מלאכי על קורא צלחת פלואורסצנטית עבור 2 שעות ב 37 °C (77 °F) ועם עירור 610 ננומטר ופליטה 655 ננומטר. לאחר סיום, לשמור את הצלחת על קרח עד הצורך.
- מיקרוגל 0.5x TBE חוצץ במשך 2 דקות 30 s או עד ~ 70 °C (70 °F). הפעל את מוצרי ה- RNA של כל באר בג'ל פולאקרילמיד TBE-Urea 12% במאגר TBE מחומם של 0.5x ב- 280 V למשך 20 דקות, או עד שחזית הצבע מגיעה לסוף. מכתימים את הג'ל בכתם חומצת גרעין בצבע ציאנין למשך 10 דקות בשייקר מסלולי לפני ההדמיה.
הערה: טבלה 19 מכילה את נוסחת התגובה עבור ג'ל דפי TBE-Urea 12% .
תוצאות

איור 5: ניתוח SDS-PAGE של ביטוי SNAP T7 RNAP ובבדיית תמלול במבחנה. (A)SNAP T7 RNAP ניתוח טיהור חלבון, SNAP T7 RNAP משקל מולקולרי: 119.4kDa. FT = זרימה דרך מהעמודה, W1 = שברי אלוטציה של מאגר כביסה המכיל זיהומים, E1-3 = שברי אלו?...
Discussion
מחקר זה מדגים גישה בהשראת ננוטכנולוגיה DNA כדי לשלוט על הפעילות של T7 RNA פולימראז על ידי צימוד covalently SNAP רקומביננט T7 RNAP עם אוליגונוקלאוטיד פונקציונלי BG, אשר שימש לאחר מכן לתכנת תגובות TMDSD. על ידי עיצוב, SNAP-תג היה ממוקם ב N-terminus של פולימראז, כמו C-terminus של סוג בר T7 RNAP קבור בתוך הליבה מבנה החלבון ועושה...
Disclosures
אין אינטרסים פיננסיים מתחרים להצהיר על ידי אף אחד מהמחברים.
Acknowledgements
L.Y.T.C מכיר בתמיכה נדיבה מהחזיתות החדשות בקרן המחקר-חקר (NFRF-E), מענק דיסקברי של המועצה למדעי הטבע והנדסה של קנדה (NSERC) ואוניברסיטת טורונטו לרפואה על ידי יוזמת עיצוב, המקבלת מימון מהקרן למצוינות במחקר הראשון בקנדה (CFREF).
Materials
| Name | Company | Catalog Number | Comments |
| 0.5% polysorbate 20 (TWEEN 20) | BioShop | TWN510.5 | |
| 0.5M ethylenediaminetetraacetic acid (EDTA) | Bio Basic | SD8135 | |
| 10 mM sodium phosphate buffer (pH 7) | Bio Basic | PD0435 | Tablets used to make 10 mM buffer |
| 10% ammonium persulfate (APS) | Sigma Aldrich | A3678-100G | |
| 100 kDa Amicon Ultra-15 Centrifugal Filter Unit | Fisher Scientific | UFC910008 | |
| 100% acetone | Fisher Chemical | A18P4 | |
| 100% ethanol (EtOH) | House Brand | 39752-P016-EAAN | |
| 10x in vitro transcription (IVT) buffer | New England Biolabs | B9012 | |
| 10x Tris-Borate-EDTA (TBE) buffer | Bio Basic | A0026 | |
| 1M Isopropyl β- d-1-thiogalactopyranoside (IPTG) | Sigma Aldrich | I5502-1G | |
| 1M sodium bicarbonate buffer | Sigma Aldrich | S6014-500G | |
| 1M Tris(hydroxymethyl)aminomethane (Tris) | Sigma Aldrich | 648311-1KG | |
| 1X Tris-EDTA (TE) buffer | ThermoFisher | 12090015 | |
| 2M imidazole | Sigma Aldrich | 56750-100G | |
| 2-mercaptoethanol (BME) | Sigma Aldrich | M3148 | |
| 3M sodium acetate | Bio Basic | SRB1611 | |
| 40% acrylamide (19:1) | Bio Basic | A00062 | |
| 4x LDS protein sample loading buffer | Fisher Scientific | NP0007 | |
| 5M sodium chloride (NaCl) | Bio Basic | DB0483 | |
| 5mM dithiothreitol (DTT) | Sigma Aldrich | 43815-1G | |
| 6x gel loading dye | New England Biolabs | B7024S | |
| agarose B powder | Bio Basic | AB0014 | |
| BG-GLA-NHS | New England Biolabs | S9151S | |
| BL21 competent E. coli | Addgene | C2530H | |
| BLUeye prestained protein ladder | FroggaBio | PM007-0500 | |
| bromophenol blue | Bio Basic | BDB0001 | |
| coomassie blue (SimplyBlue SafeStain) | ThermoFisher | LC6060 | |
| cyanine dye (SYBR Gold nucleic acid gel stain) | Fisher Scientific | S11494 | |
| cyanine dye (SYBR Safe nucleic acid gel stain) | Fisher Scientific | S33102 | |
| dry dimethyl sulfoxide (DMSO) | Fisher Scientific | D12345 | |
| formamide | Sigma Aldrich | F9037-100ML | |
| glycerol | Bio Basic | GB0232 | |
| kanamycin sulfate | BioShop | KAN201.5 | |
| lysogeny broth | Sigma Aldrich | L2542-500ML | |
| malachite green oxalate | Sigma Aldrich | 2437-29-8 | |
| N,N,N'N'-Tetramethylethane-1,2-diamine (TEMED) | Sigma Aldrich | T9281-25ML | |
| NuPAGE MES SDS running buffer (20x) | Fisher Scientific | LSNP0002 | |
| NuPAGE Novex 4-12% Bis-Tris gel 1.0 mm 12-well | Life Technologies | NP0322BOX | |
| oligonucleotide (cage antisense) | IDT | N/A | TATAGTGAGTCGTATTAATTTG |
| oligonucleotide (cage sense) | IDT | N/A | TCAGTCACCTATCTGTTTCAAA TTAATACGACTCACTATA |
| oligonucleotide (malachite green aptamer antisense) | IDT | N/A | GGATCCATTCGTTACCTGGCT CTCGCCAGTCGGGATCCTATA GTGAGTCGTATTACAGTTCCAT TATCGCCGTAGTTGGTGTACT |
| oligonucleotide (malachite green aptamer sense) | IDT | N/A | TAATACGACTCACTATAGGATC CCGACTGGCGAGAGCCAGGT AACGAATGGATCC |
| oligonucleotide (Transcription Factor A) | IDT | N/A | AGTACACCAACTACGAGTGAG |
| oligonucleotide (Transcription Factor B) | IDT | N/A | TCAGTCACCTATCTGGCGATAA TGGAACTG |
| oligonucleotide with 3’ Amine modification (tether) | IDT | N/A | GCTACTCACTCAGATAGGTGAC TGA/3AmMO/ |
| Pierce strong ion exchange spin columns | Fisher Scientific | 90008 | |
| plasmid encoding SNAP T7 RNAP and kanamycin resistance genes | Genscript | N/A | custom gene insert |
| protein purification column (HisPur Ni-NTA spin column) | Fisher Scientific | 88226 | |
| rNTP mix | New England Biolabs | N0466S | |
| Roche mini quick DNA spin column | Sigma Aldrich | 11814419001 | |
| Triton X-100 | Sigma Aldrich | T8787-100ML | |
| Ultra Low Range DNA ladder | Fisher Scientific | 10597012 | |
| urea | BioShop | URE001.1 |
References
- Cherry, K. M., Qian, L. Scaling up molecular pattern recognition with DNA-based winner-take-all neural networks. Nature. 559 (7714), 370-376 (2018).
- Qian, L., Winfree, E., Bruck, J. Neural network computation with DNA strand displacement cascades. Nature. 475 (7356), 368-372 (2011).
- Chen, Y. -. J., et al. Programmable chemical controllers made from DNA. Nature Nanotechnology. 8 (10), 755-762 (2013).
- di Bernardo, D., Marucci, L., Menolascina, F., Siciliano, V. Predicting synthetic gene networks. Synthetic Gene Networks: Methods and Protocols. 813, 57-81 (2012).
- Xiang, Y., Dalchau, N., Wang, B. Scaling up genetic circuit design for cellular computing: advances and prospects. Natural Computing. 17 (4), 833-853 (2018).
- Gould, N., Hendy, O., Papamichail, D. Computational tools and algorithms for designing customized synthetic genes. Frontiers in Bioengineering and Biotechnology. 2, (2014).
- MacDonald, J. T., Siciliano, V. Computational sequence design with R2oDNA Designer. Mammalian Synthetic Promoters. 1651, 249-262 (2017).
- Cervantes-Salido, V. M., Jaime, O., Brizuela, C. A., Martínez-Pérez, I. M. Improving the design of sequences for DNA computing: A multiobjective evolutionary approach. Applied Soft Computing. 13 (12), 4594-4607 (2013).
- Zadeh, J. N., et al. NUPACK: Analysis and design of nucleic acid systems. Journal of Computational Chemistry. 32 (1), 170-173 (2011).
- Fornace, M. E., Porubsky, N. J., Pierce, N. A. A unified dynamic programming framework for the analysis of interacting nucleic acid strands: enhanced models, scalability, and speed. ACS Synthetic Biology. 9 (10), 2665-2678 (2020).
- Wetterstrand, K. DNA sequencing costs: Data. Genome.gov. , (2020).
- Lopez, R., Wang, R., Seelig, G. A molecular multi-gene classifier for disease diagnostics. Nature Chemistry. 10 (7), 746-754 (2018).
- Pardee, K., et al. low-cost detection of Zika virus using programmable biomolecular components. Cell. 165 (5), 1255-1266 (2016).
- Yurke, B., Turberfield, A. J., Mills, A. P., Simmel, F. C., Neumann, J. L. A DNA-fuelled molecular machine made of DNA. Nature. 406 (6796), 605-608 (2000).
- Lin, K. N., Volkel, K., Tuck, J. M., Keung, A. J. Dynamic and scalable DNA-based information storage. Nature Communications. 11 (1), 2981 (2020).
- Yurke, B., Mills, A. P. Using DNA to power nanostructures. Genetic Programming and Evolvable Machines. 4 (2), 111-122 (2003).
- Zhang, D. Y., Turberfield, A. J., Yurke, B., Winfree, E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 318 (5853), 1121-1125 (2007).
- Wang, B., Thachuk, C., Ellington, A. D., Winfree, E., Soloveichik, D. Effective design principles for leakless strand displacement systems. Proceedings of the National Academy of Sciences. 115 (52), 12182-12191 (2018).
- Machinek, R. R. F., Ouldridge, T. E., Haley, N. E. C., Bath, J., Turberfield, A. J. Programmable energy landscapes for kinetic control of DNA strand displacement. Nature Communications. 5 (1), 5324 (2014).
- Cabello-Garcia, J., Bae, W., Stan, G. -. B. V., Ouldridge, T. E. Handhold-mediated strand displacement: a nucleic acid-based mechanism for generating far-from-equilibrium assemblies through templated reactions. bioRxiv. , (2020).
- Brophy, J. A. N., Voigt, C. A. Principles of genetic circuit design. Nature Methods. 11 (5), 508-520 (2014).
- Khalil, A. S., et al. A synthetic biology framework for programming eukaryotic transcription functions. Cell. 150 (3), 647-658 (2012).
- Swank, Z., Laohakunakorn, N., Maerkl, S. J. Cell-free gene-regulatory network engineering with synthetic transcription factors. Proceedings of the National Academy of Sciences. 116 (13), 5892-5901 (2019).
- Howland, S. W., Tsuji, T., Gnjatic, S., Ritter, G., Old, L. J., Wittrup, K. D. Inducing efficient cross-priming using antigen-coated yeast particles. Journal of immunotherapy. 31 (7), 607 (2008).
- Abil, Z., Ellefson, J. W., Gollihar, J. D., Watkins, E., Ellington, A. D. Compartmentalized partnered replication for the directed evolution of genetic parts and circuits. Nature Protocols. 12 (12), 2493-2512 (2017).
- Baugh, C., Grate, D., Wilson, C., Doudna, J. A. 2.8 Å crystal structure of the malachite green aptamer11. Journal of Molecular Biology. 301 (1), 117-128 (2000).
- Chou, L. Y. T., Shih, W. M. In vitro transcriptional regulation via nucleic acid-based transcription factors. ACS Synthetic Biology. 8 (11), 2558-2565 (2019).
- Lykke-Andersen, J., Christiansen, J. The C-terminal carboxy group of T7 RNA polymerase ensures efficient magnesium ion-dependent catalysis. Nucleic Acids Research. 26 (24), 5630-5635 (1998).
- Pu, J., Disare, M., Dickinson, B. C. Evolution of C-terminal modification tolerance in full-length and split T7 RNA Polymerase biosensors. Chembiochem. 20 (12), 1547-1553 (2019).
- Gardner, L. P., Mookhtiar, K. A., Coleman, J. E. Initiation, elongation, and processivity of carboxyl-terminal mutants of T7 RNA polymerase. Biochemistry. 36 (10), 2908-2918 (1997).
- Yin, J., Lin, A. J., Golan, D. E., Walsh, C. T. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nature Protocols. 1 (1), 280-285 (2006).
- Warden-Rothman, R., Caturegli, I., Popik, V., Tsourkas, A. Sortase-tag expressed protein ligation: combining protein purification and site-specific bioconjugation into a single step. Analytical Chemistry. 85 (22), 11090-11097 (2013).
- Zhang, W. -. B., Sun, F., Tirrell, D. A., Arnold, F. H. Controlling macromolecular topology with genetically encoded SpyTag-SpyCatcher chemistry. Journal of the American Chemical Society. 135 (37), 13988-13997 (2013).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved