
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
胚の脊髄から交連ニューロンの解剖と文化
* これらの著者は同等に貢献しました
要約
このビデオでは、E13ラット背側脊髄から交連神経細胞を細かく分析し、文化に方法を示しています。解離交連ニューロンの軸索成長とガイダンスの細胞および分子メカニズムを研究するのに便利です。
要約
交連ニューロンが広く胚脊髄の開発中に軸索誘導のメカニズムを調査するために使用されている。これらのニューロンの細胞体は背側脊髄に位置しており、その軸索は、胚発生時にステレオタイプ化された軌道に従っています。交連軸索は、最初にfloorplateに向かって腹側に投影する。正中線を交差した後、これらの軸索が脳に向かって前方およびプロジェクトにします。これらの各ステップは、いくつかの指導の手がかりの作用によって調節されている。高度交連ニューロンに富んだ文化を理想的に回転アッセイ、免疫化学、生化学を含む軸索の経路探索のメカニズムを、対処する多くの実験に適しています。ここでは、E13ラット背側脊髄から交連神経細胞を細かく分析し、文化に方法を説明します。最初に、脊髄が隔離され、背板はグレー解剖されています。背側組織は、トリプシン処理し、機械的破砕により細胞懸濁液にして解離している。神経細胞はポリ- L -リジンでコーティングされたガラス製カバースリップまたは組織培養皿に播種されています。 30時間後
プロトコル
1。ラット胚の背側脊髄の解剖
一般的な推奨事項
氷の上でL - 15培地を維持し、頻繁に胚を低温に保つための解剖皿に培地を変更してください。これは、組織の整合性を維持することができます。記載がない限り、すべての手順は、デュモン#5鉗子の二組で行われます。コンタミネーションを避けるために、70%エタノールですべてのツールと作業面をスプレーし、解剖の培地ボトルを閉じたまま。皿の間に胚を転送するには、カットのプラスチック製のピペットまたは穴あきスプーンを使用してください。それは、正常解剖を完了するために脊髄を(ストレッチ、ニッキング)傷つけないように重要です。
準備
- コールドL - 15培地
- L - 15の50 mLの+ 10%の熱不活化ウマ血清(HiHS)。氷上に置きます。
脊髄の解剖
- E13安楽死制度のガイドラインにしたがってCO 2チャンバーでラット(E0 =初日、次の交配の日)を妊娠すると、上演。
- 腹部に70%エタノールをスプレーしてください。鉗子と外科はさみでカットと下腹部の皮膚をつまんで引き上げます。腹腔に到達するための筋肉と腹膜の層を介してカットして繰り返します。上胸部に、腹部の側面に沿って組織を切断することによりV字型の切開を作成します。腹腔を露出するために組織を持ち上げて、引き戻す。
- 子宮は、3つの位置に取り付けられている:下部中央の腹部で、そして両方の上部の横方向の隅に。胚嚢の間の組織上つかんで子宮を持ち上げる。氷の上でL - 15で満たされたペトリ皿で子宮と場所を削除するには、結合組織をカット。
- 次のステップは、解剖顕微鏡下で行われている。胚体外組織と胚膜から胚を分離するには、、鉗子の一組で子宮嚢の間の組織をつかむ。他のペアと、表面的な膜(暗い側が胎盤)を介してカットする嚢のより透明性の側面をつまんで。優しく嚢の胎盤側を押して、胚を絞り出す。氷の上でL - 15で満たされたペトリ皿内のすべての胚と場所を削除します。
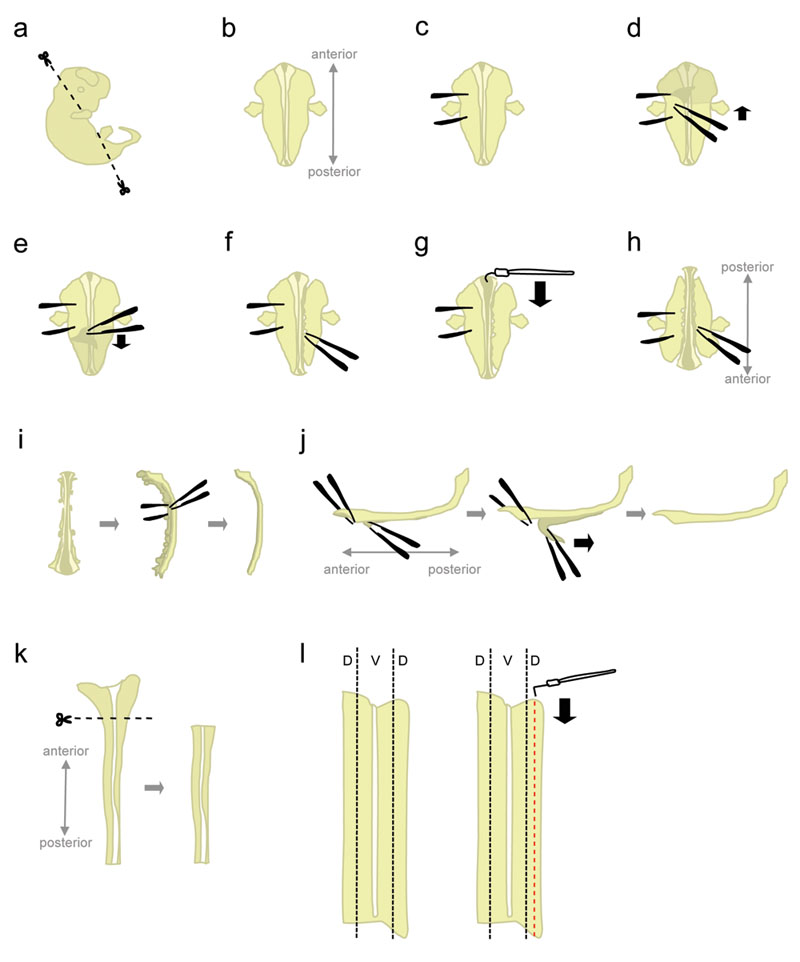
- 氷冷L - 15を充填した10cmペトリ皿に片足胚。 microscissorsを使用して、図1aに示す角度で頭部と後部の部分を切り取る。
これらの角度でカットする位置胚を"ダウン腹顔"に配置するのに役立ちます。 - 位置胚"腹側を下に"(前方の距離を指し、実験者の方を向いて後方)(図1B)。
- しっかりと(図1C)胚を"突き止める"に鉗子を使用して、片側から胚を保持。それは組織を引き裂くので、鉗子を移動しないでください。鉗子の他のペアと、皮膚を剥がし、"保持"鉗子(図1D - E)の間に領域から始まる胚の背面をカバーしています。これは、この時点では、膜(髄膜)で包まれて、脊髄を公開します。
脊髄をニッキング避けるために、側面からではなく、脊髄を上から皮膚をつかむ。 - 部分的に脊髄の右側(図1F)に組織を切り離します。保持鉗子のレベルから開始し、できるだけ近い脊髄へ、閉じた鉗子で組織を突く。その後、徐々に組織を引き裂くために鉗子を開きます。これは脊髄から後根神経節(DRGS)をデタッチする必要があり、また腹の器官を破壊する必要があります。 したがって、次のステップの間に上向きに引っ張られるからそれを防止する、接続されている 。 まま組織が 胚に重量を追加する前部と後部の両端に接続されているいくつかの組織を残す。さらに、その組織は後のステップで胚を保持するために使用されます。
- 前端から始まる、髄膜をカットし、roofplate(図1G)に沿って脊髄を開くためにフック状のタングステン針を使用してください。
- 胚を180度(後方は前方実験者の方を向いて、離れて指している)(図1H)を回します。
これは、実験者は、胚の他の側面から組織を切り離すために、同じ手を使用することができます。 - 以前に切り離された側につかんで胚を保持しており、(1.8を参照してください。)同じ方法を使用して、残りの側から組織を混乱させる。完了したら、完全に脊髄の両側に組織を切り離す。
代替:完全に左側に、残りの組織を切り離すが、右側の前部と後部の端に接続されているいくつかの組織を残す。これは、時々ステップ1.12の位置で脊髄を維持するのに役立ちます。 - その側に脊髄を置き、残りの間葉組織とDRGS(図1I)の大部分を取り除く。
- このステップでは、髄膜と脊髄は、お互いに並列した組織の二つの"枚"です。脊髄の大きい、前部、ほとんどの部分を突き止める、と脊髄CORからの髄膜の短いセグメントを剥がしD(図1J、左)。今、鉗子で全体をつかんで2つの分離されたセグメントを保持し、滑らかな、一定の動き(図1J)と髄膜剥離。
不均一な脊髄および/または髄膜層の破壊につながる"はがれ"。それはまだ髄膜に接続されている脊髄の一部を回復することは通常不可能です。 - プラスチック製のピペットを用いて、L - 15 + 10%HiHSを含むペトリ皿に分離された脊髄を転送し、氷上におきます。一般的に、すべての胚の脊髄は背側の部分を解剖する前に収集されます。
背側脊髄の解剖
- 、L - 15 +10%HiHSを含む"オープンブック"の構成で平らにペトリ皿に片足脊髄。広い、前方部分(後脳の一部を含む)(図1K)を切り取る。
- 背側組織は脊髄の外側 - ほとんどの部分(図1リットル、左)に位置しています。ストレートタングステン針でコードを固定しながら、半分の脊髄の5分の1幅であるストリップをカットしてL字型タングステン針を使用してください。 (図1リットル、右)。氷の上でL - 15 + 10%HiHSを含む15 mLのプラスチックチューブの背板を置きます。
〜12 E13胚の背側神経管のセクションでは、解離した後、メッキのために〜3.5〜4000000細胞が得られます。より多くの細胞は背側脊髄(広い背板の切断)の粗解剖を行うことによって得ることができるが、交連ニューロンの純度が低下します。
2。交連ニューロンの文化
一般的な推奨事項
特に明記しない限り、すべてのステップは、組織培養フードに無菌条件下で実行する必要があります。新鮮な培地と新鮮融解サプリメントや試薬を使用してください。解離と粉砕のステップは、+ / Mg 2 +のフリーHBSSのCa 2を最小限に+ /のMg 2 +依存性接着のCa 2で実行されます。
準備
- コーティングされたカバースリップ(ドイツDesagのガラスを使用)または組織培養プレート(下のコーティングの手順を参照してください)。
- メッキの前に、少なくとも1.5時間、組織培養インキュベーターで2平衡化した組織培養皿とCOの暖かいNeurobasalメッキのメディア(下記参照)、。
- 37 ° Cの水浴中で2.5%トリプシン
- HBSSのCa 2 + / Mg 2 +を -無料、4一℃、37少なくとも一つ℃の2本
- twoファイアーポリッシュガラスパスツールは、直径が通常の半分サイズを持つ、と半分よりわずかに小さい直径を持つものをピペット。滅菌パスツールピペットを使用してください。ピペットを発射-磨くため、わずかに直径を減少させるために先端を溶融するために(水色の炎の上部)ブンゼンのバーナーを使用してください。このステップは、組織培養フードの外側に実行されるため、組織培養のボンネットの下に配置する前に、70%エタノールでピペットをスプレー。ニューロンのより効率的な回収のため、コート解離の開始前に、または単に粉砕工程の前に血清を含む培地でパスツールピペット(メディアとピペットを記入し、30秒間保持する)。これは、粉砕中にパスツールピペットの内側に付着する細胞を防ぐことができます。
- PLLでコーティングされた皿やカバーガラスを洗浄するための滅菌MilliQ水
- HBSSで12.5%のMgSO 4ソリューション
ポリ- L -リジンコーティング
カバーガラス、24時間の酸洗浄及び(Kaechとバンカー、2006参照)メッキの前に滅菌を使用する場合。ドイツDesagのガラス製カバースリップを使用してください。
コートカバーガラスまたはプラスチック製組織培養皿上にポリ- L -リジンには:
- 組織培養のボンネットの下に、1.75から2時間では100 ug / mlとPLLソリューションの小さなドームで表面を覆う。
- MilliQ水、洗浄ごとに少なくとも5分(下の細胞解離のステップで実行できる)で2回洗浄。
- 使用するまで水に店舗。 PLLでコーティングされた表面が乾燥させないでください。
カバーガラスのコーティングのためのPLL浪費を減らすために、無菌の細菌のペトリ皿にカバースリップを配置。コートやカバースリップで液体のドームを配置することによって、カバースリップを洗う。細菌のペトリ皿は疎水性であるため、液体はガラス製カバースリップ上に残るはず。単に細胞を播種する前に組織培養皿にカバースリップを転送する。
プラスチック製組織培養皿に播種神経細胞は、密着性が通常より高い場合、このように神経突起伸長が低下される可能性があります。
解離とメッキ
- 背側ストリップをチューブの底に落ち着いていることを確認します。パスツールピペットでL - 15 +10%HiHSのほとんどを削除します。すぐに冷たい(4℃)HBSS 3mlを加えることによって一度背側神経管のストリップを洗浄。
- 背側神経管のストリップが2分間静せ、その後、パスツールピペットでHBSSを除去。
- 4.7ミリリットルの容積に温かい(37℃)HBSSを追加。 T鶏は、0.15%トリプシンの最終濃度を与えるために2.5%トリプシンの0.3 mlを加える。
- 37℃で7分間水浴中でインキュベートする。インキュベーションを通じて静かに半分の方法一度混ぜる。
- 150 U / mlの最終濃度を30μlのDNaseを(25 000 U / mL)を追加。 MgSO 4を 60μlを加え、0.15%の最終濃度のため、簡単に混ぜる。
粗解剖から組織の場合は、37℃で余分な1分間インキュベートします水浴中でCを。
この段階では、背側神経管のセクションが断片化されるべきである。背側神経管セクションが断片に開始されていない場合は、通常2.5%トリプシン株式が古いことを意味し、そして新しい株式を解凍するか、または冷HBSS with washは効果的にサンプルからHiHSが削除されませんでした。 - 4分の200 gで組織片を遠心します。
- チューブの底に液体の〜50〜100μlを残し、パスツールピペットで上清を取り除きます。
- ペレットをゆるめるために軽くチューブをフリックして、暖かいHBSS 5mlを加えて細胞を洗浄。 2分間、室温で解決しましょう。
- 5分、200 gで遠心する。
- チューブの底に液体の〜50〜100μlを残し、パスツールピペットで上清を取り除きます。
- チューブは、ペレットを緩めると、部分的に細胞を再懸濁させる優しくフリック。その後暖かいHBSS 2 mlを加える。
- ゆっくり上下4-6回ピペッティングして細胞を解離するために小さな(ハーフ直径)ファイアーポリッシュガラスパスツールピペットを使用してください。気泡を加えることは避けて、そしてチューブの側面に液体をピペット。オーバー粉薬しないでください。
- さらにゆっくりと上下3〜4回ピペッティングして細胞を解離するために最小のファイアーポリッシュガラスパスツールピペットを使用してください。気泡を加えることは避けて、そしてチューブの側面に液体をピペット。オーバー粉薬しないでください。
粗解剖から組織の場合は、解離の終了時にチューブに暖かいHBSSの余分な1 mlを加える。
細胞を解離するときに、それはすべての細胞塊及び凝集体を解離する必要はありません。上下のピペッティング停止したり、さらにピペッティングで細胞凝集体のサイズをさらに低下は見られない場合は、小さい直径でパスツールピペットに変更。 - 残りの組織片は1分間チューブで解決しましょう。それは、新しいチューブに細胞を転送する必要はありません。
- 細胞懸濁液20μlをとり、トリパンブルーの5μlを加える。 haemocytometer上のセルを数える。
神経細胞はトリパンブルー排除により、≥95%実行可能でなければなりません。 - プレートNeurobasalめっきメディアのニューロン(下記レシピ参照 )。
- 分離された神経細胞を取得するためのメッキの密度は、(低密度の文化が互いに凝集または重複するニューロンを避けるために)提案した:
- 120 000 - /ウェル、6ウェルプレート用180 000細胞
- 60 000から75 000細胞/ウェル、12ウェルプレート用
- 16〜18時間後、(下のレシピを参照)Neurobasal成長のメディアにメディアを変更する
メディアを変更する際に培養皿から培地を吸引除去するため真空ポンプを使用しないでください。ピペットを使用する。これは、ニューロンを外れ回避。
代表的な結果:
メッキ後4時間は、神経細胞はポリ- L -リジン(PLL)でコーティングされた表面に付着している必要があります。位相コントラスト照明の下、付着細胞体は典型的には、比較的平坦なと(図2A)楕円形です。よく遵守していない細胞は、料理がとても優しく側にタップされたときにわずかに移動する球体として表示されます。多くの要因が潜在的に(議論を参照)細胞の接着を妨げることができる。
in vitroで 30時間後、ほとんどのニューロンは、目に見える成長円錐(図2C、D)と軸索を延長しました。貧しい軸索の成長が観察されている場合は、Neurobasal増殖培地を新鮮な培地とサプリメントで作られていることを確認します。ニューロンは、これらの条件の少なくとも6日間健康なまま。この手順では、DCC(ヤムら2009)を発現する神経細胞の〜90%と、非常に交連ニューロンに富んだ準備を確実にもたらすことが証明されています。細胞懸濁液を調製するために使用されている背側脊髄のストリップの幅が薄いストリップを使用する高い純度で、文化の純度に影響を与えます。サンプルアプリケーションは、図3(免疫蛍光)で示されています。ヤムらの論文を参照してください。 (2009)その他の例について。

図1脊髄の解剖の手順の概略図。 D =背側、V =腹。 大きな図を見るにはここをクリック。
{kind=link}

図2分離された交連の代表結果ニューロンは、PLLでコーティングされたガラスカバースリップ上に播種。 、B)めっき後4時間、ニューロンは、表面に付着している。バー= 20μmの。 C、D)めっき後30時間は、ほとんどのニューロンは、目に見える成長円錐と軸索を延長しました。バー= 20μmの。

図3。交連ニューロンはファロイジン(F -アクチン、赤)およびDAPI(青)による核で標識F -アクチンで、γ-チューブリン(緑)を免疫染色。
レシピとコメント
Neurobasalめっきメディア
- Neurobasal
- 10%熱不活性化FBS(HiFBS)
- 2mMのL -グルタミン(200mMのストック溶液から)
オプション:ペニシリン/ストレプトマイシン系抗生物質(通常の半分の濃度を使用)
Neurobasal成長メディア
- Neurobasal
- B27(株式1 / 50希釈)
- 2mMのL -グルタミン(200mMのストック溶液から)
- オプション:ペニシリン/ストレプトマイシン系抗生物質(通常の半分の濃度を使用)
メディアが行われると、それは2週間4℃に保つことができる。少なくとも1.5時間、細胞、組織培養ペトリ皿の場所のメディアと組織培養インキュベーター内の場所をめっきする前に温度と培地のpHを平衡化する。
Neurobasal
Neurobasal培地の瓶が開かれた後、4℃で一ヶ月間保存することができます℃で暗所で。そうでない場合は細胞の生存率が低くなる1ヶ月以上開かれているNeurobasal廃棄してください。
熱不活化ウシ胎児血清(HiFBS)またはウマ血清(HiHS)
30分間水浴中° C 56における熱、熱不活性化FBSまたはHSへ。スワールボトルは約10分ごとまたはそう。 (精度は水で満たされた同じようなサイズのボトルを使用。56℃に達したとき。この時点で時間を計り始める。見て水のボトルに温度計を置いて)熱不活化FBSは、沈殿物をクリアするために遠心分離する必要があるかもしれません、と-20℃で分注し、再凍結することができます
L -グルタミン
常に各実験のためのL -グルタミンの新鮮なアリコートを解凍。
B27
B27のアリコートは、° C、長期保存のため、または4℃で一ヶ月、最大で20に保つことができる。
ディスカッション
我々は、ラット胚脊髄から交連神経細胞を細かく分析し、文化に方法を記載している。この手順は、定期的に確実に軸索ガイダンスの細胞および分子メカニズムを研究する神経細胞を準備する私たちのラボで使用されています。細胞生物学および免疫化学実験、一腹の子の解剖のために十分なニューロンを生成します。より多くの細胞がこのような多くの生化学の実験のように、必要な場合?...
謝辞
この作品は、カナダ衛生研究所(CIHR)、ピーターライード医学研究財団、Neuroengineeringのマギルプログラムからの補助金によって支えられて、フォンデルシェルシュエンサンテケベック(FRSQ)、およびイノベーションのためのカナダ基金(CFI )。セバスチャンD.ラングロワはデラルシェルシュエンサンテケベック(FRSQ)と健康研究(CIHR)のカナダの協会からフレデリックバンティングとチャールズベストカナダ大学院奨学金で修士賞フォンからマスターのトレーニング賞によってサポートされていました。我々は数字の支援のためのジェシカMTファムに感謝しています。
資料
ラット胚の背側脊髄の解剖(表Iも参照)
- E13ラットの妊娠は、上演
- 70%エタノール
- 手術用はさみ、ファイン科学ツール
- 鉗子、デュモン#5、ファイン科学ツール
- ペトリ皿
- L - 15培地
- 解剖顕微鏡、ライカ
- プラスチック製のトランスファーピペット
- Microscissors、ファイン科学ツール
- タングステン針とピンホルダー(つのフックの形をした針、1つのL形状の針、1本の直線針)、ファイン科学ツール
- 熱不活化ウマ血清(HiHS)
- 15ミリリットルプラスチックチューブ
交連ニューロンの文化(I表も参照)
- 組織培養インキュベーター(37℃、5%CO 2、湿度制御)
- ドイツDesagのガラス製カバースリップおよび/またはプラスチック製組織培養ディッシュ
- ポリ- L -リジン、シグマ
- 滅菌ミリQ H 2 O
- Neurobasal、インビトロジェン
- 熱不活化ウシ胎児血清(HiFBS)
- L -グルタミン
- B27、インビトロジェン
- ペニシリン/ストレプトマイシン系抗生物質
- 37 ° Cの水浴
- 滅菌パスツールピペット
- ブンゼンガスバーナー
- HBSS、Ca 2 +の /のMg 2 +フリー、Invitrogen社
- DNアーゼ、ワーシントン
- MgSO 4を
- 遠心
- 血球計
- トリパンブルー溶液
参考文献
- Charron, F., Stein, E., Jeong, J., McMahon, A. P., Tessier-Lavigne, M. The morphogen sonic hedgehog is an axonal chemoattractant that collaborates with netrin-1 in midline axon guidance. Cell. , 113-1111 (2003).
- Fabre, P., Shimogori, T., Charron, F. Segregation of ipsilateral retinal ganglion cell axons at the optic chiasm requires the Shh Receptor Boc. Journal of Neuroscience. 30, 266-275 (2010).
- Helms, A. W., Johnson, J. E. Progenitors of dorsal commissural interneurons are defined by MATH1 expression. Development. 125, 919-928 (1998).
- Kaech, S., Banker, G. Culturing hippocampal neurons. Nature Protocols. 1, 2406-2415 (2006).
- Okada, A., Charron, F., Morin, S., Shin, D. S., Wong, K., Fabre, P. J., Tessier-Lavigne, M., McConnell, S. K. Boc is a receptor for sonic hedgehog in the guidance of commissural axons. Nature. 444, 369-373 (2006).
- Yam, P. T., Langlois, S. D., Morin, S., Charron, F. Sonic hedgehog guides axons through a noncanonical, Src-family-kinase-dependent signaling pathway. Neuron. 62, 349-362 (2009).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved