
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
音響ナノディスペンス技術を用いたハイスループットDNAプラスミド多重化とトランスフェクション
要約
このプロトコルは、音響液滴排出技術を用いて384ウェルプレートにおける哺乳動物細胞のハイスループットプラスミドトランスフェクションについて説明する。時間がかかり、エラーが起こりやすいDNA分配と多重化だけでなく、トランスフェクション試薬の分配は、ソフトウェア駆動型であり、ナノディスペンサーデバイスによって実行されます。細胞は、次いで、これらの予め充填された井戸に播種されます。
要約
多くの生物学的研究に不可欠な細胞トランスフェクションは、正確で成功した達成のために多くのパラメータを制御する必要があります。ほとんどの場合、低スループットで実行され、さらに時間がかかり、エラーが発生しやすく、複数のプラスミドを多重化する場合はさらにそうです。私たちは、音響液滴放出(ADE)技術を使用して384ウェルプレートレイアウトで細胞トランスフェクションを行う簡単で迅速かつ正確な方法を開発しました。本研究で用いられるナノディスペンサー装置は、この技術に基づいており、ソースウェルプレートから目的地への高速での正確なナノボリューム送達を可能にします。あらかじめ設計されたスプレッドシートに従って、DNAおよびトランスフェクション試薬を分配し、多重化することができる。ここでは、ADEベースのハイスループットプラスミドトランスフェクションを実行するための最適なプロトコルを提示し、コトランスフェクション実験において最大90%の効率とほぼ100%のコトランスフェクションに到達することを可能にします。最大 1,536 個の異なるプラスミドを含むライブラリから最大 4 つのプラスミド/ウェルを管理できるユーザーフレンドリーなスプレッドシート ベースのマクロと、タブレットベースのピペッティング ガイド アプリケーションを提案することで、初期作業を拡張します。マクロは、ソースプレートの必要なテンプレートを設計し、ナノディスペンサーとタブレットベースのアプリケーション用にすぐに使用できるファイルを生成します。4ステップトランスフェクションプロトコルは、i)古典的な液体ハンドラを有する希釈剤、ii)プラスミド分布および多重化、iii)ナノディスペンサーによるトランスフェクション試薬分配、およびiv)予め充填されたウェル上の細胞めっきを含む。ADEプラスミド多重化およびトランスフェクションの記載されたソフトウェアベースの制御は、現場の非専門家でさえ、迅速かつ安全な方法で信頼性の高い細胞トランスフェクションを実行することを可能にします。この方法は、特定のセルタイプに最適な設定を迅速に特定し、より大規模かつ手動のアプローチに転置することができます。このプロトコルは、ヒトORFeomeタンパク質(ゲノムにおけるオープンリーディングフレーム[ORFs]のセット)発現やCRISPR-Cas9ベースの遺伝子機能検証などのアプリケーションを非プールスクリーニング戦略で容易にします。
概要
ここで提示される方法は、384ウェルプレート内の音響ベースの液体ナノディスペンサーを用いて、哺乳類細胞におけるDNAプラスミド多重化およびトランスフェクションを高スループットで行う方法を詳細に説明する。この最近公開された方法1は、1回の実験で384個もの独立プラスミドDNA多重化およびトランスフェクション条件を行うことを可能にし、1時間未満で単一または共トランスフェクション実験に成功し、ほぼ100%に達するトランスフェクト細胞集団内のコトランスフェクション。このプロトコルは、退屈で時間がかかり、エラーが発生しやすい手順のほとんどがソフトウェア駆動型であるため、トランスフェクションを容易にします (概要については、図 1を参照)。さらに、全体的なプロセス中の人為的ミスを回避しつつ、使いやすさを高め、現場の非専門家にとってもトランスフェクションの成功を促進するための専用ツールの開発に取り組んでいます。記述されたプロトコルには、各ウェルに最大 4 つのプラスミドの可能性を多重化する可能性を持つ 384 の独立したトランスフェクション条件を管理するために開発した「ユーザーフレンドリーな」マクロ スプレッドシートが含まれています。マクロは、入力された実験設計時にナノディスペンサーソフトウェアを駆動するために必要なファイルと、開始ストックソリューションから予想されるDNAプラスミドボリュームをロードするために、ソースプレートのテンプレートを自動的に生成します。384ウェルのソースプレートにDNAを手動で塗布するのは退屈でエラーが起こりやすいため、テンプレートに従ってDNA溶液を分配しながらユーザーを導く専用のタブレットベースのアプリケーションも開発しました。

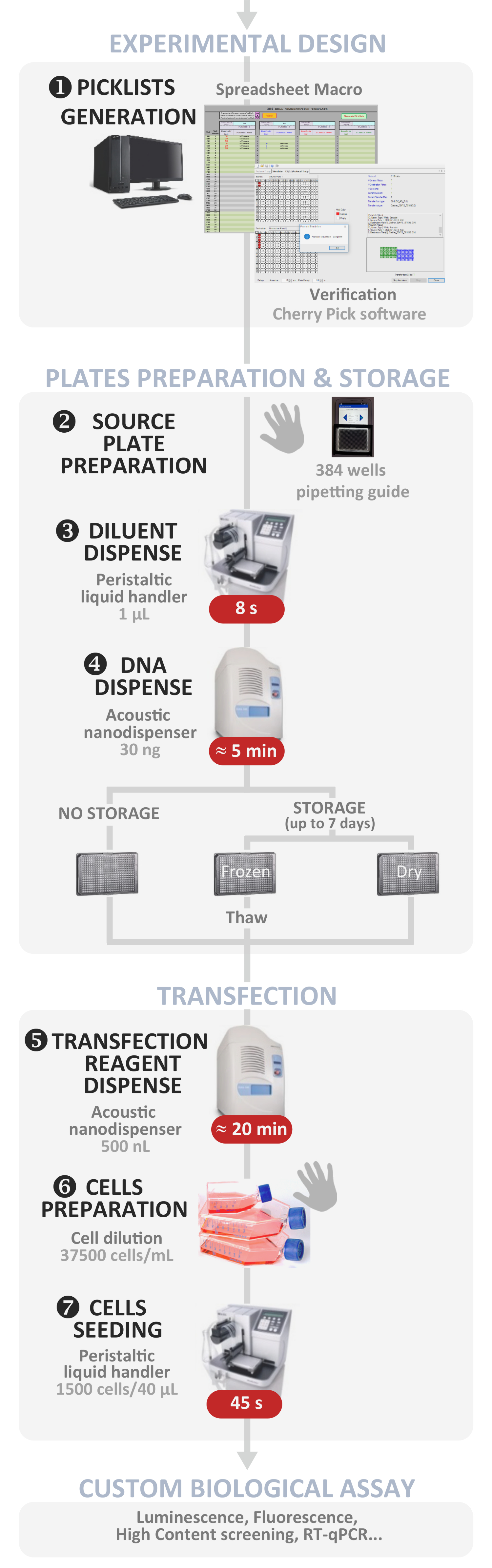
図 1: 実験的なワークフロー。最適な自動ハイスループット逆トランスフェクションプロトコルの概略表現(実験設計からカスタム生物学的アッセイまで)。手動ステップは手の記号で示され、各ステップのおおよその時間は赤いボックスに書き込まれます。この図のより大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
多くの細胞ベースの実験はプラスミドDNAトランスフェクションから始まり、トランスフェクション効率を高め、手順を容易にするために多くの専用試薬が開発されている場合でも、多くのことが行われるべきである2、3,4.DNAプラスミド細胞トランスフェクションは、初期複合体取り込み、エンドソームエスケープ、および核への細胞質輸送などの高効率に到達するためのいくつかのステップを伴う5、6。カルシウム沈殿や専用デバイス7を用いてのエレクトロポレーションやマイクロインジェクションなどの物理的な技術に加えて、現代の化学的方法は、細胞の毒性を低下させながらDNA細胞送達を高めることに焦点を当ててきた8、 9.リポソーム様複合体を形成する脂質またはカチオン性ポリマーの使用により、より最近では、非リポソーム高分子化学系はトランスフェクションをより容易かつ効率的に10にした。これらの開発にもかかわらず、細胞トランスフェクションは、これらの物理的または化学的トランスフェクションプロトコルのほとんどが、科学者が手動で各DNAトランスフェクション反応状態を準備する必要があるため、正確に実行する特定のスキルを必要とします。スループットを損なう。この問題を回避するために、逆トランスフェクションプロトコルは、化学トランスフェクション試薬11、12、13を使用して開発され、ユーザがより速い方法で複数のプラスミドをテストまたは組み合わせることを可能にする。これらのプロトコルでは、トランスフェクション試薬を用いた核酸複合体が、複合体上の細胞を播種する前に形成される。しかし、これらの逆プロトコルは、DNA溶液の手動処理と、独立した条件のそれぞれの組み合わせによって依然として制限されています。96ウェルプレート形式で行うことが可能ですが、DNAの調製と分配は退屈であり、間違いが起こる可能性があります。複数のDNAプラスミドの異なる量が必要とされ、互いに多重化されると、細胞トランスフェクションを達成するのがさらに難しくなり、時間がかかり、ヒューマンエラーは非常に避けられません。逆トランスフェクションアプローチで384ウェルプレートフォーマットにスケールアップすることは、多重化されたDNAトランスフェクション条件が少ないにもかかわらず、以下の理由により不可能な課題となります。i) 管理するDNA量、トランスフェクション試薬、または反応混合物量は、各ウェルに対して1μL未満である。ii) 384の独立した条件に対するプラスミドの多重化は極めて複雑になる。384ウェルの各々における送達もiii)非常に時間がかかり、iv)エラーが発生しやすい。確かに、期待される井戸に適切なソリューションを分配することは、既に分配されている少量は空の井戸と既に充填された井戸の間の視覚的な監視を許可しないため、管理が困難です。v)最後に、必要な分配工程を行うために必要な時間のために細胞が添加される前に蒸発によって混合物を乾燥させる危険性が高い。要約すると、ハイスループットのDNAプラスミドトランスフェクションアッセイを設定する制限要因は、アッセイの小型化であり、これは、もはや手動では処理できないが、また、ほとんど達成できないことを意味するアッセイの小型化である。古典的な前立腺液体ハンドラーによって信頼できる方法。
このようなアッセイを自動化し、高スループットを得ることが困難な証拠として、トランスフェクションを自動化する試みは、これまでに発表されているだけです:市販の液体処理装置とリン酸カルシウム沈殿14を使用した96ウェルプレートフォーマットそして、より最近では、リポレックス試薬、および280の独立したトランスフェクション15を可能にするマイクロ流体チップが、この分野で専門的なスキルを必要とします。別の方法は、アコストフォレシス、液体浮上を可能にし、流体操作および混合につながる、24〜96ウェルプレートフォーマット16でDNAトランスフェクションを行うために使用された。実現可能ですが、DNAトランスフェクション混合物と細胞を混合するには、播種前に1点ごとに60sのインキュベーションが必要とされるため、このアプローチは非常に低いスループットに苦しみます。これは、完全な96ウェルプレートの持続時間が少なくとも96分を意味します。さらに、このプロトコルは、現在市場で入手できない社内で設計され、製造されたデバイスで行われたため、生物学者全体の聴衆に対して受け入れられるものからは程遠い。それどころか、ここ数年、ソフトウェア駆動の音響ベースのディスペンス技術がナノボリュームディスペンサーデバイスで登場しました。集中した音響エネルギーを使用して、これらの装置は、ソースプレートから宛先117への2.5 nLから500 nLまでの小さな液体容積の厳密に制御された排出を可能にします。音響液滴の排出(ADE)と呼ばれるこの技術は多数の利点を有する:それは十分に自動化され、非接触、先端無し、正確、精密、および非常に再生可能であり、高いスループット18を有する。最初にジメチルスルホキシド(DMSO)ソリューションの提供に専念し、水性バッファー19を分配するように設定が強化されました。音響ナノディスペンサーは、その後、逆細胞トランスフェクションプロトコルに適しているように見え、上記の手動制限のほとんどを回避することができます。この技術を用いてプラスミドトランスフェクションの試みはこれまで述べられていなかったので、我々は最近、逆細胞トランスフェクションを行う音響ベースの分配システムの適合性を評価した。
ナノディスペンサーのスループットと使いやすさを活かし、384ウェル、シングルプレート、すなわち総DNA量とDNAトランスフェクションに影響を与える可能性のあるいくつかのパラメータをクロステストすることにより、HeLa細胞の逆トランスフェクションプロトコルを最適化しました。ソースDNA開始濃度、希釈体積、トランスフェクション試薬、および広がり細胞数。開発されたプロトコルは、細胞トランスフェクションの上述の手動制限を回避し、他の自動トランスフェクションの試みに対していくつかの利点を提示します。第1に、小型化により、DNAプラスミド製剤およびトランスフェクション試薬を保存することにより、費用対効果の高いトランスフェクション試薬を可能にします。第二に、384ウェルプレート全体のトランスフェクションが1時間未満で達成することができるので、それは手動プロトコル(初心者のためにも)よりもはるかに高スループットと再現性があります。最後に、それはソフトウェア主導であり、分配されたDNA量およびいくつかのプラスミドの多重化の制御を可能にする。確かに、ナノディスペンサーソフトウェア(材料のテーブル)のおかげで、ユーザーは、定義されたソースウェルプレートから宛先の1に分配されるボリュームを制御するための研究計画を精緻化することができます。
ここで提示されるプロトコルは、主にナノディスペンサーにアクセスし、高スループットでトランスフェクション実験を設定したい人を対象としていますが、特定のセルタイプに対するトランスフェクションパラメータを迅速に最適化したい方も対象です。このプロトコルを適用して、高スループットでいくつかのパラメータをクロステストします。実際、このナノスケールプロトコルで同定された最適化されたパラメータは、大規模かつ手動のトランスフェクション実験に移すことができることを示しました。最後に、本プロトコルで使用されるトランスフェクション試薬は、製造元に応じてDNAまたはsiRNAトランスフェクションを可能にするように、プロトコルは、遺伝子過剰発現またはノックダウンのための配列アプローチを行うことを目指す人にも興味がある。DNAで埋め込んだ宛先プレートは、有効性を損なうことなくトランスフェクションアッセイで使用する7日前まで保存することができ、この種のアプリケーションに対する次のプロトコルのもう一つの利点である。
プロトコル
1. 事前準備
- 蠕動液体ハンドラプログラムの準備
注:プロトコルの希釈および細胞分配ステップのために、使用されるプレートへの分配ヘッドの高さとステップ意図を考慮して、専用のプログラムを準備する必要があります。- 1 μL希釈分配工程では、1 μLカセットを取り付け、ステップ1.1.1.1および1.1.1.2に記載されている設定でプログラムを準備します。
- このステップでは生体材料の損傷が予想されないため、流量パラメータを[高]に調整して、最良のスループットを実現します。調剤の高さを9.6mmに調整します(使用する細胞培養プレートに従って、補足図1)、1 μLの滴が分配中に井戸の底に触れるようにします。
注:このステップは、落下するのに十分な体積に達するまで、分配ヘッド上の液滴の保持を避けるために重要です。 - プレートクリア高さを14.4mmに調整し、各行を分配した後にプレート上の分配ヘッドの自由な変位を可能にします。視覚上の蠕動液体ハンドラーの頭の高さの適切な設定を制御する:分配中に分配の先端に滴が保持されていないことを確認し、各行を分配した後に頭部の変位を可能にするのに十分な高さであることを確認してください。
注:落下保持を避けることは、分配のボリュームの精度を損なう重要なパラメータです。
- このステップでは生体材料の損傷が予想されないため、流量パラメータを[高]に調整して、最良のスループットを実現します。調剤の高さを9.6mmに調整します(使用する細胞培養プレートに従って、補足図1)、1 μLの滴が分配中に井戸の底に触れるようにします。
- 40 μLセルサスペンションを分配する場合は、10 μLカセットを取り付け、手順1.1.2.1-1.1.2.2に記載されている設定でプログラムを準備します。
- 流量パラメータを低に調整して、低速で細胞を分配し、せん断ストレスや井戸底部への衝撃の大きさから細胞への潜在的な損傷を促進しないようにします。分配の高さを11.43 mmに調整する(使用される細胞培養プレートによれば、補足図1)、調剤プロセス中に井戸の底部に対する細胞の影響を下げるのに十分な高さであるが、上の液滴の保持を避けるのに十分低い分配ヘッド。各行を分配した後、プレート上の分配ヘッドの自由な変位を可能にするために、プレートのクリアな高さを16 mmに調整します。
- 視覚上の蠕動液体ハンドラーの頭の高さの適切な設定を制御する:分配中に分配の先端に滴が保持されていないことを確認し、各行を分配した後に頭部の変位を可能にするのに十分な高さであることを確認してください。
注: ドロップ保持を回避することは、信頼性の低いセル番号を分配することにつながる重要なパラメータです。
- 1 μL希釈分配工程では、1 μLカセットを取り付け、ステップ1.1.1.1および1.1.1.2に記載されている設定でプログラムを準備します。
- DNAプラスミド調製(古典的なミニプレップ抽出プロトコル)
- LB培地で形質転換DH5α細菌株を125μg/mLアンピシリン選択抗生物質(材料表)で一晩37°Cで、軌道シェーカー上で穏やかな攪拌(200rpm)下で補充した(材料の表)。
- 培養の2mLを収穫し、6,000×gで5分間遠心分離して細胞をペレットし、上清を廃棄する。
- RNase A(材料の表)を含む250 μLの再懸濁バッファーで細胞ペレットを再懸濁する。250 μLのリシスバッファーを追加し、メーカーの指示に従って室温で5分間インキュベートします。
- 300 μL の中和バッファー (材料の表)を追加し、まもなく vortexting を加えて、ライシス反応を停止します。11,000 x gで5分間チューブを遠心分離します。
- 2 mLのコレクションチューブに新しいプラスミドミニカラム(材料のテーブル)を置き、11,000 x gで1分間遠心分離することによってカラムの上清をデカントします。
- フロースルーを破棄し、ミニカラムをコレクションチューブに戻します。
- メーカーの指示に従って、500 μLのオプションの洗浄バッファー(材料の表)と遠心分離機を1分間1分間1分間洗浄します。
- フロースルーを破棄し、プラスミドミニカラムを回収管に戻します。
- 製造業者の指示に従って、11,000 x gで1分間エタノールと遠心分離機を補充した洗浄バッファー(材料表)の700 μLを追加します。
- フロースルーを破棄し、プラスミドミニカラムとその収集チューブを11,000 x gで2分間、シリカ膜を乾燥させます。
- 乾燥プラスミドミニカラムを新しい1.5mLチューブに入れ、60°Cで予め温めた蒸留水の30μLを加え、室温で2分間インキュベートし、11,000 x gで1分間遠心分離します。
- プラスミドミニカラムを廃棄し、精製されたDNAプラスミドを含む溶出物を保持します。
- 微量分光光度計(材料表)を用いて、ELITのDNA濃度を測定する。
- 分光光度計をオンにし、DNA測定設定を選択します。
- 分光光度計のサンプリングアームを上げ、水のピペット1μLを測定台座に上げて、ブランクキャリブレーションを行います。
- サンプリングアームを下げて、ブランク測定を開始し、完了を待ちます。
- サンプリングアームを上げ、上下の台座からサンプルを拭きます。
- それを測定するために下の台座にDNA溶液のピペット1 μL。
- サンプリングアームを下げ、DNA濃度測定を開始し、完了を待ちます。
- サンプリングアームを上げ、上下の台座からサンプルを拭きます。
- さらなるDNA濃度測定のために、ステップ1.2.13.5-1.2.13.7を繰り返します。
- 測定が完了したら、使用するまでDNA溶液を4°Cに保存します。
2. ADEベースのディスペンスを駆動するピックリストの実験的な設計と生成
注:専用の「ユーザーフレンドリー」スプレッドシートマクロは、DNA量を管理し、384ウェルプレート形式で最大4つのプラスミドを混合するために開発されました。入力された実験設計に基づいて、このマクロはナノディスペンサーによるADEベースのDNAトランスフェクションプロトコルを駆動するために必要なファイルを生成します。これらのファイルを生成するには、図 2に示すように、いくつかのフィールドをテンプレート シートに入力する必要があります。

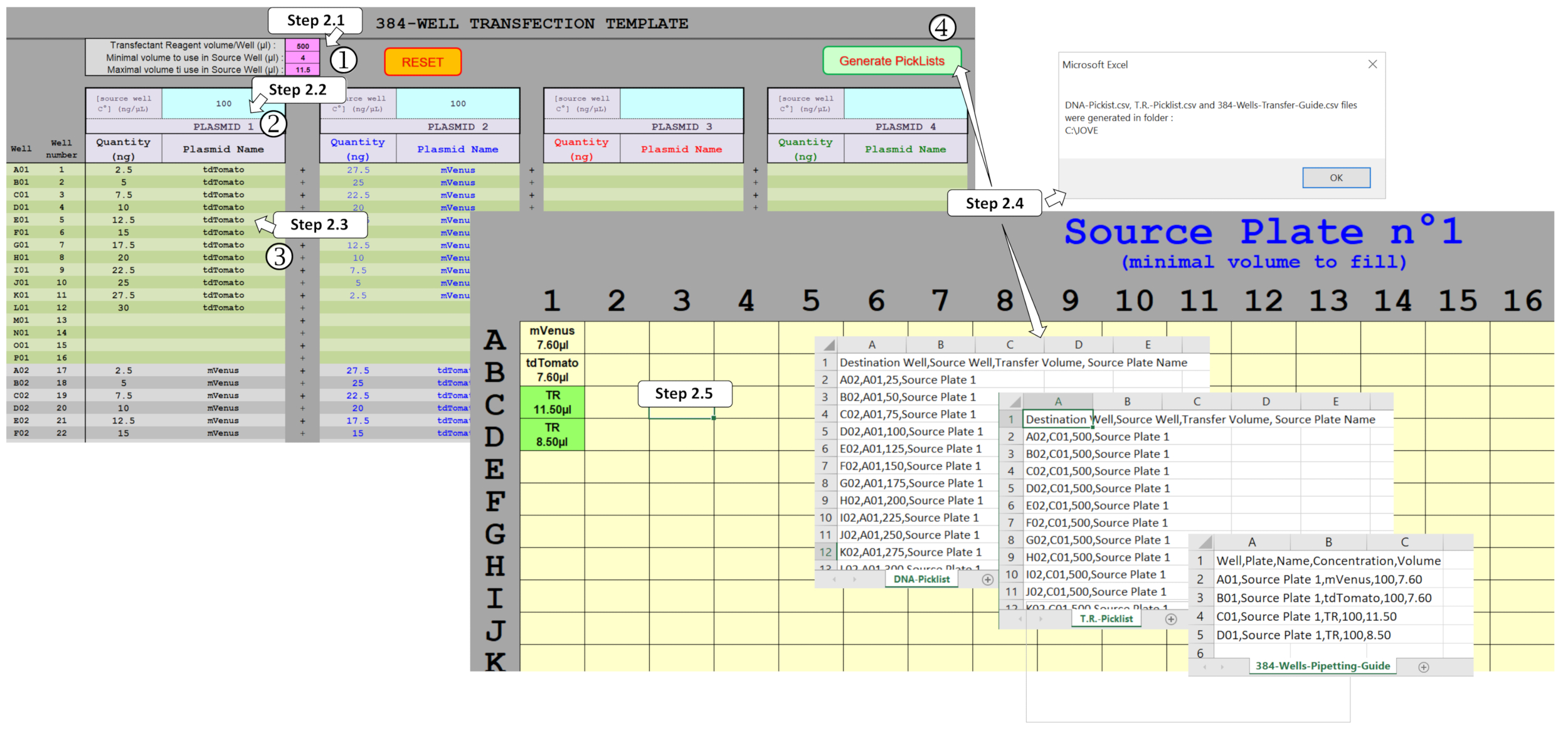
図 2: スプレッドシート マクロを使用して ADE ディスペンスを駆動するピクリストの生成。いくつかのパラメータを充填する必要がある、すなわち(1)トランスフェクション試薬(TR)およびソースプレートで使用される最小/最大容積、(2)ソースプレートに分配される初期プラスミド濃度、および(3)予想プラスミド量を含む全プレート設計、および384ウェルのそれぞれに多重化。(4)ピックリストの生成アクティベーションにより、異なるフィールドを検証し、適切に塗りつぶすと、DNAとTR分配のピックリストと必要なソースプレートテンプレートが自動的に生成されます。この図のより大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
- ピンクのフィールドにナノディスペンサープロトコルパラメータを入力します。トランスフェクション試薬(TR)混合値を500 nLに設定します。ソースプレートウェルの最小体積値を4 μLに設定し、ソースプレートウェルの最大体積を11.25 μLに設定します。
注:ここで使用されるナノディスペンサーは、ADEの1回の実行で最大500 nLしか転送できません。これらのピンクのフィールドは推奨値であらかじめ入力されていますが、ユーザーのニーズに応じて変更できます。 - 基になるDNAに対応する青色のフィールドに100 ng/μL DNA開始濃度を入力します。
注: この値は、以前に定義された最適な濃度ですが、異なるユーザーニーズに合わせて変更できます。 - グレー/グリーンフィールドに目的の DNA 量を入力します。384ウェルの量とプラスミド名を入力し、同じプラスミドが複数のウェルで使用されている場合は、同じスペルを確保します。
- ソース プレートの設計、ピクリスト ファイル、およびピペッティング ガイド ファイルを生成します。ピックリストの生成をクリックすると、マクロが対応するシートで収集されたデータから DNA-Picklist.csv、T.R.-Picklist.csv、および 384-Wells-Pipetting-Guide.csv ファイルを生成できます。要求された場合は、ナノディスペンサーで処理できないエラーまたはボリュームを示すオレンジ色で塗りつぶされたセル値を修正します。
- ソース プレートシートからテンプレートを印刷します。プラスミド名とウェルを埋めるために最小限の体積が示されています。同様に、次に次のウェルに充填する必要があるトランスフェクション試薬混合物量は、TRとして示され、緑色で強調表示されます。
3. 384ウェルピペッティングガイドアプリケーションを用いてDNAソースプレート調製
- 蒸留水を使用して、貯蔵されたDNAプラスミドをステップ1.2.14から100 ng/μLに希釈します。
- 384 ウェル グリッドをプレートの寸法に調整する:タブレットで 384 ウェル ピペッティング ガイド アプリケーションを開きます (図3)。下画面のグリッド上のソースプレートを配置し、左上のキャリブレーションメニューで、[+ or-(または赤いカーソルを使用する)] をクリックして、グリッドとウェルのサイズを拡張または縮小し、緑色の井戸をプレートの4つのコーナーウェルに調整します。.

図 3:384ウェルピペッティングガイドアプリケーションの使用。(1) プレートサイズに384ウェルグリッドのキャリブレーション;(2) 両面テープを使用してタブレットにユニバーサル3Dプリントプレートアダプタのマウント。(3) アダプター上のプレートの配置。(4) 格子の変位を取り付けられたプレートに中央に配置します。(5)キャリブレーションステップのロック。(6) 384ウェルピペッティングガイド.csvファイルを開きます。(7)ファイルリストを与えると、アプリケーションは、期待されるソースプレート名、試薬(DNAまたはトランスフェクション試薬)、濃度、および体積をターゲットウェルに分配し、1つずつ照射されることを示します。(8)左右の矢印ボタンを使用すると、ユーザーはピペッティングガイドに従って、スプレッドシートのマクロソースプレートテンプレートに従って試薬を簡単に分配することができます。この図のより大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
- 両面テープを使用して、3Dプリントプレートアダプタを画面に取り付け、分配中のソースプレートの動きを避けます。必要に応じて、回転矢印と上下/右/左ボタンを使用してキャリブレーションされたグリッドを移動し、画面上のグリッドをプレート位置に調整します。グリッドとウェルサイズが適切にキャリブレーションされ、配置されたら、ロックキャリブレーションボックスにチェックを入れます。
- FILEをクリックし、384ウェルピペッティング ガイド.csvファイルを開きます。画面の指示に従って、期待されるプレートの適切な標的先によく対応する白色の示された濃度で示されたプラスミドの示された体積を手動で分配する。DNA分配プロセスに戻るか、さらに進むには- または+矢印を使用します。最初のトランスフェクション試薬溶液をロードする際に分配を停止します。
- DNAの分配が終わったら、アダプターからソースプレートを取り外します。複数のソースプレートを充填する必要がある場合は、アダプタに新しいソースプレートを置き、ディスペンスの指示に従います。DNAの分配が完了したら、DNA充填ソースプレート(2分間1,500xg)を遠心分離して、適切な液体の平準化を確保し、ADEベースの転送の不正確さにつながる気泡を除去します。
4. ペリスタル液体ハンドラベースの1 μL希釈剤(宛先プレート内)
注: 生物学的安全キャビネットで手順 4.1-4.5 を実行します。
- 1 μLカセットヘッドをスプレー消毒剤(材料の表)でスプレーして消毒し、この溶液が先端ホルダーに入るようにします。吸収紙に残留消毒剤を吸収する。1 μLカセットを蠕動液取り扱い装置に取り付けます。デバイスの電源を入れ、カセットタイプの設定が正しい(1 μL)、プレートフォーマット(384ウェル)を確認します。
- チューブの内膜全体を消毒する:チューブオーガナイザー(8本のチューブを一緒に保持)を滅菌容器に挿入し、70%アルコールの5 mLでそれを埋めます。蠕動液ハンドラのプライミング機能を使用して、まずチューブ内のアルコールを洗い流し、5 mLの蒸留水と5mLの無血清培地(Dulbeccoの改変イーグル培地[DMEM])を100U/mLで補って洗い流します。ペニシリン連鎖マイシン;材料のテーブル)、連続して同じ容器に充填。すべての先端から液体の流れを目視で検査して、先端が詰まっていないことを確認してください。
- 新しい無菌容器に10mLの無菌培地を充填し、チューブオーガナイザーをダイビングすることにより、無血清培地でチューブをプライムします。蠕動液ハンドラのプライムボタンを約10s押します。もう一度、すべての液体の流れを目視で検査して、先端が詰まらないようにしてください。
- 1 μLの希釈剤でプレートを充填します。滅菌384ウェル培養プレート(宛先)を蠕動液ハンドラプレートキャリアに置き、蓋を取り外します。
- 384ウェルプレートの各ウェルに1 μLを分配するために、予備プログラムを実行します。分配時間は約8sです。次に、384ウェルプレートの蓋を交換します。
注:または、このステップは、マルチチャンネルマイクロピペットを使用して、安全キャビネットで手動で処理することができます。
5. 手動で分配されたボリュームを制御するための調査のパフォーマンス
注: 詳細については、図 4を参照してください。
- ナノディスペンサープログラムを実行し、診断タブに移動し、ソースプレートアウトボックスをチェックし、プレートホルダーにソースプレートをロードし、プレートに入るためにオンにします。プロンプトが表示された場合は、384LDV_AQ_B2を選択して、ナノディスペンサーを水性バッファーディスペンスモードに設定し、[OK]を押します。
- [その他]メニューで[アンケート]を選択し、[起動]をクリックします。分析し、移動ボタンをクリックするプリフィルドウェルを選択します。測定されたボリュームが予想されるボリュームと一致していることを確認し、転送を回避するため、12 μL を超えるボリュームがウェルにロードされていないことを確認します。

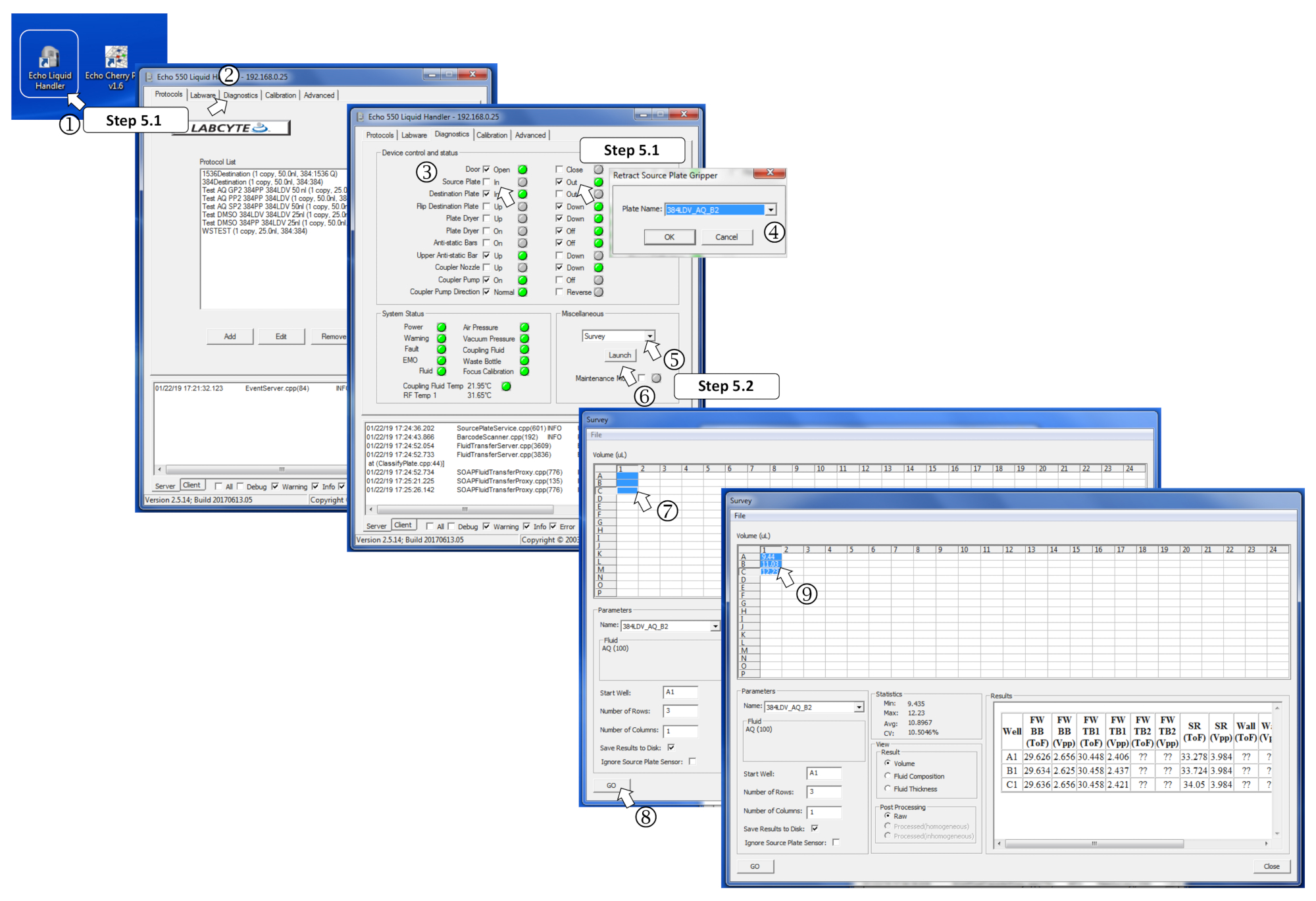
図 4: 測量ソフトウェア パラメータの定義。(1) ナノディスペンサープログラムを起動します。(2) [診断]タブを開く (3) ソース プレートにチェックを入れてソース プレートを挿入し、で.(4) プロンプトが表示された場合は、メニュー内のソースプレートタイプを定義します。(5) [その他] ボックスで、ドロップダウン メニューで[調査]を選択します。(6) 起動をクリックして調査プログラムを起動します。(7) 測定するあらかじめ充填された井戸を選択します。(8) [移動] をクリックして分析を開始します。(9) 調査を行うと、測定量は対応する選択された井戸に書き込まれます。この図のより大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
6. ADE駆動DNA分配を宛先プレートに
- ピックリスト ソフトウェアを実行し、384 ウェルのソースと宛先プレートの種類をそれぞれ 384_LDV と Greiner 384PS_781096 に設定します (図5)。384LDV_AQ_B2を選択して、デバイスを水性バッファディスペンスモードに 設定し、"転送スループットの最適化" をオフにします。

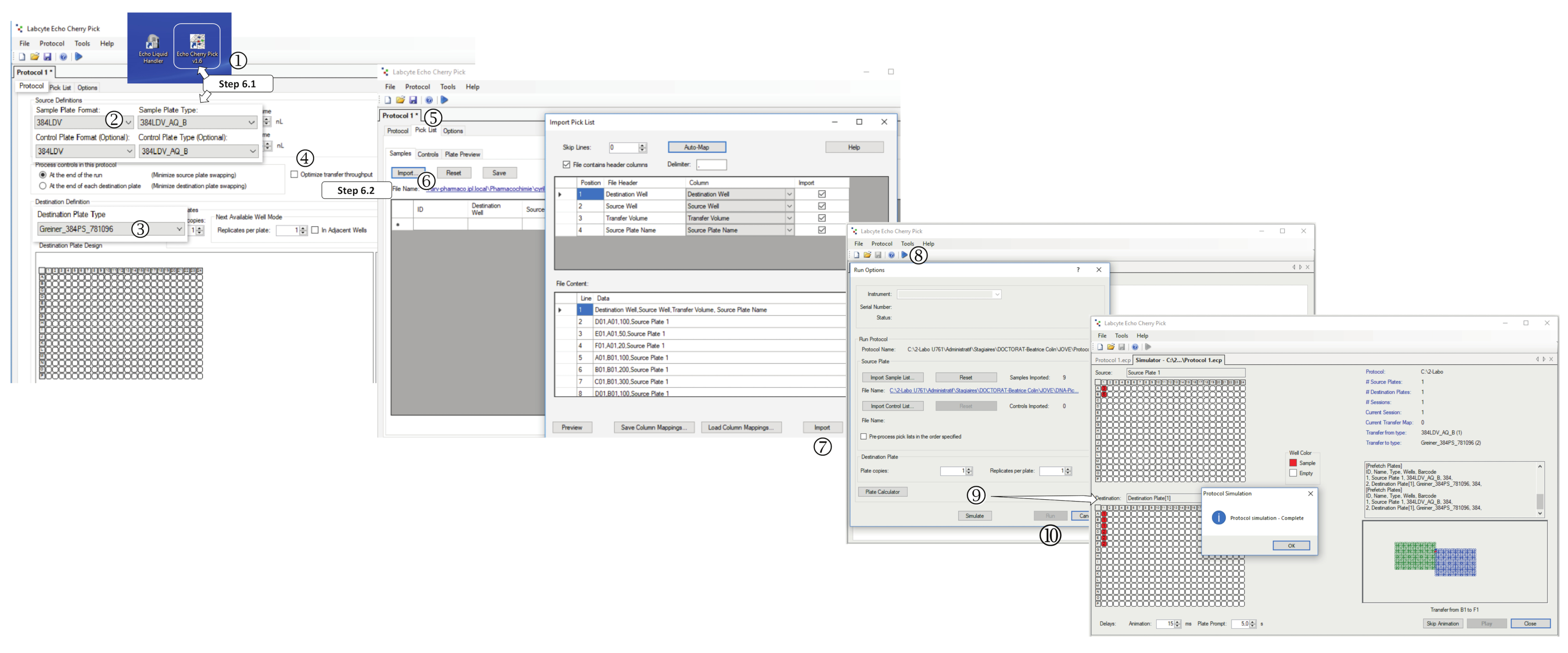
図 5: ピックリストベースの分配のパフォーマンス。(1) ナノディスペンサーソフトウェアを起動します。[プロトコル]タブで、(2)サンプルプレート形式、(3)宛先プレートタイプ、(4)を「転送スループットを最適化する」を選択します。(5) [選択リスト]タブを選択します (6)インポートをクリックし、適切な *.csv ファイル (DNA ピックリストまたは T.R.ピックリスト) を選択します。(7) 選択したら、[インポート]をクリックします。(8) [再生]をクリックし、プロトコルを保存します。(9) [シミュレート]をクリックして塗布シミュレーションを実行するか、(10) [実行]をクリックしてプログラムされたディスペンスを開始します。この図のより大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
- [リストを選択]タブを選択し、[インポート]をクリックし、DNA-Picklist.csvファイルを選択します。[再生]をクリックし、プロトコルを保存します。[シミュレート]をクリックして、プログラムされたディスペンスのシミュレーションを実行し、選択リストが予想される実験計画と一致することを確認します。完了したら、をクリックします。
- [再生]をクリックし、[実行]をクリックして、ディスペンス プログラムを開始します。
注:塗布時間は、選択されたボリュームと実験設計の分配の総数に応じて、完全な384ウェルプレートの約5〜20分です。 - あるいは、希釈剤とDNAで満たされたプレートが最大7日間乾燥または凍結保存を処理できるため、ここでプロトコルを一時停止します。乾式保管の場合は、プレートを室温でベンチで乾燥させ、同じ方法で保管します。解凍および遠心分離機(2分間1,500 x g)は、トランスフェクションステップ(セクション7)で使用する前に、貯蔵されたプレートを凍結しました。
7. ADE駆動トランスフェクション試薬調剤
- バイオセーフティキャビネットでは、無血清培地中のリポポリプレックストランスフェクション試薬を1倍の最終濃度に一時的に希釈する。Vortexは、マクロによって設計された定義済みのソースプレートに従って、このトランスフェクション試薬ミックスを直ちに分配し、ステップ3.4で説明するように予備に設けた384ウェルピペッティングガイドアプリケーションを使用します。
注:遠心分離後にトランスフェクションが気付かないように、試薬を装填したら、ソースプレートを遠心分離しないでください。 - 12 μLを超えるボリュームによるディスペンスエラーを避けるために、セクション5に記載されている「測量」を実行して、ソースプレートのすべての手動で充填されたTRウェルの体積を制御します。
- [リセット]をクリックして、ピックリスト ソフトウェアの DNA ピックリストのサンプル リストをクリアし、デバイス パラメータが引き続き水性バッファーと、手順 6.1 のように使用されるソースプレートと宛先プレートタイプに設定されていることを確認します。
- [インポート]をクリックし、TR-Picklist.csv ファイルを選択します。[再生]をクリックし、プロンプトが表示された場合はプロトコルを保存し、(これはオプションで強く推奨されます)プログラムされたトランスフェクション試薬混合物のシミュレーションを実行し、をクリックしてディスペンスの適切な設計を行います。ボタンをシミュレートします。完了したら、をクリックします。
- [再生]を クリックし、[実行]ボタンをクリックしてディスペンス プログラムを開始します。
注:TR混合物の500 nLを分配するとき、分配時間は完全な384ウェルプレートのために20分未満である。 - メーカーのプロトコルで示されているようにDNAにTRを加えた後、室温で15〜30分をインキュベートする。
8. 蠕動液体ハンドラベースの細胞分配
- 細胞を分配するための蠕動液ハンドラを準備します。アニオスプレーサーフ29消毒剤でスプレーし、紙に残骸を吸収することにより、10 μLカセットヘッドを消毒します。蠕動液ハンドラ装置にカセットを取り付け、カセットタイプの設定を10μLに変更し、プレートフォーマットが384ウェルに設定されていることを確認します。
- ステップ4.2で前述したように、10 μLカセットチューブを消毒します。無菌容器にチューブオーガナイザーをダイブし、70%アルコールの5 mLでチューブをフラッシュし、その後、蒸留水の5 mLで、最後に、5 mLの無血清培地で、連続して同じ容器に充填され、各チューブが空になるまで。
- 細胞懸濁液を分配する準備をする。コンフルエントHeLa細胞B10培養皿から、1xリン酸緩衝生理食べ物(PBS)溶液で細胞を1x洗浄し、トリプシン/EDTAで細胞を37°Cで5分間解離します。
- 顕微鏡下で細胞解離を確認し、10mLの完全培地(DMEMは10%の胎児ウシ血清と100U/mLペニシリン連鎖筋痛を補充し、培養皿に材料の表を参照)を追加してトリプシン/EDTA作用を停止します。50 mLチューブで細胞を収穫し、マラセス細胞または自動細胞カウンターを使用して、顕微鏡下の細胞をカウントします。
- 完全な384ウェルプレート用の完全な培地(すなわち、1,500セル/40 μL)の37,500細胞/mLの濃度でHeLa細胞懸濁液の少なくとも25 mLを調製し、チューブプライミングと40 μL/ウェル分配を確保します。
- 細胞を分配するには、新しい滅菌容器を調製した細胞懸濁液で充填し、分配の細胞密度の不正確さにつながる堆積を避けるためにそれをかき混ぜる。このソリューションにチューブオーガナイザーを挿入し、セルサスペンションが分配ヘッドからフラッシュし始めるまで、プライムボタンを押します。すべての液体の流れを目視で検査して先端が詰まっていないことを確認し、各チューブにセルサスペンションが搭載されていることを確認します。
- DNAとTRで満たされた384ウェルの宛先プレートを蠕動液体ハンドラプレートキャリアにロードし、蓋を取り外します。完全な384ウェルプレート(すなわち、1,500細胞/ウェル)に40 μLの細胞懸濁液を分配するために予備プログラムを実行します。分配時間は約8sです 384ウェルプレートの蓋を交換してください。
注:または、40 μLセルサスペンションは、マルチチャンネルマイクロピペットを使用して手動で分配することができます。
9.カスタム生物学的アッセイ(細胞トランスフェクション効率モニタリング)
注:実験の実験設定と意図に従って、発光、蛍光、高含有量スクリーニング、および逆転転写定量ポリメラーゼ連鎖反応(RT-qPCR)に必要な方法を使用します。プロトコルのこのセクションでは、細胞トランスフェクション効率は、自動蛍光顕微鏡および画像解析によって評価される。
- 水飽和雰囲気で5%CO2でプレートを37°Cでインキュベートし、適切なタンパク質発現まで。
注:ここでは、TdTomato-およびmVenus発現プラスミドを使用して、トランスフェクション効率を監視するためにHeLa細胞に48時間のインキュベーション時間が使用されます。 - プレートを反転して培養培地48hポストトランスフェクションを取り出し、蠕動液ハンドラ(10μLカセット)を用いて10%ホルマリンの30μL/ウェルを加え、室温で15分間インキュベートする。
- プレートを反転してホルマリンを取り除きます。その後、1x PBS溶液で0.1 ng/mLのホークストを希釈して室温で15分間細胞をインキュベートする。
- ホルマリン溶液インキュベーション工程の6.9pHによって失われた高蛍光シグナルを回収するために、pH=8に調整された1x PBSの80 μLで細胞を3xを15分間洗浄する。
- 自動蛍光顕微鏡を使用して、10倍の目的と適切な発光フィルタセット(4',6-diamidino-2-フェニリンドール]、dSRed、およびフルオレセイン)で2つまたは3つの蛍光チャネル(Hoechst、tdTomato、およびmVenus)の画像を順次取得します。イソチオシアネート[FITC])。
- トランスフェクション効率を評価するには、画像解析ソフトウェアを使用して、核染色に基づくスクリプト解析を使用してトランスフェクション効率を決定します。
結果
fADE技術が自動逆トランスフェクションプロトコルに使用できるかどうかを判断するために、赤色蛍光tdトマト発現プラスミドを用いて蛍光顕微鏡による細胞トランスフェクション効率をモニタリングした。まず、最良のトランスフェクションパラメータを決定するために、異なる希釈量とDNAの総量をクロステストしました。希釈体積は、DNA液滴が一度分配されると、?...
ディスカッション
特定の細胞株に対する正確なハイスループットトランスフェクション法の確立と最適化は、科学者がこのセクションで説明するいくつかの重要なパラメータに従う必要があります。HeLa細胞用に最適化されたこれらの設定はHEK細胞に対しても効率的であることが証明されたため、プロトコル全体で推奨値から始めることを強くお勧めします。しかし、最良のパラメータは細胞株およびトランス...
開示事項
著者は何も開示していない。
謝辞
著者らは、この記事の研究、著作者、および/または出版物のための次の財政的支援の領収書を開示しました:インサーム、リール大学、リールパストゥール研究所、コンセイユ・レジオナル・デュ・ノール、およびPRIM-HCV1と2(Pôle de Rechercheインタージシプリンエア・シュル・ル・メディカメント、アジェンス・ナショナル・デ・ラ・レチェルシュ(ANR-10-EQPX-04-01)、フェデラー(12001407(D-AL)イマジネーションス・バイオメッド)および欧州共同体(ERC-STG INTRACELLTB n° 260901)。著者らは、S.モウリュー博士、B.ヴィルマグネ博士、R.フェルー・クレメント博士、H.グルート博士に対し、原稿の批判的なレビューと訂正に感謝したいと考えている。
資料
| Name | Company | Catalog Number | Comments |
| 384LDV Microplate | Labcyte | LP-0200 | |
| 384-well Microplate μClear Black | Greiner | 781906 | |
| Ampicilin | Sigma | A9393-5G | Selection antibiotic for bacteria transformed with ampicilin expressing vector |
| Android Tablet | Samsung | Galaxy Note 8 | used to guide the user while the source plate manual dispense |
| Aniospray Surf 29 | Anios | 2421073 | disinfectant to clean the MicroFlo head |
| Columbus software | Perkin Elmer | image analysis software | |
| Dulbecco's Modified Eagle Medium (DMEM), high glucose, GlutaMAX Supplement, pyruvate | Thermo Fisher Scientific | 10566032 | cell culture medium |
| Echo Cherry Pick 1.5.3 software | Labcyte | Software enabling ADE-based dispenses by the Echo550 device from a *.csv file; nanodispenser software | |
| Echo550 | Labcyte | ADE-based dispenser | |
| Fetal Bovine Serum | Thermo Fisher Scientific | 16000044 | to add in cell culture medium |
| Formalin solution, neutral buffered, 10% | Sigma-Aldrich | HT501128-4L | to fix cell |
| HeLa cells | ATCC | HeLa (ATCC® CCL-2™) | |
| Hoechst 33342, Trihydrochloride, Trihydrate | Thermo Fisher Scientific | H3570 | 10 mg/mL Solution in Water |
| INCell Analyzer 6000 | GE Healthcare | 29043323 | automated laser-based confocal imaging platform |
| LB medium | Thermoischer Scientific LB Broth Base (Lennox L Broth Base)®, powder | 12780052 | culture medium for bacteria growth |
| Lysis Buffer (A2) | Macherey-Nagel | 740912.1 | Buffer from the NucleoSpin Plasmid kit used to prepare plasmid from bacterial culture |
| MicroFlo 10µL cassette | Biotek Instruments Inc | 7170013 | to use with the Microflo Dispenser |
| MicroFlo 1μL cassette | Biotek Instruments Inc | 7170012 | to use with the Microflo Dispenser |
| MicroFlo Dispenser | Biotek Instruments Inc | 7171000 | peristaltic pump-based liquid handler device |
| Microvolume spectrophotometer | Denovix | DS-11 Spectrophotometer | Measure the DNA concentration of samples |
| mVenus plasmid | mVenus cDNA was cloned by enzymatic restriction digestion and ligation in Age1/BsrG1 sites of the tdTomato-N1 plasmid | Vector type: Mammalian Expression, Fusion Protein: mVenus | |
| Neutralization Buffer (A3) | Macherey-Nagel | 740913.1 | Buffer from the NucleoSpin Plasmid kit used to prepare plasmid from bacterial culture |
| NucleoSpin Plasmid kit | Macherey-Nagel | 740588.50 | used to prepare plasmid from bacterial culture |
| Optimal-Modified Eagle Medium (Opti-MEM) Medium | Thermo Fisher Scientific | 31985070 | |
| optional Wash bufferWash Buffer (A4) | Macherey-Nagel | 740914.1 | Buffer from the NucleoSpin Plasmid kit used to prepare plasmid from bacterial culture |
| orbital shaker | incubated large capacity shaker | 444-7084 | Used to grow bacteria under gentle agitation and 37°C |
| Penicillin-Streptomycin | Thermo Fisher Scientific | 15140122 | 10,000 U/mL |
| Phosphate-Buffered Saline | Thermo Fisher Scientific | 10010001 | |
| Plasmid mini-columns | Macherey-Nagel | 740499.250 | Silica membrane mini-column to prepare plasmid from bacterial culture |
| Resuspension Buffer (A1) | Macherey-Nagel | 740911.1 | Buffer from the NucleoSpin Plasmid kit used to prepare plasmid from bacterial culture |
| RNAse A | Macherey-Nagel | 740505 | Enzyme from the NucleoSpin Plasmid kit used to prepare plasmid from bacterial culture |
| tdTomato-N1 plasmid | Addgene | Plasmid #54642 | Vector type: Mammalian Expression, Fusion Protein: tdTomato |
| TransIT-X2 Dynamic Delivery System | Mirus Bio | MIR 6000 | |
| Wash Buffer (AW) | Macherey-Nagel | 740916.1 | Buffer from the NucleoSpin Plasmid kit used to prepare plasmid from bacterial culture |
| 3D printer | Creality | CR10S | used to print the plate adapter |
| Blender Software | https://www.blender.org/ Free software under GNU General Public License (GPL). | version 2.79b | used to design the plate adapter |
参考文献
- Colin, B., Deprez, B., Couturier, C. High-Throughput DNA Plasmid Transfection Using Acoustic Droplet Ejection Technology. SLAS Discovery: Advancing Life Sciences R & D. , 2472555218803064 (2018).
- Mirus Bio. . Optimising Transfection Performance. , (2019).
- Thermo Fisher Scientific. . Factors Influencing Transfection Efficiency | Thermo Fisher Scientific - FR. , (2019).
- Boussif, O., et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proceedings of the National Academy of Sciences of the United States of America. 92 (16), 7297-7301 (1995).
- Figueroa, E., et al. A mechanistic investigation exploring the differential transfection efficiencies between the easy-to-transfect SK-BR3 and difficult-to-transfect CT26 cell lines. Journal of Nanobiotechnology. 15 (1), 36 (2017).
- Kirchenbuechler, I., Kirchenbuechler, D., Elbaum, M. Correlation between cationic lipid-based transfection and cell division. Experimental Cell Research. 345 (1), 1-5 (2016).
- Zhang, Z., Qiu, S., Zhang, X., Chen, W. Optimized DNA electroporation for primary human T cell engineering. BMC Biotechnology. 18 (1), 4 (2018).
- Cao, D., et al. Transfection activity and the mechanism of pDNA-complexes based on the hybrid of low-generation PAMAM and branched PEI-1.8k. Molecular bioSystems. 9 (12), 3175-3186 (2013).
- Bos, A. B., et al. Development of a semi-automated high throughput transient transfection system. Journal of Biotechnology. 180, 10-16 (2014).
- Colosimo, A., et al. Transfer and expression of foreign genes in mammalian cells. BioTechniques. 29 (2), 314-318 (2000).
- Villa-Diaz, L. G., Garcia-Perez, J. L., Krebsbach, P. H. Enhanced transfection efficiency of human embryonic stem cells by the incorporation of DNA liposomes in extracellular matrix. Stem Cells and Development. 19 (12), 1949-1957 (2010).
- Sabatini, D. M. . Reverse transfection method. , WO2001020015A1 (2001).
- Raymond, C., et al. A simplified polyethylenimine-mediated transfection process for large-scale and high-throughput applications. Methods (San Diego, CA). 55 (1), 44-51 (2011).
- Junquera, E., Aicart, E. Recent progress in gene therapy to deliver nucleic acids with multivalent cationic vectors. Advances in Colloid and Interface Science. 233, 161-175 (2016).
- Woodruff, K., Maerkl, S. J. A High-Throughput Microfluidic Platform for Mammalian Cell Transfection and Culturing. Scientific Reports. 6, 23937 (2016).
- Vasileiou, T., Foresti, D., Bayram, A., Poulikakos, D., Ferrari, A. Toward Contactless Biology: Acoustophoretic DNA Transfection. Scientific Reports. 6, 20023 (2016).
- Hadimioglu, B., Stearns, R., Ellson, R. Moving Liquids with Sound: The Physics of Acoustic Droplet Ejection for Robust Laboratory Automation in Life Sciences. Journal of Laboratory Automation. 21 (1), 4-18 (2016).
- Grant, R. J., et al. Achieving accurate compound concentration in cell-based screening: validation of acoustic droplet ejection technology. Journal of Biomolecular Screening. 14 (5), 452-459 (2009).
- Sackmann, E. K., et al. Technologies That Enable Accurate and Precise Nano- to Milliliter-Scale Liquid Dispensing of Aqueous Reagents Using Acoustic Droplet Ejection. Journal of Laboratory Automation. 21 (1), 166-177 (2016).
- Day, R. N., Davidson, M. W. The fluorescent protein palette: tools for cellular imaging. Chemical Society Reviews. 38 (10), 2887-2921 (2009).
- Zielinski, D., Gordon, A., Zaks, B. L., Erlich, Y. iPipet: sample handling using a tablet. Nature Methods. 11 (8), 784-785 (2014).
- Brunner, S., et al. Cell cycle dependence of gene transfer by lipoplex, polyplex and recombinant adenovirus. Gene Therapy. 7 (5), 401-407 (2000).
- Nii, T., et al. Single-Cell-State Culture of Human Pluripotent Stem Cells Increases Transfection Efficiency. BioResearch Open Access. 5 (1), 127-136 (2016).
- Noonan, D. J., Henry, K., Twaroski, M. L. A High-Throughput Mammalian Cell-Based Transient Transfection Assay. Signal Transduction Protocols. 284, 051-066 (2004).
- . . Transfection | TransIT Transfection Reagents | Mirus Bio. , (2015).
- American Type Culture Collection. . General protocol for transfection of stem cells, primary cells, and continuous cell lines with ATCC TransfeX Transfection Reagent. , (2017).
- American Type Culture Collection. . Transfection Reagents for Nucleic Acid Transfer into ATCC Cells. , (2017).
- de Los Milagros Bassani Molinas, M., Beer, C., Hesse, F., Wirth, M., Wagner, R. Optimizing the transient transfection process of HEK-293 suspension cells for protein production by nucleotide ratio monitoring. Cytotechnology. 66 (3), 493-514 (2014).
- Promega. . FuGENE® 6 Transfection Reagent. , (2019).
- Olden, B. R., Cheng, Y., Yu, J. L., Pun, S. H. Cationic polymers for non-viral gene delivery to human T cells. Journal of Controlled Release: Official Journal of the Controlled Release Society. 282, 140-147 (2018).
- Park, E., Cho, H. B., Takimoto, K. Effective gene delivery into adipose-derived stem cells: transfection of cells in suspension with the use of a nuclear localization signal peptide-conjugated polyethylenimine. Cytotherapy. 17 (5), 536-542 (2015).
- Wood, R. W., Loomis, A. L. The physical and biological effects of high-frequency sound-waves of great intensity. The London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science. 4 (22), 417-436 (1927).
- Mamat, U., et al. Eliminating Endotoxin at the Source - A Novel Competent Cell Line with Modified Lipopolysaccharide for Low-Endotoxin Plasmid Production. , (2014).
- Ivanova, N. V., Kuzmina, M. L. Protocols for dry DNA storage and shipment at room temperature. Molecular Ecology Resources. 13 (5), 890-898 (2013).
- Lesnick, J., Lejeune-Dodge, A., Ruppert, N., Jarman, C. . High-Precision Cell Dispensing with the Labcyte Echo® Liquid Handler. , (2017).
- Yang, X., et al. A public genome-scale lentiviral expression library of human ORFs. Nature Methods. 8 (8), 659-661 (2011).
- Peng, J., Zhou, Y., Zhu, S., Wei, W. High-throughput screens in mammalian cells using the CRISPR-Cas9 system. The FEBS journal. 282 (11), 2089-2096 (2015).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved