
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
の単離と精製ショウジョウバエ末梢ニューロン
要約
このビデオ、記事では、の単離および精製のための方法を提示ショウジョウバエ末梢神経。単離された細胞から得られたRNAは、容易にマイクロアレイ解析などのダウンストリームアプリケーションに使用することができます。
要約
市販
プロトコル
ショウジョウバエ末梢の神経細胞の磁気ビーズソート上の一般的なコメント(議定書の完成のための総タイミング:2.5〜3時間)
きれいな、RNaseフリーの環境を維持するための標準的なラボの手順は、RNA分解を防ぐために、常に観察する必要があります。
ショウジョウバエ幼虫のクチクラを切開し、細胞解離バッファーに格納されている場合、末梢神経細胞は表皮からデタッチする最後のセルのいずれかです。我々は、このプロパティを悪用し、そのような筋肉と前のDAニューロンを単離することに脂肪のようなクチクラから非特定の細胞の大部分を削除するには、このプロトコルを設計している。
練習すれば、全体のプロトコルが正常に約2.5時間以内に完了することができます。実験が始まる前に抗体被覆ビーズの製造が完了しておく必要があります。
1。結合細胞用磁気ビーズの準備:
このステップは、前の実験の開始までに完了する必要があります。必要になるまで準備したビーズを用意し、4℃で保存することができます。
- 新しいPBS500μlにそれを再懸濁し、強い磁場にするたびにそれを沈殿させダイナビーズM - 280ストレプトアビジンコーティングビーズをPBSで3倍の100μlを洗う。
- 最終的に希釈していないビオチン化ラット抗マウス- CD8a抗体(抗体の濃度は100μg/ mlの場合)の100μlに直接ビーズを懸濁します。
- 沈降を防ぐために時折、軽度にボルテックスしながら、氷上で1時間混合物をインキュベートする。 [ダイナビーズM - 280を1μlのビオチン化抗体の0.05から0.10μgをバインドすることもできます]。
- として過剰な抗体を除去する手順1.1で説明したビーズ - 抗体混合3回洗浄する。磁気ビーズは、現在抗体でコーティングして使用できる状態にされています。 4で100μlの1X PBSにビーズ - 抗体混合物を保管° Cを使用するまで。
2。 (10-15分):幼虫の選択と洗浄
- 30〜50歳のマッチ三齢幼虫を選択し、1Xリン酸の1〜1.2ミリリットルと1.6ミリリットルマイクロ遠心チューブの中に置いてください(PBS)緩衝生理食塩水。 (3〜5分)
- それぞれ1秒間チューブと最大の設定で渦、それを三回を閉じます。
- ファイアーポリッシュパスツールピペットを使用すると、完全に上清を捨てる。洗浄(2.1)繰り返し、上清はどんな食べ物の粒子や破片の目に見えて明らかになるまで渦(2.2)3-4回繰り返します。
- 簡単に言うと70%エタノール1mlで洗浄を繰り返し、上清を捨てる。
- ddH2O 1mlで二回幼虫を洗って、上清を捨てる。
- 簡単に説明するのRNase AWAY 1mlで再度洗浄を繰り返し、上清を捨てる。
- のRNase AWAYの完全な除去を確実にするためにddH2O 1ml中に幼虫を3回洗浄する。
3。解剖:(10-12分)
- Sylgardコーティングされた35ミリメートルペトリ皿の中央に10月12日幼虫を置きます。少し離れてお互いにそれらを置きます。 [ 重要なステップ:適切な発達段階にしないと思われる任意の幼虫を破棄]
- すべての幼虫のために微細な解剖ハサミを使って幼虫の前方先端を切り開く。
- 幼虫インサイドアウト反転鈍いデュモン第5ピンセットを使用する。幼虫のクチクラの内側後端までずっとone forcepを挿入します。クチクラ(図1a)(それを容易にするためにSylgardの表面にキューティクルを押してみてください)の後端をつかむために一緒に鉗子の先端をつまんで。鉗子の2番目のペアを使用して、裏返し幼虫のクチクラを押してください。 [ 重要なステップ:数回の細胞の分離実験を試みる前にこの方法を実践してみてください。すべての軟部組織が容易に解離するためのソリューションにさらされていることを保証するために、幼虫が完全に反転して取得してください。]
- 3月4日の幼虫を解剖した後、1.6 mlのマイクロ遠心チューブに新鮮な、氷冷PBS(チューブを氷上に置く)にすぐにそれらを転送する。
- 必要なすべての幼虫が(このプロトコルは30〜40の幼虫)に収集されるまで3.4に手順3.1を繰り返します。 [ 重要なステップ:。それに応じて10〜20%の解離と解離時の損失、および計画を先取り]
4。緩く付着した非特異的な細胞の除去:(2-3分)
[このステップでは、脂肪体とCNSとして緩く付着した非特異的な組織の清算を支援します。]
- 反転幼虫のキューティクルを含む1.6ミリリットルのマイクロチューブを取り出し、新鮮な氷冷PBSの約700から800μlの上清を交換してください。
- パルス渦マイクロ遠心チューブフルスピードで5回(1パルス当たり3秒)。
- 上清を捨て、約700〜800μlの新鮮な、氷冷PBSに交換してください。
- ステップ4.2と4.3を3回繰り返します。
- レス新鮮な、氷冷PBS 400μlの幼虫のキューティクルをuspend
5。単一の細胞懸濁液に組織を解離:(18-20分)
[ 重要なステップ:オーバー解離は乏しい細胞収率と低い細胞の生存率につながる細胞表面マーカーの損失を引き起こす可能性があります。幼虫組織は機械的解離(超音波処理、douncing)、酵素的解離(トリプシン、コラゲナーゼ等)またはその両方の組み合わせによって解離させることができる。これらの幼虫の組織が解離することは困難であるので、我々は両方の機械的および酵素的解離の組み合わせが最良の結果をもたらしたことがわかった。]
- PBS 400μlに懸濁し幼虫のクチクラに1XリベラーゼBlendzyme 3(28 W nsch単位/バイアル)を1.5μlを添加する。
- 最大の設定で1秒ごとに(これは、溶液中にキューティクルから緩く接着細胞を除去する必要があります)のための渦は、溶液を2〜3回。
- 5分間室温(22〜25 ° C)での解をインキュベートする。 [ 重要なステップ:インキュベーション時間が大幅に最終的な細胞選別の効率に影響を与える。推奨されるインキュベーション時間は、組織を緩めるための十分なはずです。 15分を超えないようにしてください。]
- フルスピードでのパルスごとに2秒間パルスボルテックスチューブは最大設定で20〜30回。これは、溶液中に筋肉や他の組織を解放する必要があります。各ステップで蛍光ステレオ顕微鏡下で溶液から少量のサンプルを検査する。 (経験では、ある者は、直接蛍光対応のステレオ顕微鏡下のマイクロチューブを観察することによって、解離のレベルを決定することができるはずです)
- 新鮮な、氷冷PBSで幼虫のキューティクル2-3回洗浄し、最終的に新鮮な、氷冷PBSを含む1%BSA500μlに再懸濁します。
- 2mlのKontes組織グラインダーと大クリアランス乳棒、プリコートPBS溶液でと短いが、BSA溶液を廃棄リンス後、1%BSAで組織グラインダーと乳棒のガラスの表面に付着した幼虫のクチクラを避けるために。その後、ファイアーポリッシュパスツールピペットを使用して、BSAでコーティングされた組織グラインダーにステップ5.5からキューティクルを転送する。 [ 重要なステップ:細胞の損傷/溶解を防ぐために、数分間氷上に置くことによってプレクール組織グラインダー/乳棒]。
- (約20〜30ストローク)泡を避け、ゆっくりと着実なストロークで組織をダウンス。 [ 重要なステップ:特に細胞が溶解することが、ゆっくりと着実にダウンス]
- 細胞解離のレベルを評価するために、きれいなキムワイプの組織と組織グラインダーの外壁を拭いて、そして蛍光ステレオ顕微鏡下でそれを点検。ニューロンは、キューティクルから切り離されているはず、と溶液中で見ることができます。これは難しいことが判明した場合、代わりにソリューションの小さなサンプルをピペッティングし、蛍光ステレオ顕微鏡下に観察します。細胞解離の良い指標は、幼虫のクチクラからニューロンが存在しないことです。細胞は単一の細胞懸濁液を得るまでは、1つがまだキューティクルに接続されている神経細胞を観察する、または不完全解離細胞を観察する場合、さらにダウンス。
- ファイアーポリッシュパスツールピペットで5回は、ファイアーポリッシュパスツールピペットで10回に続いて、標準的な先端の直径の約50%に縮小ソリューションをひいて粉にすると、標準的な先端の直径の約25%に縮小した。 [ 重要なステップ:強制粉砕は、細胞を損傷する恐れがあります。ステップ間の細胞を監視し、それに応じて手順を調整する]。
- 30μmのセルのフィルターを通してソリューションをフィルタリングし、1.6 mlのマイクロ遠心チューブに細胞ろ液を集める。得られた溶液は、単一の細胞懸濁液から構成され、現在は磁気細胞選別のための準備ができているはず。
6。磁気ビーズ細胞選別:(45〜75分、抗体のインキュベーション時間に依存)
- 細胞懸濁液の500μlの(ステップ5.10)に抗体被覆磁性ビーズの15μlを添加する。その後の細胞のアイソレーションのために必要になるまで残りの抗体結合した磁気ビーズを保存することができます。
- 時折手で混合し、氷上で30-60分間抗体被覆磁性ビーズで細胞をインキュベートする。 [ 重要なステップ:インキュベーションより高い温度または長い時間では、非特異的抗体の結合になることがあります。]
- 2分間の強力な磁場でのマイクロチューブを置きます。未結合ビーズと一緒にすべて肯定的に選択したセルは、チューブの側面に分離されます。
- 徐々に細胞ペレットを乱さないように確認しながら、上清をピペットで。
- 残りの非特定のセルを削除するには、新鮮な、氷冷したPBSで細胞を3〜4回洗浄する。
- 新鮮な、氷冷PBS30μlの細胞を再懸濁します。
- 細胞のおおよその純度および収率に、細胞のSUSPのピペットを5μl血球計算板の研磨面上にensionと蛍光ステレオ顕微鏡下で表示されているすべての蛍光細胞を数える。また、非蛍光細胞と不純物の兆候の量を確認してください。一般的にサンプルは非常に蛍光を発する細胞を濃縮されます。
7。磁気ビーズソートされた細胞からのRNA単離:(60 - 75分)
- 、ペレットの磁場で細胞をカウントした後、上清を廃棄し、PicoPure™RNAアイソレーションキット(Molecular Devices社)からの抽出緩衝液20μlを加える。細胞の数に応じていずれかの抽出緩衝液の高いボリュームを追加する必要があります。
- 抽出バッファーで細胞ペレットの混合を可能にするため、最高速度でボルテックスチューブ。
- 30分間42℃でチューブをインキュベートします。
- (ステップ7.5を参照)前にRNAのカラム精製に磁気ビーズの除去を確実にするために、チューブが簡単にペレット〜2分間磁性ビーズ2,000(X)gで遠心分離される。チューブは、その後、ペレットを保持するために強力な磁場に置かれ、その上清を新しいマイクロ遠心チューブに転送されます。
- 抽出し、カラムはPicoPureのRNA抽出キットの製造業者の指示に従ってRNAを精製する。 DNase処理は省略可能であり、分析の要件に記載のRNA精製中の列に対して実行できます。最後に、使用直前まで-80℃での溶出バッファーとストア° Cの少量(11〜30μL)にバインドされたトータルRNAを溶出させる。必要に応じて1μlのアリコートをバイオアナライザ2100(Agilent Technologies社)にトータルRNAの品質を評価するために使用されることがあります。
代表的な結果:
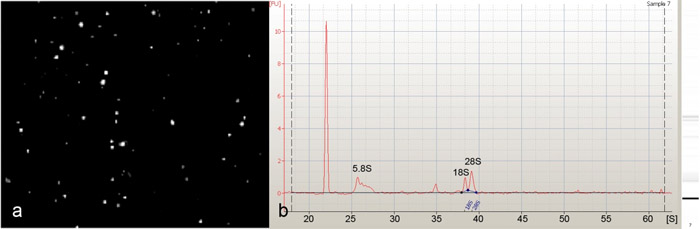
磁気ビーズ並べ替えは、 ショウジョウバエ DAニューロン(図1)を単離するために使用されました。これらの孤立したDAニューロン(図2a)から精製したRNAは、Agilent 2100バイオアナライザ(Agilent Technologies社)で解析するとシャープ5.8S、18Sおよび28SリボソームRNAのピークの存在によって示されるように、優れた品質であることがわかった(図2b)。 30〜40三齢幼虫で始まる我々は、PPK - GAL4ドライバー、および使用して1500年から2000年DAニューロン(クラスI、II、III及びIV)のパンを使用して平均300から500クラス- IV DAニューロンに分離することができたDAニューロン特異的GAL4 21から7ドライバ。私たちの単離された細胞の神経細胞特異的な濃縮を評価するために我々は2つの神経細胞の遺伝子特異的マーカーを(elavとfutsch)を使用して定量的逆転写PCR(定量RT - PCR)を行った。これらの分析は、我々のプロトコル(図3)を使用して、フロースルーに比べて、DAニューロンのための高度に特異的な濃縮を示すマーカー遺伝子の両方の重要な倍濃縮を明らかにした。最後に、パン- DAニューロンとクラスIV DAニューロンの両方から分離されたRNAを、アジレントキイロショウジョウバエ全ゲノムオリゴマイクロアレイ(4 × 44K)(図4)の転写発現のプロファイリングを実行するために使用されました。これらの分析は、以前に未知の分子と潜在的にDAニューロンの開発において重要な機能的な役割を果たしている推定上のシグナル伝達経路の広いスペクトルに加えてDAニューロン樹状突起の形態形成の数々の以前に関与する調節因子を同定した。 DAニューロンの開発、特に樹状突起形態形成を媒介するこれらの以前に未知の分子の潜在的な役割(複数可)を評価するために設計された研究は、現在進行中です。

図1: ショウジョウバエ DAニューロンの磁気ビーズソーティングの回路図DAニューロン特異的GAL4をもつ三齢幼虫をマッチング()年齢、UAS - mCD8 - GFPレポーター遺伝子を公開する、インサイドアウト反転幼虫のクチクラによって解剖されています解離バッファーへのPNSは、氷冷PBSで格納。(B)酵素解離が幼虫のクチクラを含む溶液にリベラーゼBlendzyme 3を添加することにより行われる。(c)の幼虫の組織が ボルテックス、粉砕の組み合わせにより、さらに解離してとdouncing脂肪体、腸やCNSなどの非特異的に標識された組織を除去する。(D、E)次に、細胞を30μmのセルのフィルターを使用してフィルタリングされます。解決策は、上皮、筋肉や神経細胞などの異なる細胞型の単一の細胞懸濁液が含まれています。(F)抗マウスCD8a抗体コーティングされたダイナビーズM - 280は、細胞懸濁液に加え、30〜60分間氷上でインキュベートする。( g)を磁気ビーズには、GFPの融合タンパク質。(H、I)磁気ビーズコーティングされた細胞は、強力な磁場でソリューションを配置することによって分離されているタグ付きマウスCD8を発現しているDAニューロンに結合する。上清を捨て、そして細胞は、(j)は Hで、その結果、任意の残留非特定のセルを削除するには、3回洗浄しighly DAニューロンの集団を精製した。してくださいここをクリックして図1の拡大バージョンを参照すること。
{kind=link}

図2:細胞解離と磁気ビーズソーティングによって分離された積極的に選択、GFP蛍光クラス- IV DAニューロンの(a)の代表画像。ニューロンの結果として人口が非常にほとんどあるいはまったく汚染細胞の不純物とクラスIV DAニューロンの濃縮されると決定した。(b)の磁気ビーズから分離したトータルRNAのAgilent 2100バイオアナライザ(アジレントテクノロジー社)電気泳動では、DAニューロンをソート、5.8S、18S、および28S rRNAsの存在によって示されるように、優れたトータルRNAの品質を示す。してくださいここをクリックして図2の拡大バージョンを参照すること。
{kind=link}

図3:分数を介して分離されたDAニューロンの神経マーカーの遺伝子発現の定量RT - PCR解析(GAL421 - 7、UAS - mCD8 - GFP)との流れは三連で実施した。 two神経細胞特異的なマーカー遺伝子(elavとfutsch)の発現レベルを定量RT - PCRにより評価した。これらの分析から得られた値は、内在性コントロール(rp49)に正規化され、そして分数フロースルーで観察されたものと相対的なレベルはΔΔCτの方法6を用いて算出した。分数を通る流れと比較してelavとfutsch両方が大幅に分離されたDAニューロンの集団で濃縮した。

図4:マイクロアレイのイメージファイルというラベルの代表クラス- IV DAニューロン特異的Cy3標識。ここに示されている磁気ビーズソーティングにより精製し、クラスIV DAニューロンから分離されたのCy3 - labeldトータルRNAとハイブリダイズアジレントキイロショウジョウバエ全ゲノムオリゴマイクロアレイ(4 × 44K)です。してくださいここをクリックして図4の拡大バージョンを参照すること。
{kind=link}
Access restricted. Please log in or start a trial to view this content.
ディスカッション
ここで紹介するプロトコールは、磁気ビーズ細胞選別の戦略を用いてショウジョウバエの三齢幼虫のクチクラの内側の表面に強固に付着末梢神経細胞の単離および精製のために最適化されています。我々は特にショウジョウバエのDAニューロンを分離するためにこのプロトコルを使用している一方で、開発の幼虫または蛹の段階で表皮に付着する他の細胞型の分離にこのプロトコル(例えば?...
Access restricted. Please log in or start a trial to view this content.
謝辞
我々は、博士に感謝する。塩原-ティンニン本研究で使用されてこない系統を提供するための月とウェスGrueber。著者らはこの研究の支援(DNC)とジョージメイソン大学の学長のオフィス(EPRI)のためにトーマスF.とケイトミラーJeffressメモリアルトラストを認める。
Access restricted. Please log in or start a trial to view this content.
資料
機器
- (10)ネオジウムブロックマグネット(K&J Magnetics、Inc。製、カタログ番号B444B)、代わり™-2(Invitrogen社、カタログ番号123 - 21D)を使用することができますdynamagを。
- 大クリアランス乳棒付きKontesガラスティッシュグラインダー、2ミリリットルの作業能力、(カタログ番号#885300〜0002)
- セルのフィルタ、例えば、MACSプレセパレーションフィルター(ミルテニーバイオテク社、カタログ番号130-041-407)
- 35ミリメートルペトリ皿Sylgard(ダウコーニング社、カタログ番号3097358〜1004)でコーティング
- 解剖ツール:デュモン5号鉗子の2ペア(ファイン科学のツール、カタログ番号11251から20。)
- Vannas春顕微解剖用はさみ(ファイン科学のツール、カタログ番号15000〜08)
- 使い捨てチップとピペット(P - 1000、P - 100、P - 10)
- 細胞と見積もりの純度をカウントする標準血球計数器
- 標準直径の火ポリッシュのヒントとパスツールピペットは、標準の直径の約50%に縮小し、粉砕のための標準的な直径の約25%に縮小
- テーブルトップマイクロ遠心機(1-16,000(X)g)の
- 渦
- 蛍光ステレオ顕微鏡(ライカMZ16FAはこのプロトコルで使用されていた)
参考文献
- Corty, M. M., Matthews, B. J., Grueber, W. B. Molecules and mechanisms of dendrite development in Drosophila. Development. 136, 1049-1061 (2009).
- Parrish, J. Z., Emoto, K., Kim,, Jan, Y. N. Mechanisms that regulate establishment, maintenance, and remodeling of dendritic fields. Ann. Rev. Neurosci. 30, 399-423 (2007).
- Lee, T., Luo, L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron. 22, 451-461 (1999).
- Grueber, W. B. Projections of Drosophila multidendritic neurons in the central nervous system: links with peripheral dendrite morphology. Development. 134, 55-64 (2007).
- Song, W., Onishi, M., Jan, L. Y., Jan, Y. N. Peripheral multidendritic sensory neurons are necessary for rhythmic locomotion behavior in Drosophila larvae. Proc Natl Acad Sci USA. 104, 5199-5204 (2007).
- Livak, K. J., Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) methods. Methods. 25, 402-408 (2001).
Access restricted. Please log in or start a trial to view this content.
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved