In Vitro Differentiation of Mature Myofibers for Live Imaging
In This Article
Summary
Muscle cells are among the most complex eukaryotic cells. We present a protocol for the in vitro differentiation of highly mature myofibers that allows for genetic manipulation and clear imaging during all developmental stages.
Abstract
Skeletal muscles are composed of myofibers, the biggest cells in the mammalian body and one of the few syncytia. How the complex and evolutionarily conserved structures that compose it are assembled remains under investigation. Their size and physiological features often constrain manipulation and imaging applications. The culture of immortalized cell lines is widely used, but it can only replicate the early steps of differentiation.
Here, we describe a protocol that enables easy genetic manipulation of myofibers originating from primary mouse myoblasts. After one week of differentiation, the myofibers display contractility, aligned sarcomeres and triads, as well as peripheral nuclei. The entire differentiation process can be followed by live imaging or immunofluorescence. This system combines the advantages of the existing ex vivo and in vitro protocols. The possibility of easy and efficient transfection as well as the ease of access to all differentiation stages broadens the potential applications. Myofibers can subsequently be used not only to address relevant developmental and cell biology questions, but also to reproduce muscle disease phenotypes for clinical applications.
Introduction
Skeletal muscle composes up to 40% of the human body weight1. Muscle-associated disorders represent an immense health and economic burden2. How this highly complex and organized tissue is formed, maintained, and regenerated constitutes an extensive and well-established research field. Depending on the specific scientific interest, the most suited approach can range from simple myotube cultures to complex in vivo models3-6.
The goal of this protocol is to provide an in vitro system that allows for the monitoring of myogenesis through live imaging and immunofluorescence. Compared to traditional approaches, this system offers a very complete and dynamic insight into the mouse myogenic process. Cells can be followed from the myoblast stage to the mature, multinucleated myofiber displaying transversal triads and peripheral nuclei7. This maturation level can be achieved using regular cell culture equipment, without the need for complex stimulatory or mechanical apparatuses. Although some successful in vitro systems have been reported8,9, to our knowledge, this is the only protocol generating mature mouse myofibers with T-tubules transversally paired with Sarcoplasmic Reticulum (SR). Thus, this in vitro system can be used to study the molecular mechanisms of triad formation, which are still poorly understood10.
A further advantage of using this system is the availability of validated mouse-targeted resources, such as antibodies, drugs, and RNAi tools. The relatively simple protocol does not require laborious steps, highly skilled manipulation, or expensive and dedicated equipment. Matured myofibers start appearing after 5 d of culture differentiation7, displaying contractility coupled with calcium sparks (unpublished data). In one week, the different developmental stages of one of the most complex cells in the mammalian body can be studied in combination with a variety of in vitro assays.
Protocol
NOTE: One mouse yields sufficient myoblasts for approximately two 35 mm dishes or two live-imaging dishes, so plan mattings, dissection, and coating (step 2.6) accordingly. Since myoblasts are isolated through sequential centrifugations and preplating, the protocol should be done in batches of 5 - 10 animals.
All procedures involving animal subjects were approved by the Animal Ethics Committee at Instituto de Medicina Molecular and University Pierre et Marie Curie
1. Dissection of Neonatal Mice Hind-limb Muscles
- Prepare all solutions in advance (Materials Table) and sterilize by filtration (0.22 µm filter). Make sure all media are at 37 °C before addition to the cells, except the formulations containing basement membrane matrix (e.g., Matrigel).
- Sterilize the dissection material (one each of: curved scissors, straight scissors, regular forceps, and fine-tip forceps) and the work bench by wiping them with 70% ethanol.
- Prepare a 100 mm Petri dish with 5 mL of Dulbecco's Phosphate Buffered Saline (DPBS) for muscle collection and keep it on ice until the mincing step.
- Decapitate P6 - P8 mice with straight scissors and sterilize the skin with 70% ethanol.
- Make an incision in the back skin and pull it gently towards the hind limbs until it is removed, completely exposing the hind-limb musculature.
- Use the forceps to remove fat tissue without damaging the muscles.
- To remove the dorsal hind-limb muscles, keep the limb stretched and bend the paw to expose the heel tendons. Use the curved scissors to separate muscle from bone, starting from the tendons, by gently sliding and cutting upwards. Excise the muscles and place them in iced DPBS.
- Isolate the quadriceps by pinching the muscle with fine-tip forceps and cutting around it without damaging the femur or the knee joint.
- After dissecting all animals, proceed to a sterile laminar flow cell culture hood, where all the following steps should be performed.
2. Myoblast Isolation
- Remove the excess of DPBS. Mince the tissue with sterilized curved scissors in order to obtain a uniform mass.
- Collect the minced tissue in a 50 mL conical centrifuge tube using 5 mL of digestion mix and incubate it with agitation at 37 °C for 90 min.
- Stop the digestion by adding 6 mL of dissection medium and centrifuge the suspension for 5 min at 75 x g to pellet the remaining tissue.
- Carefully collect the supernatant. Make sure to not collect tissue debris. Centrifuge it at 350 x g for 5 min; resuspend it in 5 mL of dissection medium.
- Filter the cell suspension through a 40 µm cell strainer. Add 25 mL of dissection medium and preplate it in a 150 mm dish for 4 h in a cell culture incubator (37 °C and 5% CO2) to allow the fibroblasts to adhere.
- While preplating, coat dishes with 500 µL of basement membrane matrix diluted 1:100 in cold IMDM for 1 h at RT. Wash once with DPBS and plate the cells immediately (step 2.8) or leave with growth medium until plating.
- After preplating, collect the supernatant and centrifuge it at 350 x g for 10 min.
- Resuspend it in growth medium and count the cells on a hemocytometer. Adjust the volume so that between 150,000 and 250,000 cells are plated per basement membrane matrix-coated dish. Keep the cells in a cell culture incubator.
3. Myofiber Differentiation
NOTE: After 3 d, the cells should start to fuse and form myotubes at around 70% confluency (Figure 1B).
- At this point, transfect the cells, if desired, with a siRNA or DNA of interest. If the cells are not to be transfected, change directly to differentiation medium and skip to step 3.4.
- Transfect with transfection reagents following the manufacturer's instructions. Incubate the cells for 5 h with siRNA-lipid complexes (20 nM + 1 µL of reagent) or DNA-lipid complexes (1 µg + 1 µL of reagent). Optimize the siRNA and DNA concentrations if necessary.
- Wash them once with differentiation medium and then switch to new differentiation medium.
- The following day, dilute the basement membrane matrix 1:2 in ice-cold differentiation medium. Remove the existing medium and add 200 µL of ice-cold matrix to each dish.
- Incubate for 30 min in a cell culture incubator.
- Supplement the differentiation medium with agrin (100 ng/mL) and carefully add 2 mL to the cells.
- Carefully change half of the medium every 2 d, always supplementing with agrin to a final concentration of 100 ng/µL.
- Monitor cell differentiation and viability. Depending on a variety of factors (such as FBS and chicken embryo extract origins), the cells might take between 5 - 10 differentiation d to reach full maturation (Figure 2).
4. Immunostaining in Glass-bottom Dishes
- For immunostaining, at any time-point of interest, wash the cells once with DPBS and fix them with 4% PFA at RT for 10 min.
- Wash them twice with DPBS. At this point, the cells can be stored at 4 °C.
- Permeabilize them with 0.5% Triton X-100 for 5 min at RT.
- Wash them twice with PBS and block with blocking solution for 30 min at RT.
- Incubate them with primary antibody diluted in blocking solution O/N at 4 °C.
- Wash 3x with DPBS for 5 min at RT.
- Incubate them with the secondary antibody and 0.2 µg/mL of DAPI for 1 h at RT.
- Wash 3x with DPBS for 5 min at RT.
- Add 200 µL of mounting medium and proceed to imaging.
Representative Results
The extent of myofiber development is mostly determined by the purity and viability of the isolated myoblasts. The adhesion, proliferation, and fusion capacity can be used to empirically access those parameters (Figure 1 A, B). At proliferation D2, myoblasts should have adhered and should display the typical fusiform shape. Proliferation is expected to happen extensively at this stage, leading to spontaneous myotube formation the following day (Figure 1B).
Cell confluency might need slight adjustments. It should be increased if myoblasts take more than 3 d to proliferate and fuse. It should be decreased if myofibers are not allowed to grow and elongate relatively straight due to their density. Confluency typically decreases from the center to the periphery of the dish, so the best myofibers should be found towards the outer regions.
Myotubes will quickly elongate and display multiple centrally aligned nuclei (Figure 1C). By D5, some cells start acquiring striations and moving their nuclei to the periphery. The number of myofibers with mature characteristics will increase with time as well as with cell thickness (Figure 1D).
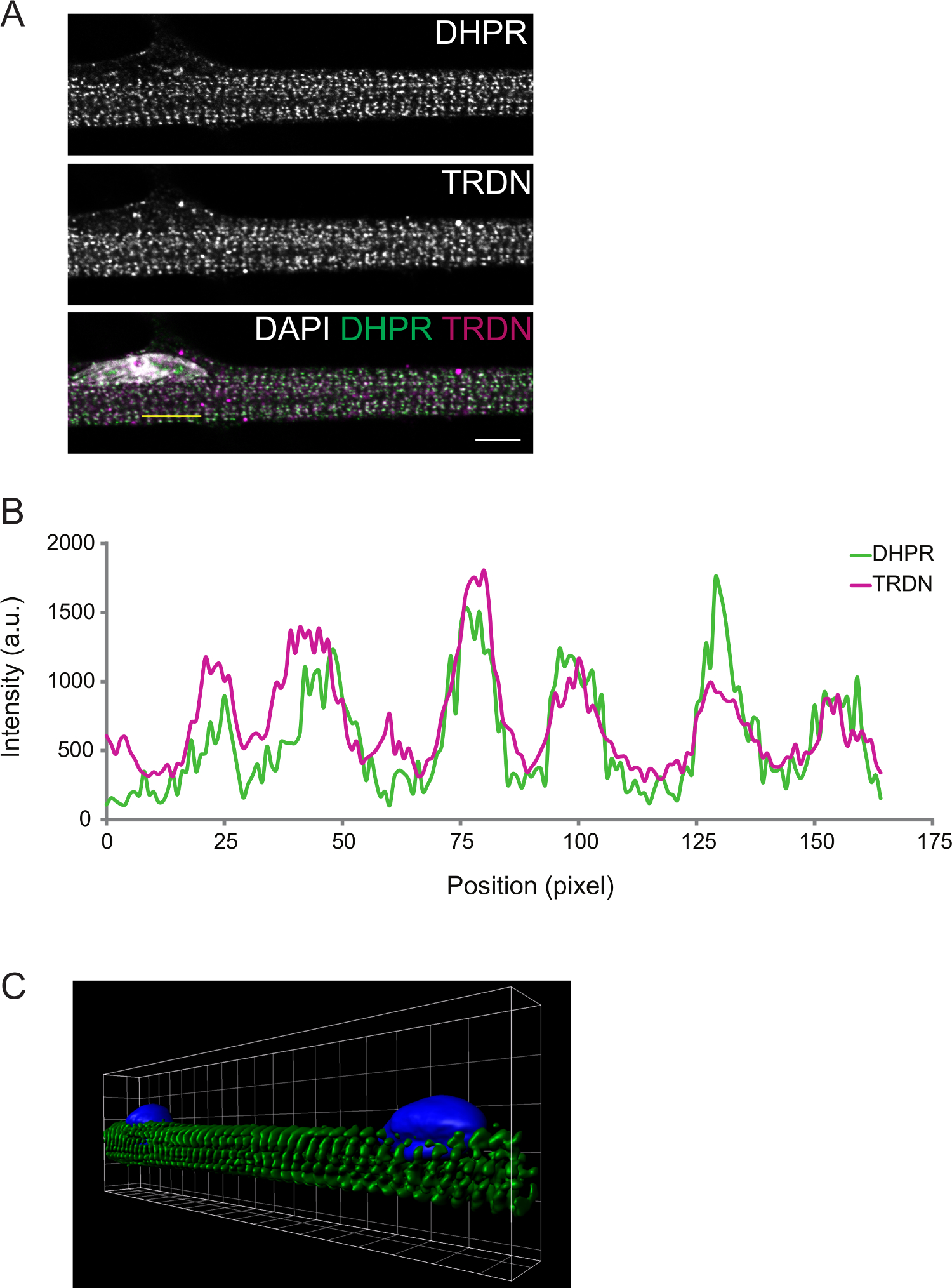
The degree of differentiation can be further observed by immunofluorescence. Myofibers fixed at differentiation D8 present transversal triads. This can be confirmed by imaging components of the T-tubules (DPHR) and the SR (triadin), which are expected to colocalize at the triads (Figure 2).
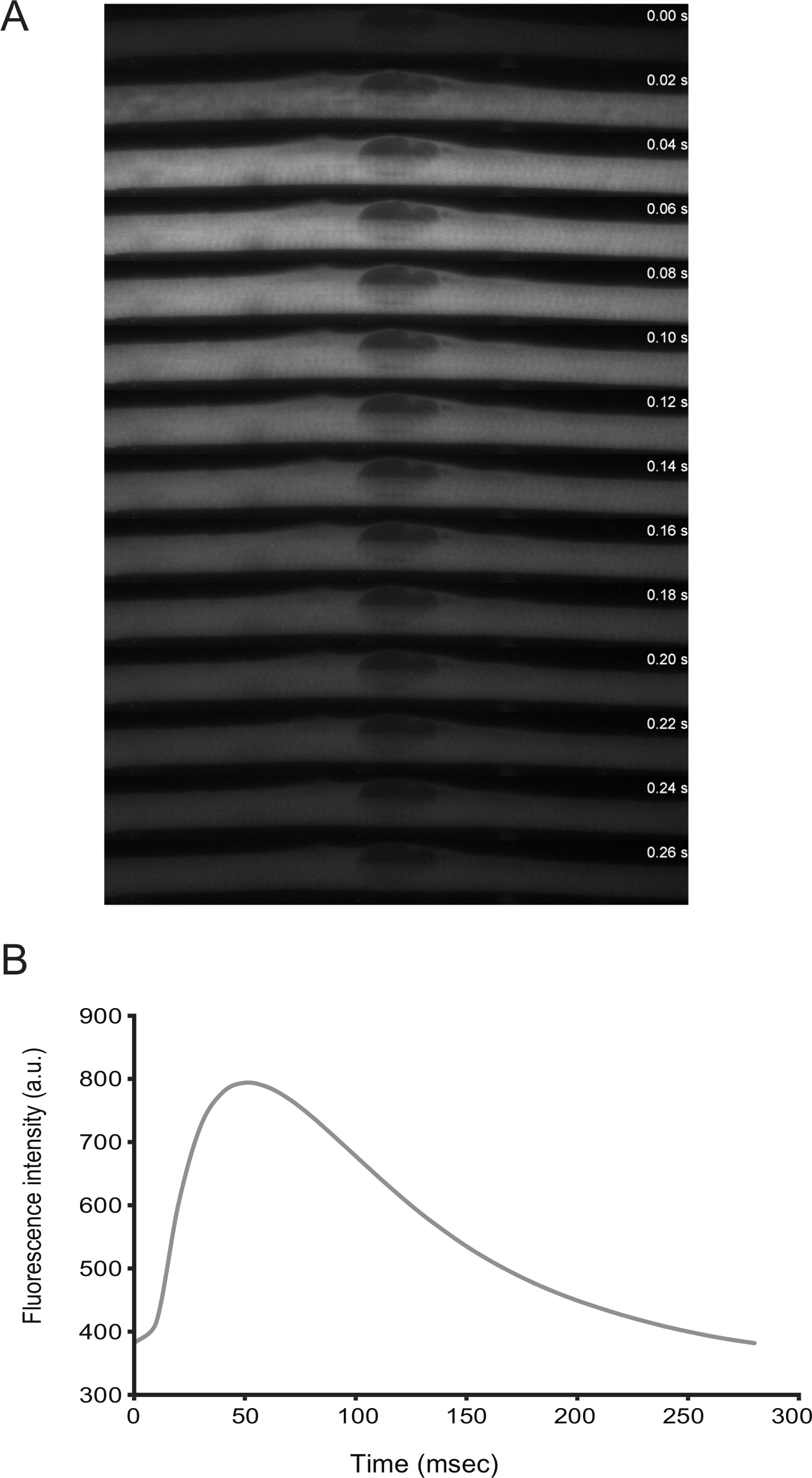
The functionality of myofibers can be addressed by live imaging. From differentiation D3 onwards, the cells display spontaneous twitching. By transfecting a calcium sensor (e.g., GCaMP6f11), it is possible to observe that the contractions are coupled with calcium peaks (Figure 3).
Using this system, we were able to identify a novel molecular pathway that is disrupted in centronuclear myopathies and myotonic dystrophies, which can therefore be a novel target for innovative molecular therapies7. We have also adapted this method to study the development of the neuromuscular junction (NMJ)12. Through the coculture with rat spinal cord explants, we have described a role for dynein in NMJ formation13.

Figure 1: Developmental Stages of the Myoblast Culture. A) At proliferation D2, myoblasts have adhered and started proliferating. B) At proliferation D3, a confluency of 60 - 80% is reached, and myoblasts start fusing spontaneously. C) At differentiation D3, myotubes containing centrally located nuclei are predominant. D) From differentiation D5 onwards (e.g., day 8), myofibers start exhibiting striations and peripheral nuclei and begin to thicken. Scale bar: 50 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Representative Confocal Image of a D8 Myofiber Immunostain. A) Immunostaining for dihydropyridine receptor (DHPR, top panel) and triadin (TRDN, middle panel). An overlay of the DHPR, TRDN, and DAPI channels shows colocalization of the triad components. B) An intensity profile of the yellow line drawn in A. C) A 3D image of volume rendering of myofibers stained for α-actinin (green) and DAPI (blue). Scale bar and grid width: 5 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Live Imaging of Calcium Levels in Myofibers with Spontaneous Twitching. A) High-speed time-lapse (20 ms frames) microscopy of a calcium spark in a twitching myofiber. Calcium was detected through the expression of GCaMP6f (Addgene plasmid #40755). B) Quantification of the fluorescence intensity over time for the calcium sensor in panel A. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The use of this protocol for the cultivation of primary myoblasts gives rise to a special niche that greatly nurtures the development of myofibers. This is partially due to other cell types that are also present in very small numbers. A balance between myoblast concentration and culture purity must be achieved. A good cell culture also depends on the quality of the products used for the medium formulation. All products derived from animal sources should be thoroughly tested. In our experience, the digestion conditions should also be monitored.
As usual for primary cultures, experimental variability can be higher than in studies with isolated fibers or immortalized myoblasts. This variability can be diminished by the standardization of medium and digestion components, mice age and size, and the time points for culture manipulation and results collection. Nevertheless, the advantage of scrutinizing in real time the intricate mechanisms necessary for myofiber development greatly surpasses the variability drawback.
This protocol confers the advantages of in vitro approaches without compromising cell differentiation. Myofibers mature until triads are formed and contractions are coupled to calcium sparks. These functional outputs can be accessed in different experimental conditions. Furthermore, there can be many technical variations made to the protocol. Myoblasts can be harvested from neonatal mice with mutations of interest relating to muscle development. Cells can be lysed for biochemical analysis at different differentiation time points. Calcium indicators can be added to the culture to follow its dynamics. Optogenetic constructs can be used to enforce certain signaling pathways or to induce specific local responses. Finally, the myofibers can be cocultured with other cells types to study their interactions.
Acknowledgements
This work was supported by the European Research Council (ERG) and EMBO installation (ERG) and by a PhD fellowship from the Fundação para a Ciência e Tecnologia (MP).
Materials
| Name | Company | Catalog Number | Comments |
| Dispase II | Gibco | 17105041 | |
| Collagenase type V | Sigma-Aldrich-Aldrich | C9263 | |
| IMDM, Glutamax supplemented | Gibco | 31980022 | |
| Matrigel Growth reduced factor | Corning | 354230 | protein concentration of the lot should be around 10mg/ml and endotoxin result should be <1.5 |
| Chicken Embryo Extract | MP biomedical | 2850145 | it is also possible to prepare in the lab (Danoviz ME, Yablonka-Reuveni Z. Methods Mol Biol (2012)) |
| Recombinant rat agrin | R&D systems | 550-AG-100 | |
| Penicillin-Streptomycin | Thermo Fisher Scientific | 15140122 | |
| Horse Serum | GE Healthcare | 11581831 | |
| Lipofectamine RNAiMAX | Invitrogen | 13778-150 | used to transfect siRNA |
| Lipofectamine LTX | Invitrogen | 15338-100 | used to transfect DNA |
| Lipofectamine 2000 | Invitrogen | 11668030 | used to transfect siRNA plus DNA |
| DPBS | Gibco | 14190094 | |
| Fetal Bovine Serum | Eurobio | CVFSVF0001 | |
| Cell strainer | Corning | 21008-949 | |
| Fluorodishes | World precision instruments | FD35-100 | dishes used to cultivate cells for live imaging |
| Triton X-100 | Sigma-Aldrich | T9284 | |
| 16% PFA (Paraformaldehyde) | Science Services GmbH | E15710 | |
| Goat Serum | Sigma-Aldrich | G9023 | |
| BSA (Bovine Serum Albumine) | Sigma-Aldrich | A7906 | |
| Saponine | Sigma-Aldrich | 47036 | |
| DAPI | Sigma-Aldrich | D9542 | use at 200ng/ul |
| Fluoromount-G | SouthernBiotech | 0100-01 | |
| Name | Company | Catalog Number | Comments |
| Solutions and Media | |||
| Digestion Mix | in DPBS 5 mg/ml collagenase 3.5 mg/ml dispase sterile filtered, can be stored in working aliquotes for 2 weeks at -20 °C | ||
| Dissection Medium | in IMDM Glutamax supplemented 10% FBS 1% Penicillin-Streptomycin sterile filtered | ||
| Growth Medium | in IMDM Glutamax supplemented 20% FBS 1% Chicken Embryo Extract 1% Penicillin-Streptomycin1% Penicillin-Streptomycin sterile filtered | ||
| Differentiation Medium | in IMDM Glutamax supplemented 2% Horse Serum 1% Penicillin-Streptomycin sterile filtered | ||
| Blocking Solution | in DPBS 10% Goat Serum 5% BSA add 0.1% saponine when incubating with primary and secondary antibodies |
References
- Janssen, I., Heymsfield, S. B., Wang, Z., Ross, R. Skeletal muscle mass and distribution in 468 men and women aged 18-88 yr. J Appl Physiol. 89 (1), 81-88 (2000).
- Manring, H., Abreu, E., Brotto, L., Weisleder, N., Brotto, M. Novel excitation-contraction coupling related genes reveal aspects of muscle weakness beyond atrophy-new hopes for treatment of musculoskeletal diseases. Front Physiol. 5, 37 (2014).
- Yaffe, D., Saxel, O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 270 (5639), 725-727 (1977).
- Pasut, A., Jones, A. E., Rudnicki, M. A. Isolation and Culture of Individual Myofibers and their Satellite Cells from Adult Skeletal Muscle. J Vis Exp. (73), (2013).
- Meng, H., Janssen, P. M. L., et al. Tissue Triage and Freezing for Models of Skeletal Muscle Disease. J Vis Exp. (89), (2014).
- Demonbreun, A. R., McNally, E. M. DNA Electroporation, Isolation and Imaging of Myofibers. J Vis Exp. (106), (2015).
- Falcone, S., Roman, W., et al. N-WASP is required for Amphiphysin-2/BIN1-dependent nuclear positioning and triad organization in skeletal muscle and is involved in the pathophysiology of centronuclear myopathy. EMBO Mol Med. 6 (11), 1455-1475 (2014).
- Flucher, B. E., Phillips, J. L., Powell, J. A. Dihydropyridine receptor alpha subunits in normal and dysgenic muscle in vitro: expression of alpha 1 is required for proper targeting and distribution of alpha 2. J Cell Biol. 115 (5), 1345-1356 (1991).
- Cooper, S. T., Maxwell, A. L., et al. C2C12 co-culture on a fibroblast substratum enables sustained survival of contractile, highly differentiated myotubes with peripheral nuclei and adult fast myosin expression. Cell Motil Cytoskeleton. 58 (3), 200-211 (2004).
- Al-Qusairi, L., Laporte, J. T-tubule biogenesis and triad formation in skeletal muscle and implication in human diseases. Skelet Muscle. 1, 26 (2011).
- Vilmont, V., Cadot, B., Ouanounou, G., Gomes, E. R. A system to study mechanisms of neuromuscular junction development and maintenance. Development. , (2016).
- Vilmont, V., Cadot, B., Vezin, E., Le Grand, F., Gomes, E. R. Dynein disruption perturbs post-synaptic components and contributes to impaired MuSK clustering at the NMJ: implication in ALS. Sci Rep. 6, 27804 (2016).
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2024 MyJoVE Corporation. Tutti i diritti riservati