
Method Article
早期着床後のマウス胚への細胞やDNAの正確ローカライズ転送する方法
要約
We demonstrate a method for grafting cultured cells into defined sites of early mouse embryos to determine their in vivo potential. We also introduce an optimized electroporation method that uses glass capillaries of known diameter, allowing the precise delivery of exogenous DNA into a few cells in the embryos.
要約
マウス初期胚の操作や文化は、このモデルシステムの価値を高める強力で利用アンダー主に技術です。逆に、細胞培養物は広く発生生物学の研究において使用されてきました。しかし、in vitroで培養された細胞が真に生体内の細胞型で表現するかどうかを決定することが重要です。開発中の貢献の評価に続いて胚に細胞を移植すると、in vitroで培養した細胞の可能性を決定するための有用な方法です。本研究では、ex vivoで培養した早期の着床後のマウス胚の定義された部位に細胞を移植する方法について説明します。また、高いトランスフェクション効率と低い細胞死の両方に外来DNAを受けた細胞の数の正確な位置と調整を可能にすること、既知の直径のガラスキャピラリーを使用して最適化されたエレクトロポレーション法をご紹介します。任意のSPを必要としないこれらの技術、ecialized機器、培養細胞亜集団におけるコミットメントの分析および細胞分化にin situで遺伝子操作の効果を可能にする、原腸陥入し、可能な初期の器官形成期のマウス胚の実験操作をレンダリングします。
概要
細胞培養物は広く発生生物学の研究において使用されてきました。マウス胚性幹細胞(ESC)の胚盤葉上層幹細胞(EpiSCs)は、インビトロで 、3つすべての胚葉に分化し、初期の哺乳類の胚発生における細胞分化のために有用なモデルであることができます。これらの細胞株の誘導は 、インビトロ操作及び初期胚パターン形成中に動作してローカライズされたシグナル伝達事象および転写ネットワークの詳細な調査のための機会を開きました。しかし、培養物中で実行される任意の操作のインビボでの関連性を決定する重要なままです。着床前胚由来のマウスESCのin vivoでの電位は、着床前胚(桑実胚または胚盤胞)1に戻って、それらを導入することによって評価されています。しかし、着床後胚における胚盤葉上層の細胞を表すEpiSCsは、着床前胚2,3で効率的に統合することはできません。私たちのprevio着床後の胚4に移植されたときに、私たちの調査結果は、EpiSCsが効率的にキメラを生成し、すべての胚葉に貢献できることを示しました。このように、in vitroで培養した細胞を評価する最良の方法は、 生体内でそれらに対応する環境にそれらを導入することです。
エレクトロポレーションは 、in vivo 実験とin vitro実験の両方で標的細胞に外因性分子を送達するために広く用いられている方法です。電気エネルギーは、外因性のデオキシリボ核酸(DNA)またはリボ核酸(RNA)が細胞に侵入することを可能にする細胞膜の孔を多数生成することができます。この手法の最大の課題の一つは、高electrotransfection効率5,6に最適な細胞生存率を組み合わせることです。胚組織中の核酸の電気穿孔のため、金電極は、最も一般的に広い空間範囲7-9の細胞の標的化を可能にする、使 用されているメッキ。もっとロカを達成するためにlized遺伝子移入、針状電極は、焦点電界10,11を達成するために利用されています。この方法を使用して、著者らは、エレクトロポレーション後、細胞は30〜60の周囲の11の DNA構築物を取り上げたことを示しました。それにもかかわらず、正確にエレクトロポレーションした細胞の数を調整することは、固定幅の電極との困難なままと思われます。キャピラリーエレクトロポレーション技術は、単一細胞12-14にプラスミドを送達するために使用されてきました。しかし、この手法は、ex vivoでの胚するプラスミドをエレクトロポレーションするために適用されていません。最近では、マイクロデバイスは、早期の着床後のマウス胚15で局所的エレクトロポレーション数遠位臓側内胚葉細胞(未満4細胞)に報告されています。しかし、このデバイスは効率的にex vivoで外胚葉および中胚葉をターゲットにできるかどうかまだ不明です。
本研究では、早期の投稿で、細胞および遺伝子機能を評価するために2つの新規な方法を記載しています-implantation胚。まず、 それらのインビボの可能性を評価するために、マウス初期胚において定義された部位へのin vitroで培養した細胞を移植する方法を示します。移植された細胞とその子孫の統合は、すべての遺伝子のタグ(例えば、緑色蛍光タンパク質(GFP)によって標識、さらに組織特異的タンパク質の免疫染色によって調べることができる4。第二 に、我々は正確にDNAを送達するための改良された方法を説明しますエレクトロポレーションを介した胚におけるローカライズサイトへ。そうではなく、針状の電極を使用するよりも、我々は先の細いガラスキャピラリー内部の細いワイヤを挿入し、この変更は、高効率と限られて少数の細胞にDNAを送達することができることを実証細胞死はまた、我々は、異なる開口サイズのガラスキャピラリーを用いて、我々は、エレクトロポレーションした細胞の数を制御することができることを示す。したがって、我々は、この方法は、少数Oを含む初期胚のパターニングを研究するために非常に有用であることができると信じF細胞。
プロトコル
動物(科学的手順)に指定されている全ての動物実験は、プロジェクトのライセンス番号4435分の60の下、英国内務省規則に基づき(1986)法を実施しました。特定の発達段階で胚を収集するには、時限交配は、O / Nを設定しました。膣プラグを見つける日の正午は、胚日(E)0.5と命名しました。
1. ex vivoでの文化のためのE7.5またはE8.5着床後胚を解剖
- 頚椎脱臼により妊娠中の雌マウスを生け贄に捧げます。
- ピンセットでそれを保持し、はさみを使用して子宮を分離し、M2培地で満たされた30ミリメートル皿に置きます。
- 慎重に微細な鉗子の二組を使用して、子宮筋層を引き裂きます。
- 胚体外空洞を穿刺しないように注意して、脱落膜をはがし。
- ピンセットでそれをつまん、ゆっくりと胚からそれを分離することによってライヘルト膜を除去します。
- 解剖の下で胚をチェック実体顕微鏡は、卵黄嚢、羊膜および栄養膜円錐が完全であることを確認します。
- 氷上で30ミリメートルのプラスチックシャーレの蓋の上に( '氷のプラットフォーム')に部分的に寒胚をピペットと場所を使用して、M2のきれいな皿に胚を転送します。
- 必要に応じて、最大1.5時間氷プラットフォーム、 例えば 、メディアの準備や小ロットの操作中に、M2に格納胚〜RTで3-4の胚。
注意:回復とげっ歯類の胚の解剖は、以前7,8,16詳細に記載されています。
2.胚培養培地を準備
- たて市販のラット血清のいずれかを解凍(仕様についてはグランビル・ジョーンズら 13を参照)、またはラット血清は、熱が56℃で30分間不活性化し、1ml中で凍結させたコップやコックロフト16に応じて、社内で準備-80℃でアリコート
注意:市販のラット血清が許容されます血清は、社内で作成しているが24-36時間の培養期間、48時間までの培養期間に優れた、私たちの経験では、あります。 - 新たに準備過剰( 例えば 、10 ml)をグラスゴー最小必須培地(GMEM)、1%非必須アミノ酸(NEAA)、2mMのL-グルタミンおよび1mMピルビン酸ナトリウムからなる定義サプリメント。
- 培養されようとしている各段階の胚の数に応じて必要とされる培地の体積を計算する(第3章を参照)。 50%の培地を構成するために定義されたサプリメントとラット血清を混合(1:1(v / v)のラット血清:定義されたサプリメント)および/または75%培地(3:1(v / v)のラット血清:定義サプリメント)。
- 0.45μmのフィルターを通して胚培養培地をパスし、万IU / mlのペニシリンおよび10μg/ mlストレプトマイシンを追加します。
注意:解凍したてのL-グルタミンおよびピルビン酸ナトリウム溶液が、ex vivoでの培養中の胚の開発のために重要です。
- 静置培養を24時間37℃で、空気中5%CO 2を供給インキュベーターで4ウェルプレートを使用して:E7.5胚のため。 50%の培地1ml中のウェルあたり2胚までの文化。
- E8.5胚の場合:24時間(胚あたり50%の培地の1ミリリットル)37℃で空気中5%CO 2を連続ガス発生を組み込んだ35回転/分で回転するローラー培養装置8を使用しています。
- E9.5胚の場合:あたり75%の培地の24時間(1 mlの37℃で5%CO 2、40%のO 2、55%のN 2が供給ガスを組み込む35回転/分で回転するローラー培養装置を使用胚)。
注:3時間内の培養胚M2における長期間に悪影響発達に影響を与えるとして、マウスを安楽死させた後。 ex vivoでの胚培養の方法のためにコップやコックロフト16を参照してください。
4. GraftinE7.5またはE8.5マウス胚にグラム培養細胞
- 物理的に遍在20-200μlのピペットチップを使用して、6ウェル培養プレートから、GFPを発現し、胚を含む30 mmディッシュに配置しEpiSCsをこすり
注:胚における細胞塊を挿入するために、細胞を物理的に廃棄ではなく、トリプシン処理する必要があります。 - 口のピペットを作るために吸引チューブに手で引っ張っグラフトキャピラリーを取り付けます。
- 静かにグラフトキャピラリーにサイズ> 20細胞の1つ以上の細胞塊を描画するために、口のピペットを吸います。
- 静かに部分的に大きな塊を分散させるために、細胞を吹き消します。
- 〜10〜20細胞を含有する一つの細胞塊を選択し、再移植のキャピラリーに吸う、キャピラリの開口部に近いそれを維持。小さな部分にブレークアップを回避するために、繰り返し毛細血管の内と外の細胞塊を移動しないように注意してください。
- ピンセットで所定の位置に緩く胚を持ち、移植を挿入開口部を作成するために、関心のある領域へのキャピラリー。
- 静かに胚に提出10-20細胞の短い文字列を残して、グラフト毛細血管から塊を追放。
- 胚の所望の数のために移植する手順を繰り返します。利便性のために3-4胚のバッチサイズを使用してください。
- カメラで解剖顕微鏡蛍光化合物を用いてグラフト化された胚は、過剰な光と熱への胚の暴露を避けるために最小限に撮影時間を保ち、画像、M2培地の同じ皿に胚を残します。
注:これらは、カメラや顕微鏡と同様に、蛍光体の性質と蛍光強度の仕様に依存しますように経験的にイメージング時間を決定します。 - すぐにイメージング後(セクション3を参照)培地平衡化を事前にM2培地の最小量とpastetteに胚を転送します。
注:慎重に、移植後の胚の形態を検討します。唯一の文化intacトンの胚。
5.手作りエレクトロ材料および装置のセットアップ(エレクトロポレーション実験の前に次のことを準備します):
- DNA注入ピペットの場合:水平マイクロピペットプラーを使用して、DNA注入ピペットを引き出します。インジェクションピペットは、胚の空洞にDNAを注入する際、組織の損傷を避けるために、開口部10μm未満で細かいチップを持っている必要があります。
- ガラスキャピラリーエレクトロポレーションの場合:20または30ミクロンのいずれかの内径にDNA注入ピペットの開口部をカットするマイクロフォージを使用しています。キャピラリーエレクトロポレーションのための胚に接触しているときに細胞の損傷を避けるために、ガラスキャピラリーの先端をきれいにカットし、シャープ、壊れたエッジを含まないようにする必要があります。
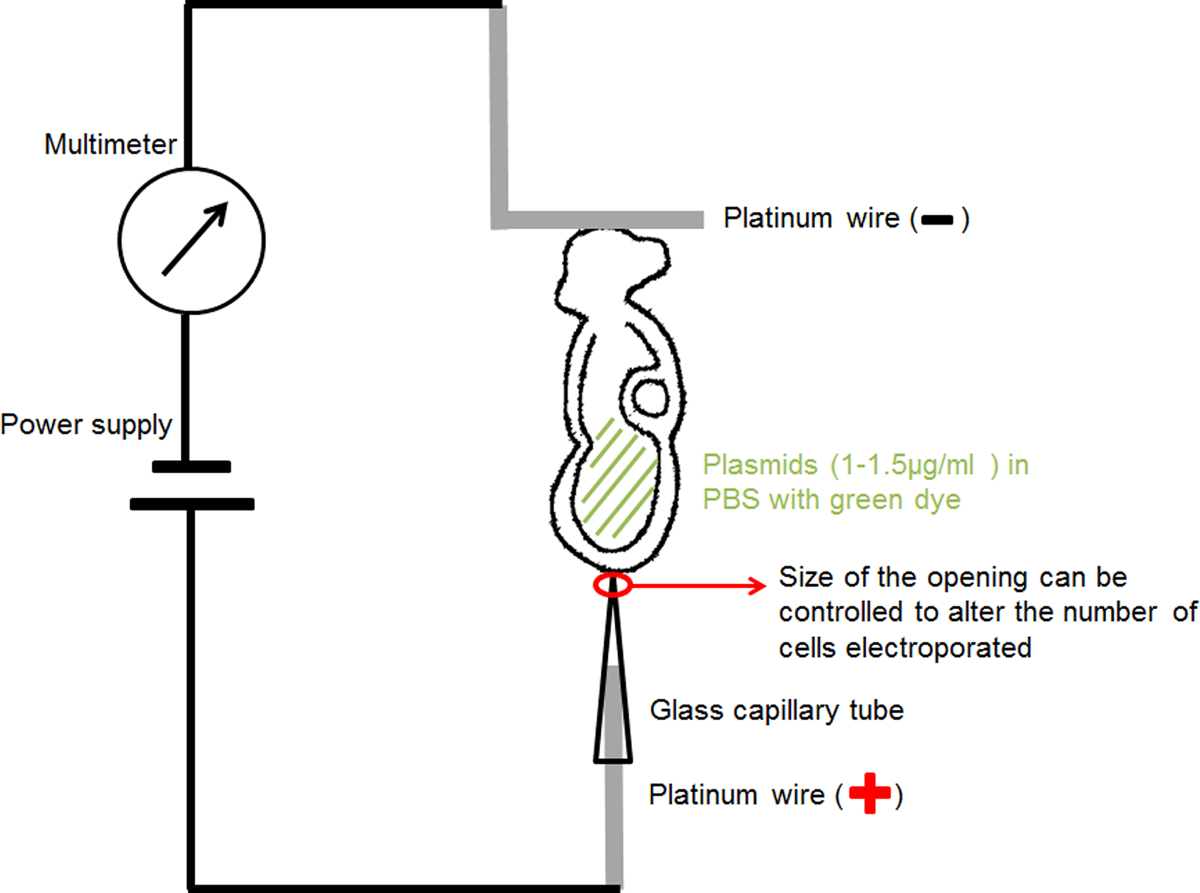
- 手作りキャピラリ電極(陽極)( 図1)のための:選出を集中する20または30ミクロンの直径の一定の開口部を有するエレクトロポレーション、ガラスキャピラリーに直径0.2mmの白金線を挿入します現在RICと胚の関心の小さな領域にプラスミドDNAを提供します。
- 手作りのL字型の電極(陰極)( 図1)のための「L」の長さは1mm程度の水平部と、「L」形状の作成0.2 mmの直径の白金線を曲げます。
- 薄い絶縁電線にそれぞれ白金電極を取り付け、絶縁テープで覆われたマイクロインジェクション針ホルダに挿入します。
- 標準のマイクロマニピュレーション機器ホルダーの針ホルダーをマウントします。
- 図1に示すような回路を接続し、電源の陽極にキャピラリー電極を接続します。マルチメータのアノードにL字型の電極を接続します。電源の陰極にmulimeterのカソードを接続してください。
6.エレクトロE7.5またはE8.5マウス胚
- トップ1-2 mm以内にPBSでエレクトロポレーションのガラスキャピラリーをする記入し、straiを挿入それは毛細管の底に達するまで、ガラスキャピラリーにGHT白金電極(陽極)。
- PBSを充填した30 mmのペトリ皿の表面上にL字型の電極(陰極)を固定します。
- PBSで満たされたエレクトロポレーション皿にM2培地から胚を転送します。
- 胚の羊膜腔に横胚盤葉上層から注射針を挿入します。それが完全にいっぱいになるまで空洞に:空気圧ピコポンプを使用して、DNA溶液(GFPまたはpCAG-GFP 0.01%緑の食品着色染料と1〜1.5μgの/ mlのpCAG-Creを)を注入します。 E7.5、E8.5胚は、5未満μlのDNA溶液は、1つの胚のために必要とされます。指標として緑色の色素を使用して、胚を破裂しないように注意してください。
- 慎重に電極間の胚を配置し、DNAが送達される正確な位置にキャピラリ電極を移動します。
注:胚の配向は、DNAをエレクトロポレーションされる領域に依存します。 - Electropo6パルス、各パルスの間に1秒間隔で50ミリ秒の持続時間のそれぞれで200ボルト(V)を使用して胚を評価します。
- 胚を転送するエレクトロポレーションの直後に、培養培地を事前に平衡化しました。必要に応じて、次の胚のためのプロセスを繰り返します。
注:滅菌容器に培地を追加し、事前に平衡化培地に培養インキュベーターに入れて。 - エレクトロポレーションした細胞を培養後2時間を検出するために、pastetteを使用して、M2培地のきれいな30ミリメートルペトリ皿に胚を移します。画像を解剖顕微鏡、蛍光化合物を用いて、ステップ5.9のように胚。
- イメージング直後pastetteを用いて培養に戻す胚を転送します。
- エレクトロポレーション(オプション)10分後に2時間37℃で:(胚培養培地中200 1)エレクトロポレーションによって引き起こされる死細胞を検出するために、遠赤細胞膜不透過性の核色素で胚を染色
- エレクトロポレーションした細胞をカウントするには、エンブリーを修正4℃で2〜4時間、4%パラホルムアルデヒド(PFA)で、OSは、紫外線または遠赤色蛍光核対比染色し、共焦点顕微鏡画像(オプション)を使用して胚で核を染色します。
注意:PBS中に長すぎる放置しておくと胚成長が悪影響を受けます。したがって、各胚のエレクトロポレーションに要する時間が最小化される(胚あたり<5分)を確保します。
結果
グラフト
普遍的にEGFP(たmESC からインビトロで誘導されたE6.5の胚盤葉上層由来R04-GFP、およびC2、)4を表現 EpiSCsは手動で培養皿から掻き取り、E7.5胚の異なるサイト( 図2A)に移植しました。胚は、ex vivoで培養し、24時間後に分析しました。ドナー細胞の分布を蛍光顕微鏡により評価しました。ドナー細胞が組み込まれている場合、それらは増殖し、その誘導体は、宿主胚( 図2B)内に分散されます。これは、ホスト胚( 図2Aおよび2B)に効率的に組み込まれた10-16細胞を含有する移植片は、しかしながら、より多くの細胞を移植することは、より良いキメリズムを生じないことが観察されました。その代わりに、移植された細胞は、組み込まれていない塊( 図2Cおよび2D)を生産しました。
エレクトロポレーション
まで胚の特定部位へ:私たちのエレクトロポレーションシステムの効率ssess、我々はGFP発現プラスミド(GFP pCAG-GFPおよびpCAG-Creを)を配信しました。以前の研究11と一致して、GFP +細胞を、エレクトロポレーション(図2Eおよび2G)後の胚1〜2時間で検出されました。後半原始線条期胚に遠位胚盤葉上層の細胞をエレクトロポレーションした場合には、標識された細胞は、培養中の24時間(図2Eおよび2F)後の神経外胚葉に貢献しました。この結果は、原腸形成期の胚17における胚盤葉上層細胞の既知の運命マップによく対応しています。 GFP発現プラスミドをE8.5(2-5体節)で原始線条に電気穿孔した場合も同様に、GFP +細胞は、近軸中胚葉(図2Gおよび2H)後半原始線条18の既知の運命マップと一致貢献しました。また、我々は、エレクトロポレーションした細胞(図2I-K)から3つのすべての胚葉への貢献を認められ、エレクトロポレーションの手順は 、インビボでの細胞の挙動を損なわないことを示唆しています。しかし、我々はまた、胚盤葉上層(E7.5)または原始線条細胞(E8.5)を標的とした一方で、いくつかの内胚葉細胞は、(図3C及び表1)を電気穿孔したことに気づきました。
キャピラリ電極を使用することの主要な利点の一つはエレクトロポレーションした細胞の数は、単に、その開口径を変化させることにより、制御できることです。エレクトロポレーションした細胞の数を決定するために、胚は、エレクトロポレーション後2時間を固定し、共焦点顕微鏡でホールマウントで画像化しました。 GFP +細胞の数は、手動で焦点のzスタックでカウントしました。表1は、所定の段階のために、多くの細胞によるDNA取り込みの20〜30μmの結果から、ガラスキャピラリーの開口サイズが増加することを示します。単一の開口サイズは(E8.5対E7.5)段階との間で比較したところ、多くの細胞は、後段に電気穿孔されることが見出されました。この効果は、E8.5での羊膜の空洞中に存在するDNAのより高い濃度に起因し得ます。 DNA溶液が緑色食品色素と混合しているので、我々は羊膜腔でDNA濃度を評価するために緑色を使用することができます。顕微鏡では、E8.5胚と比較した場合、DNA注入後の緑色のE7.5胚の空洞内の非常に軽量であることは明らかです。 DNA溶液の濃度は、同じE7.5及びE8.5の胚に注入したが、それらはサイズが大きいので、より多くのDNA溶液を完全に充填するために、E8.5胚の羊膜空洞でした。注射針を引き抜く後、羊膜腔からDNA溶液の漏れをある程度は常に存在し、穿刺孔以前胚における羊膜腔の大きさに比べて大きいので、比例より漏れがあった可能性がありますE8.5胚よりE7.5から、低いDNA濃度につながります。トランスフェクトされた異なる数の細胞はまた、異なる段階での細胞の異なる直径又は誘導貫通電圧(ITV)のしきい値によるものである可能性があります。
エレクトロポレーションの欠点は、関連した細胞死です。従来の金と同様にメッキまたは針状電極は、キャピラリ電極を用いたエレクトロポレーションは、細胞死を引き起こします。エレクトロポレーション後の標的領域は、細胞死のある程度は、この領域で発生している必要があることを示し、隣接する領域 ( 図3Aおよび3B)と比較して色が濃く現れました。さらに、エレクトロポレーション手順によって引き起こされる死細胞の数を決定するために、胚は蛍光細胞膜不透過性核染料で染色しました。死細胞の核は膜不透過性遠赤色蛍光色素で標識しました。染色は、このキャピラリー電気穿孔技術はエレクトロポレーションのみ部位 ( 図3D及びTa近くの死細胞の数が少ないことになることが確認されBLE 1)。
私たちは、死んだ細胞はエレクトロポレーションサイトに表示されているが、GFP +細胞と死細胞は、互いに(図3Eおよび3F)から最も排他的であることに気づきました。 E8.5で尾横胚盤葉上層はpCAG-GFPおよび20μmでのガラスキャピラリー開口部、GFP +細胞の数が多いエレクトロポレーションしたときまた、培養48時間( 図3Gおよび3H)後に検出されました。まとめると、これらの結果は、大部分のGFP +細胞をさらに培養中にまだ生存しているエレクトロポレーション後2時間を検出示唆しています。
私たちは24時間ex vivoでの培養後に、GFP +細胞の数を獲得しました。シックス胚を20μmの直径の毛管開口部を使用して、E7.5でpCAG-GFPでエレクトロポレーションしました。 107±31 GFP +細胞/胚が検出された(平均±SD)。培養開始(2時間)であるので、9細胞(平均あたりの胚にエレクトロポレーションしました trong>表1)、これはエレクトロポレーションした細胞は、2時間以内に3~4分裂を受けたことを示唆しています。 E8.5胚へE7.5から倍加時間平均セルは離れて腹側ノード19,20のものとすべてのセルで約6-7時間です。これは、エレクトロポレーション手順は、正常な細胞増殖を阻害しないことを示唆しています。

エレクトロポレーションの設定を示す図1の回路図である。その羊キャビティ内の胚を含むDNA溶液を、二つの電極の間に配置しました。選択されたパラメータの入力電流を矩形波パルス発生器(電源)によって提供されました。マルチメータは、胚を流れる電流を検出するために、直列に接続した。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}

図2.ホスト胚におけるグラフトまたはエレクトロポレーションした細胞の分布。ホールマウント胚(グレースケール)(A)の明視野画像に(AH)GFP蛍光のオーバーレイ(緑)が10-16 GFP + EpiSCsは後半の先端領域に移植しました-streak期胚(C)GFP + EpiSCsの大きな塊は、中期ストリーク段階の胚の遠位領域に移植された。(BとD)に示すホスト胚におけるEpiSCs由来細胞(緑)の分布を(培養における24時間後のAおよびC)。宿主胚中に分散(B)GFP +細胞を、ドナー細胞の正しい組込みを示唆する。(D)より大きな細胞塊を移植は、宿主胚における取り込まれていない塊の形成をもたらしました。 (EK)の pCAG-Creを:GFPプラスミドELE野生型胚の特定の領域にctroporated。初期の芽期胚(E)または 2-5体節期胚(G)の原始線条の先端領域をエレクトロポレーションは、2時間の手順の後、これらの地域におけるGFP +細胞が得られました。 (FおよびH)文化の中で24時間後、ホスト胚におけるGFP +細胞の分布、エレクトロポレーションした細胞は、神経外胚葉(黒矢印)に寄与することを示す(F)と 、近軸中胚葉(白矢印)(H)。(IK)エレクトロポレーションした細胞は、神経外胚葉(I)を生じることができることを示すGFP +細胞のためのDAB免疫染色、中胚葉(J)と文化の中で24時間後に内胚葉(JおよびK)。スケールバー(AH)= 250ミクロン;スケールバー(IK)が100ミクロンを=。注: 図1Aおよび1Bは、私たちの以前の出版物4から転載されています。HREF = "https://www.jove.com/files/ftp_upload/53295/53295fig2large.jpg"ターゲット= "_空白">この図の拡大版をご覧になるにはこちらをクリックしてください。

エレクトロポレーション後の胚における GFP +細胞 と死細胞 の図3の分布 (AC)pCAG-Creを:GFPプラスミドは、E8.5(2-5体節期)の胚(キャピラリー開口サイズの尾横胚盤葉上層細胞にエレクトロ:20μm)を、(A)2時間処置後、標的領域は、胚の他の部分に比べて暗い色(白矢印)を示しました。ことを示す挿入図は、エレクトロポ地域の拡大。(B)明視野画像(グレースケール)は、エレクトロポレーションした細胞(緑)を示す緑色蛍光チャネルを重ね示しています。(C)共焦点のzスライス尾横胚盤葉上層の細胞を標的としたときに、2つの内胚葉細胞(緑)も、プラスミドを取り上げました。 。細胞核は(DF)pCAG-Creを赤で表示されます:GFPプラスミドは、E8.5胚のノードの尾側の側面に電気穿孔した(毛細管開口サイズ:30ミクロン)。胚を2時間培養しました。エレクトロポレーションした細胞は赤で緑と死んだ細胞に示されている。(D)は、エレクトロポレーション領域は、GFP +細胞ならびに死細胞の両方が含まれています。白いボックス内の領域は、さらに、共焦点顕微鏡で分析しました。 zスタックの手動カウントが33 GFP +細胞およびこの分野で23死んだ細胞が存在することを示しました。 2つだけの細胞の共焦点のzスライスの両方のフルオロフォアのための正の両方であった。(EおよびF)のXYZビューD示すGFP +細胞における白箱入り地域からは、死細胞から分離されています。核は青色で表示されている(GおよびH)pCAG-Creを:GFPプラスミドは、いくつかのセルにエレクトロポレーションしましたE8.5胚の尾側方外胚葉におけるLS(キャピラリー開口サイズ:20ミクロン)と2(G)、48(H)時間のex vivo培養注後に撮像された:(H)胚を培養した後、二つに切断されました。頭と心の領域が削除されました。スケールバー(A、B、D、GおよびH)= 250ミクロン。スケールバー(C、EおよびF)=100μmである。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}
| キャピラリーチューブの開口部の直径 | 胚段階 | エレクトロポレーション効率:なし。 2時間/総なした後、GFP +細胞を含有する胚。エレクトロポレーション胚(24または48時間の培養後に正常に開発していない。GFP +胚)の | SD±胚あたりのGFP +細胞の平均数(N =なし。検討した胚の) | の号SD±各胚あたりのGFP +内胚葉細胞(N =なし。検討した胚の) |
| 20μmの | E7.5(LS-LB) | 7/9(7) | 9±3(n = 4)は | 4±2(N = 4) |
| 30μM | E7.5(LS-LB) | / 15(12)13 | 17±2(N = 4) | 6±1(N = 4) |
| 20μmの | E8.5(2-5体節) | 12/13(10) | 21±4(N = 4) | 11±4(N = 4) |
| 30μM | E8.5(2-5体節) | 2/2(2) | 33及び26(N = 2) | 14及び16(N = 2) |
pCAG-Creを表1エレクトロ効率:マウス胚におけるGFPプラスミド。
略称:LS、後半原始線条段階; LB:後期芽段階。胚に応じてステージングされダウンズとデイヴィス12
ディスカッション
グラフト
細胞移植実験のための重要なステップは、塊の崩壊を回避するために、理想的には、単一の操作でセルのコヒーレント文字列の挿入です。この手法は、口のピペットを制御する際にいくつかの練習が必要です。ドナー細胞は、宿主でよく組み込む場合、その誘導体は、胚中に分散されます。さらに分散し、ドナー由来の細胞を宿主に適切に区別するかどうかを判断するには、免疫染色は、胚切片上で行うことができます。ドナー細胞は、ホスト環境との互換性がない場合、それらのいずれかが検出された(これらは胚から排出されるように)、または培養後の胚に組み込まれていない凝集塊を形成することはできません。分散した細胞及び細胞塊の両方が観察された場合、これは、あまりにも多くの細胞を移植したと周囲の宿主細胞と相互作用することができない過度のドナー細胞は塊形成をもたらしたことを示してもよいです。この場合、付加的なグラフトは含みます細胞の少数を行うことができます。
技術グラフト細胞の主要な制限は、48時間より長い期間にわたってマウスのex vivo培養が達成されていないので、それは細胞の完全なin vivoでの可能性を決定することができないということです。しかし、超音波誘導細胞注射と組み合わせた場合、それは子宮内の胚に培養細胞を転送することも可能です。要約すると、細胞移植する実験は、広く我々のグループで使用されているし、私達に様々な細胞型4,21,22 の in vivoでの可能性についての貴重な手がかりを与えています。これは、初期の着床後の胚におけるin vitroで培養された細胞のインビボでの可能性を評価するために一般的な有用性の技術です。
エレクトロポレーション
この研究において、我々は、私は、それが胚盤葉上層を標的とするためにキャピラリー電気穿孔技術を使用することが効率的であることが示されているがtは、意図的に、このような内胚葉細胞などの他の胚葉を標的とすることも可能です。キャピラリー電気穿孔法のための重要なステップは、PBSをマウス初期胚のために非常に次善のであるため、各胚(胚あたり<5分)をエレクトロポレーションするのにかかる時間を最小限に抑えることです。我々のデータは、上記の胚におけるほとんどの地域では、エレクトロポレーションは、胚の成長に影響を与えない、ことを示しています。しかし、ノード内のエレクトロポレーションは、発達異常を引き起こし、胚の早期死亡につながりました。これは、重要なシグナルセンター23を形成する細胞の損傷または死にそうです。従って、この領域は、この技術で回避しなければなりません。さらに注意すべき点は、胚盤葉上層または原始線条細胞を標的とした一方で、いくつかの内胚葉細胞はまた、エレクトロポレーションし、結果の項で述べたように、ということです。 DNAは、胚盤葉上層の上皮下の隙間から内胚葉に達したためと考えられます。内胚葉は、上皮細胞で構成され、目の経験であり、ESE細胞はDNAを取るために高い傾向があります。運命のマッピングのためにこの技術を適用する場合したがって、それは最初にDNAを取り込みどのセル評価することが重要です。
GFPプラスミドは、効率的に本研究で示されたエレクトロポレーションパラメータを使用して配信することができ、他のDNA構築物の効率が変化し、個々の最適化が必要な場合があります。また、pCAG-GFPおよびpCAG-Creをがが、ことに留意すべきです。プラスミドをトランスフェクトすることが困難であることが判明している場合、DNA濃度、エレクトロポレーション電圧またはパルスの数の変化を行うことができます。
限定された細胞死との胚では非常に少数の細胞にGFPプラスミド:要約すると、我々の最適化されたキャピラリー電気穿孔システムを効率的かつ再現GFPまたはCreをを提供することができます。この方法は、高価な、または非常に特殊な装置を必要としないので、それは、細胞追跡研究のためまたは異所性発現または条件削除oの効果を試験において非常に有用であることができます初期胚中のF遺伝子は、エレクトロポレーション場合はフロックス条件変異対立遺伝子を保有する胚で行われます。したがって、このエレクトロポレーション技術は、ローカライズされた野生型胚の環境の文脈でセルごとにセル固有の因子の役割を理解するための有用な機能的なツールを提供します。
開示事項
The authors have no conflicts of financial or other interest to declare.
謝辞
We thank Filip Wymeersch and Anestis Tsakiridis for comments on the manuscript, staff in the SCRM animal unit for help with animal maintenance and Prof. Stuart Forbes for immunohistochemistry reagents. This work was supported by MRC grant Mr/K011200/1 and the China Scholarship Council
資料
| Name | Company | Catalog Number | Comments |
| Forceps | Dumostar | T5390 | |
| Dissecting stereomicroscope | Zeiss | Stemi 2000-C | |
| Stereomicroscope system with fluorescence | Nikon | AZ100 | |
| Inverted microscope with a digital camera | Olympus | Olympus BX61 | |
| Inverted confocal microscope | Leica Microsystems | Leica TCS SP8 | |
| Low melting point agarose | Life Technologies | 16520-050 | |
| Pasteur pipettes | Fisher Scientific | 11397863 | |
| 30mm Petri dishes | Fisher Scientific | 121V | |
| 4-well plates | Thermo scientific | 179820 | |
| M2 medium | Sigma-Aldrich | M7167 | |
| Phosphate Buffered Saline (PBS) | Life Technologies | 10010015 | |
| Paraformaldehyde (PFA) | Sigma-Aldrich | P6148 | |
| Pipettes | NICHIRYO | Nichipet | |
| tips | Greiner Bio One | 685280 | |
| Cell culture incubator | SANYO | MCO-17AIC | |
| Roller culture apparatus | BTC Engineering | ||
| Syringe filters 0.45µm, sterile | Sigma-Aldrich | 10462100 | |
| Glasgow Minimum Essential Medium (GMEM) | Sigma-Aldrich | G5154 | |
| non-essential amino acids (NEAA) | Life Technologies | 11140050 | |
| L-glutamine | Fisher Scientific | SH30549.01 | |
| Sodium pyruvate solution | Fisher Scientific | SH30239.01 | |
| Penicillin and Streptomycin 10.000UI/ml | Lonza | DE17-602E | |
| Gas Cartridge for Portable Meker Burner | COLEMAN | COLEMAN 250 | |
| Thin Wall Borosilicate Capillary Glass with Fillament, OD 1.0 mm, ID 0.78 mm | Harvard Apparatus | 640798 | |
| Aspirator tube assemblies for calibrated microcapillary pipettes | Sigma-Aldrich | A5177-5EA | |
| Flaming/Brown micropipette puller | Sutter Instrument Company | P-97 | |
| Microforge | De Fonbrune | BS030301 | |
| Pneumatic pico pump | World Precision Instruments | PV830 | |
| Microloader tips | Eppendorf | 5242956.003 | |
| ECM 830 square wave pulse generator | BTX | 45-0002 | |
| Green food coloring dye | Sigma-Aldrich | C.I. 42053 | |
| A far-red cell membrane-impermeable nuclear dye | Biotium | 40060-T | |
| pCAG-Cre:GFP | Addgene | #13776 | |
| pCAG-GFP | Addgene | #16664 | |
| Multimeter | Excel | XL830L | |
| Micromanipulators | Leitz | ||
| 0.2mm diameter platinum wire | Agar Scientific | E404-2 | |
| Anti-GFP antibody | Abcam | ab13970 | |
| Goat anti-Chicken IgY, HRP | Santa Cruz | sc-2428 | |
| Liquid DAB+ Substrate Chromogen System | Dako | K3467 | |
| 4',6-diamidino-2-phenylindole (DAPI) | Life Technologies | D21490 | |
| A far-red fluorescence nuclear counterstain | Life Technologies | T3605 |
参考文献
- O'Hagan, A. R., Morton, R., Eid, N. Loss of asthma control in pediatric patients after discontinuation of long-acting Beta-agonists. Pulmonary med. , 894063 (2012).
- Brons, I. G., et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature. 448, 191-195 (2007).
- Tesar, P. J., et al. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature. 448, 196-199 (2007).
- Huang, Y., Osorno, R., Tsakiridis, A., Wilson, V. In Vivo differentiation potential of epiblast stem cells revealed by chimeric embryo formation. Cell rep. 2, 1571-1578 (2012).
- Sadik, M. M., et al. Scaling relationship and optimization of double-pulse electroporation. Biophys. J. 106, 801-812 (2014).
- Kaestner, L., Scholz, A., Lipp, P. Conceptual and technical aspects of transfection and gene delivery. Bioorg. Med. Chem. Lett. , (2015).
- Soares, M. L., Torres-Padilla, M. E., Zernicka-Goetz, M. Bone morphogenetic protein 4 signaling regulates development of the anterior visceral endoderm in the mouse embryo. Dev. Growth Differ. 50, 615-621 (2008).
- Pierreux, C. E., Poll, A. V., Jacquemin, P., Lemaigre, F. P., Rousseau, G. G. Gene transfer into mouse prepancreatic endoderm by whole embryo electroporation. JOP. 6, 128-135 (2005).
- Falk, J., et al. Electroporation of cDNA/Morpholinos to targeted areas of embryonic CNS in Xenopus. BMC Dev. Biol. 7, 107 (2007).
- Davidson, B. P., Tsang, T. E., Khoo, P. L., Gad, J. M., Tam, P. P. Introduction of cell markers into germ layer tissues of the mouse gastrula by whole embryo electroporation. Genesis. 35, 57-62 (2003).
- Khoo, P. L., Franklin, V. J., Tam, P. P. Fate-Mapping Technique: Targeted Whole-Embryo Electroporation of DNA Constructs into the Germ Layers of Mouse Embryos 7-7.5 Days Post-coitum. CSH protocols. 2007. , pdb.prot4893 (2007).
- Tawk, M., Bianco, I. H., Clarke, J. D. Focal electroporation in zebrafish embryos and larvae. Methods Mol Biol. 546, 145-151 (2009).
- Haas, K., Jensen, K., Sin, W. C., Foa, L., Cline, H. T. Targeted electroporation in Xenopus tadpoles in vivo--from single cells to the entire brain. Differentiation. 70, 148-154 (2002).
- Nolkrantz, K., et al. Electroporation of single cells and tissues with an electrolyte-filled capillary. Anal. Chem. 73, 4469-4477 (2001).
- Mazari, E., et al. A microdevice to locally electroporate embryos with high efficiency and reduced cell damage. Development. 141, 2349-2359 (2014).
- Copp, A. J., Cockroft, D. L. . Postimplantation mammalian embryos : a practical approach. , (1990).
- Tam, P. P., Behringer, R. R. Mouse gastrulation: the formation of a mammalian body plan. Mech. Dev. 68, 3-25 (1997).
- Wilson, V., Beddington, R. S. Cell fate and morphogenetic movement in the late mouse primitive streak. Mech. Dev. 55, 79-89 (1996).
- Tzouanacou, E., Wegener, A., Wymeersch, F. J., Wilson, V., Nicolas, J. F. Redefining the progression of lineage segregations during mammalian embryogenesis by clonal analysis. Dev Cell. 17, 365-376 (2009).
- Bellomo, D., Lander, A., Harragan, I., Brown, N. A. Cell proliferation in mammalian gastrulation: the ventral node and notochord are relatively quiescent. Dev. Dynam. 205, 471-485 (1996).
- Tsakiridis, A., et al. Distinct Wnt-driven primitive streak-like populations reflect in vivo lineage precursors. Development. 141, 1209-1221 (2014).
- Gouti, M., et al. In vitro generation of neuromesodermal progenitors reveals distinct roles for wnt signalling in the specification of spinal cord and paraxial mesoderm identity. PLoS biology. 12, e1001937 (2014).
- Beddington, R. S. Induction of a second neural axis by the mouse node. Development. 120, 613-620 (1994).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved