
Method Article
腫瘍抗原特異的T細胞の生成にヒト誘導多能性幹細胞を用いた
* これらの著者は同等に貢献しました
要約
本稿では、OP9/DLL1共培養系を用いて機能性腫瘍抗原特異的人工多能性幹細胞由来CD8αβ+単陽性T細胞を生成する方法について説明する。
要約
インビトロでの機能的なT細胞の生成と拡張は、臨床応用の広い範囲につながることができます。そのような使用の1つは、進行癌の患者の治療のためである。高度に濃縮された腫瘍抗原特異的T細胞の養子T細胞移植(ACT)は、一部の患者において転移癌の耐久性的な退行を引き起こすことが示されている。しかし、膨張の間、これらの細胞は疲れ果てたり老化したりして、生体内でのエフェクター機能と持続性を制限する。人工多能性幹細胞(iPSC)技術は、未分分化腫瘍抗原特異的T細胞の多数のインビトロ生成につながることでこれらの障害を克服し得る。ヒトiPSC(hiPSC)は、T細胞が出発細胞として使用される場合に元のT細胞受容体(TCR)ゲノム再構成を保持するリンパ球を含むあらゆるタイプの体細胞に分化する能力を有する。従って、ヒト腫瘍抗原特異的T細胞をhiPSCにリプログラミングし、続いてT細胞系統に再分化し、若返った腫瘍抗原特異的T細胞を産生する可能性を有する。ここで説明する方法は、OP9/DLL1共培養系を用いてhiPSCから腫瘍抗原特異的CD8αβ+単一陽性(SP)T細胞を生成する方法である。この方法は、インビトロT細胞系統生成のための強力なツールであり、再生医療および細胞ベースの治療に使用するためのin vitro由来T細胞の開発を容易にします。
概要
生理学的利点に加えて、T細胞は多くの潜在的な治療用途を有する。インビトロにおけるT細胞の生成と拡張は、疾患モデリングおよび治療検証だけでなく、遺伝性および後天性免疫不全状態(すなわち、ウイルス免疫不全およびリンパ枯渇二次性の治療源)に使用することができる。化学療法または移植)および癌の根絶のために。この後者の品質は、進行癌1の患者の治療のための養子T細胞移植(ACT)の開発につながっている。
ACTは、患者の腫瘍を切除し、腫瘍浸潤性リンパ球(TI)を抽出し、TIをex vivoに拡張し、次いで、拡張された細胞を患者2に再注入する。転移性癌の一部の患者にとって効果的な治療様式であることが示されている。 残念ながら、すべての患者がこの療法に反応するわけではありません。以前の報告では、転送されたセル3、4、5、6、7、8、9の区別状態が大きな数値の使用が示されています。高度に濃縮された癌抗原特異的T細胞10の、および転写後のT細胞の持続性は、すべて、より耐久性のある応答13、14と相関している。従って、ACTが抗腫瘍応答を惹起しない場合、一部は癌抗原特異的T細胞の収率が低い、反応性クローンの枯渇および損失につながる非効率的なエクスビボ膨張、または転移後の持続性の欠如によるものである可能性がある。.これらの障害は、インビトロ15、16における未分化癌抗原特異的T細胞の大量生成によって克服され得ると仮定されている。
造血幹細胞/前駆細胞(HSPC)は、インビトロT細胞生成のための従来の供給源であるが、この方法は、単一のドナー1から回収することができる少数の細胞によって制限される。胚性幹細胞(ESC)はT細胞を産生することも示されているが、収率は17度と低く、臨床応用には非効率的である。さらに、T系統細胞は初期の発達段階でT細胞受容体(T眼)の確率的遺伝子組換えを経験するので、HSPCまたはESCを使用して、それ以上ゲノムを持たずに抗原特異的T細胞の純粋な集団を生成することはできないTCR遺伝子導入のような修飾。
これらの注意点を克服する1つのアプローチは、TIをヒト人工多能性幹細胞(hiPSC)に再プログラムすることであり、これはin vitro T細胞生成のための無限の供給源を提供する可能性がある。癌抗原特異的TIをhiPSCに再プログラムし、T細胞系統に再分化できることが示されており、これは元のT細胞18、19と同じT細胞受容体(TCR)遺伝子再構成を保持する。 個々の患者腫瘍は固有の突然変異プロファイルを有し、かつ非常に少数の癌抗原が患者20の間で共有することが示されているので、この詳細はACTにとって重要である。したがって、hiPSC由来T細胞のインビトロ生成の供給源として癌抗原特異的TIを用いると、転移性癌患者のパーソナライズされた治療のための新しい戦略を提供し得る。
ここで詳細に提示するプロトコルは、OP9/DLL1共培養系を用いてhiPSC由来T系統細胞を機能的抗原特異的CD8αβ+単一陽性(SP)T細胞に区別するためのプロトコルである。この方法は、hiPSC、造血前駆体、胚性幹細胞のインビトロT細胞分化のための強力なツールであり、再生医療および細胞ベースの治療におけるさらなる応用を行う。
プロトコル
1. マウス胚性線維芽細胞(MEF)におけるヒトiPSC(hiPSC)の培養
注:hiPSCを培養するための代替方法は、ゼラチンで事前にコーティングされた6ウェルプレートへの播種、ゼラチンタンパク質混合物、組換えラミニン511、またはhiPSC膨張に使用される他の任意の細胞外マトリックスへの播種を含むがこれらに限定されないが使用できる。ヒト多能性幹細胞培養のために特別に製剤化された定義済み媒体を用いて培養する。
-
MEFの培養
- 10cm細胞培養ペトリ皿を4mLの0.1%ゼラチンでコーティングし、37°Cで30分間インキュベートする。
- 4 x 106照射されたMEFのバイアルを37°MeFメディアの10 mLに素早く解凍する(DMEM + 10% FBS + 1xペニシリン・ストレプトマイシン + 1x L-グルタミンサプリメント)。4 °Cで5分間300 x gで遠心分離機。上清を吸引し、MEF媒体の9mLで細胞ペレットを再中断する。
- ゼラチンコーティングされた皿をインキュベーターから取り出します。ゼラチンを吸引し、MEFメディアの7 mLを追加します。メフ懸濁液のプレート3 mL(ステップ1.1.2から)をゼラチンコーティング皿に。皿を左右にロックし、前面から背面にロックして、MEFを皿の上に均等に分配します。8-36時間37°Cでインキュベートする。
-
MEF の hiPSC の受け渡し
注:データは、HLA-A*02:01の文脈で特にマート1ペプチドを認識する長期培養黒色腫TILに由来するMART-1 iPSCを用いて生成された。- コロニーの直径が0.8~1.2mmの場合の通路hiPSC。通過する前に、ステレオ顕微鏡でhiPSCコロニーをチェックし、200°L先端のプラスチックエッジを使用して培養物との差別化の領域を除去します。
- 吸引使用媒体を使用し、10 mL hiPSC媒体(ヒトES培養媒体[材料表]+10ng/mLヒト塩基性線維芽細胞増殖因子[hbFGF])を10μM ROCK阻害剤で補充した。
- セル培養皿を片手に持ち、使い捨てセルパフォージングツールを片方に回します。ローラーブレード全体が培養皿に触れ、圧延中に均一な圧力を維持するように十分な圧力を加えます。
- 培養皿を90°回転させ、ステップ1.2.3を繰り返します。顕微鏡でプレートを見て、チェッカーで表示されるコロニーの適切な切断を視覚的に確認します。200°Lピペットを使用して穏やかな機械的フラッシュによって切断されたコロニーを取り外します。
注:3分後にカットコロニーが皿に再び付着し始め、フラッシングによって均質な大きさのコロニーを剥離することが困難になるため、機械的なフラッシングによるカットコロニーの剥離はローラーでコロニーを切断した直後に行う必要があります。 - 10μM ROCK阻害剤を補充した新鮮なhiPSCメディアの10 mLで、MEFの新しい10cm皿(hiPSCパッシング前に8-36時間をめっき)にカットコロニーの350-600塊を転送します。37°Cでインキュベートする。
注: 600 束は約 1.0 x 106 MART-1 iPSC を表し、35 日目に 0.5 -1.0 x 106 DP セルが生成されます。ただし、期待される数値は、開始細胞株の効力および培養条件によって異なる。 - 翌日、使用済みメディアを吸引し、新鮮なhiPSCメディアを10mL追加します。hiPSC の成長率に応じて、1 ~ 2 日ごとに hiPSC メディアを変更します。
2. hiPSCとの共培養のためのOP9/DLL1細胞の調製
- OP9培地中の培養OP9/DLL1細胞[α-最小必須培地(α-MEM)+20%胎児ウシ血清(FBS)+1xペニシリン・ストレプトマイシン]を37°CでOP9/DLL1細胞が共奏に達すると、吸引媒体を吸引し、1xマグネシウム、カルシウム、およびフェノール赤フリーリン酸緩衝生理食塩水(PBS)の5mLで1回洗浄する。
- PBSを吸引し、0.05%トリプシンEDTAの2 mLを追加します。 37°Cで5分間インキュベートする。次いで、4mLのOP9メディアを加え、ピペットによって細胞層を機械的に解解き、単一細胞懸濁液を作る。
- 細胞の塊を避けるために、100 μmのセルストレーナーを通して50 mL円錐形チューブに細胞懸濁液を移します。4 °Cで5分間300 x gで遠心分離機。OP9メディアの上清を吸引し、12 mLで再中断。
- 6つの新しい10 cm細胞培養ペトリ料理のそれぞれに8 mLのOP9メディアを追加します。ステップ2.3から新しい10cm皿にOP9/DLL1細胞懸濁液のプレート2 mL。皿を左右にロックし、その後、皿の上にOP9 /DLL1の均等な分布を確保するために、前面から背面にロックします。
- 37°Cでインキュベートする。細胞が合流に達すると、2~3日ごとに通過を繰り返します。
注:OP9/DLL1細胞の十分な凍結ストックを作成し、4-6週間ごとに新しいストックを解凍することが重要です。
3. CD8αβ+シングルポジティブ(SP)T細胞へのhiPSCのインビトロ分化
- hiPSCとの共培養の1週間前にゼラチン化OP9/DLL1料理を準備します。0.1%ゼラチン溶液を調製するには、5mLの室温(RT)組織グレードのストックゼラチン溶液を500mLのPBSに添加します。
- コート3新しい10cm細胞培養ペトリ皿は、0.1%ゼラチンの1皿あたり4 mLを加えることによって。 37°Cで30分インキュベートする。
- ゼラチンを吸引し、各皿に8 mLのOP9メディアを加えます。OP9/DLL1の1つのコンフルディッシュ(上記のセクション2で行うように)を3つのゼラチン事前コーティングされた料理に通過させます。
- 4日後、ゼラチンのOP9/DLL1の10cm皿に10mLのOP9メディアを加えて、1皿あたり合計20mLのメディアを加えます。
- 7 - 8日後に、OP9/DLL1コンフルエント料理(差別化日0)でhiPSC共培養を開始します。
- MEF上のhiPSCのコンフルエント10センチ皿からメディアを費やしました。OP9 メディアを 10 mL 追加します。手順 1.2.3 および 1.2.4 で行った使い捨てセルパフォージング ツールを使用して、hiPSC コロニーをカットしてデタッチします。
- 200 μL ピペットを使用して、10 cm のゼラチン化済み OP9/DLL1 皿 (ステップ 3.1) に切断されたコロニーの 350 ~ 600 束を転送します。培養皿を左右にロックし、その後、コロニーの均等な分布を確保するために、フロントツーバック。
注:あるいは、あらかじめ形成されたhiPSC胚体(EB)または小さな束懸濁液が使用され得る。 しかしながら、使い捨てセルパソエーションツールまたはEB形成システムの使用は、均一なサイズのhiPSC塊を生成することが好ましい。
- 1日目に、吸引は使用されたメディアを吸引し、新鮮なOP9メディアの20 mLに置き換えます。OP9/DLL1 で 1 日間共培養された hiPSC 塊は、小さな丸い単層コロニーとして表示されます (図 1)。
- 5日目に、10 mLの使用済みメディアを吸引し、新鮮なOP9メディアを10mL追加します。hiPSCコロニーは、多層暗い中心によって特徴付けられた原始的な中皮に分化し始めます。
- 9日目に、10 mLの使用済みメディアを吸引し、新鮮なOP9メディアを10mL追加します。この時点で、多層中心構造はドーム状の形状に進化し、周辺ネットワークのような領域が明らかになり始めます。
- 13日目に造血前駆細胞(HpC)を収穫する(図1)。13日目のhiPSC由来の構造は、ドーム状領域のネットワークに囲まれた暗い中央オルガノイドによって特徴付けられ、造血帯(HZs)を代表して、ヒト胚性幹細胞由来造血前駆体を囲むよう以前に報告された21.
注:ドーム状の構造の存在は、暗い中心がない場合でも、成功したプロセスを示しています。HPCの生産不能は、OP9/DLL1の品質の低下、FBSロットの品質、OP9/DLL1に播種されたiPSC塊のコンフルエンシー(350-600塊が最適)、および/または造血前駆体を生成するiPSCラインの効力の変動が原因である可能性があります。- 吸引は、カルシウムとマグネシウム(HBSS)で修飾された1xフェノール赤フリーハンクスのバランスのとれた塩溶液の5 mLで使用されたメディアと洗浄1x。
- HBSSを吸引し、HBSSの10 mLに5000単位/mLコラゲラーゼIVの250 μLを加えます。コラゲラーゼIVで37°Cでインキュベートし、5mLのPBSで1回洗浄します。
- PBSを吸引し、0.25%トリプシンEDTAの5 mLを追加します。37°Cで20分間インキュベートします。次いで、4mLのOP9メディアを加え、ピペットによって細胞層の解ソを解除し、単一細胞懸濁液を作る。
- 100μmのセルストレーナーを介して50 mL円錐管に細胞懸濁液を移します。 4 °Cで5分間300 x gで遠心分離機。OP9メディアの上清を吸引し、10mLで再中断する。
- 新しいゼラチン化10cm細胞培養ペトリ皿にプレートセル懸濁液(ステップ3.1.1および3.1.2を参照)。37°Cで45分間インキュベートします。次いで、穏やかなピペットによって非付着細胞を収集する。
- 収集した細胞懸濁液を100μmの細胞ストレーナーを介して50mL円錐管に移す。 4 °Cで5分間300 x gで遠心分離機。分化媒体の10mLで上清を吸引し、再サスペンド[OP9メディアは5ng/mLヒト幹細胞因子(hSCF)、5 ng/mLヒトFlt3リガンド(hFLT3L)、および5 ng/mLヒトインターロイキン7(hIL-7)]。
- セルサスペンションを新しい10cm OP9/DLL1コンフルディッシュにプレートします。
- 16日目に細胞を通す。
- 穏やかなピペットによって非付着細胞を機械的に取り外し、100μmの細胞ストレーナーを通してろ過する。4 °Cで5分間300 x gで遠心分離機。上清を吸引し、10 mLの分化媒体で再中断する。
- セルサスペンションを新しい10cm OP9/DLL1コンフルディッシュにプレートします。
- ステップ3.8を繰り返して、その後5〜7日ごとに非付着細胞の受け渡しを続ける。
- 35日目に、CD4+CD8+ダブル陽性(DP)集団を濃縮し、刺激してCD8αβ+SP T細胞を産生する(図2)。
- 穏やかなピペットによって非付着細胞を機械的に取り外し、100μmの細胞ストレーナーを通して濾過し、細胞塊を除去します。メーカーのプロトコルに従ってCD4磁気ビーズ分離によってCD4+細胞集団を豊かにします。
注:CD4磁気ビーズを使用する根拠は、CD4−CD8−DN細胞を培養物から除去することにあるが、これらは刺激22後にCD4+CD8+DP細胞の直接殺傷を引き起こすことが実証されている。 - ノイバウアー血球計とトリパンブルー色素を使用して、生きたCD4濃縮細胞を数えます。 総濃度0.5 x 106セル/mLでOP9メディアにサスペンド。細胞懸濁液のアリコート1mL(0.5x106細胞)をコンフルエントOP9/DLL1の組織培養平底24ウェルプレートの各ウェルに。
- 100 IUヒトインターロイキン2(hIL-2)、5 ng/mL hIL-7、500 ng/mL抗ヒトCD3抗体、および2μg/mL抗ヒトCD28抗体を加え、37°Cで培養する。
- 刺激後4日目から7日目に、分子分析用の細胞を採取(図3)、またはペプチドパルス抗原提示細胞(APC)と共培養する。
- 穏やかなピペットによって非付着細胞を機械的に取り外し、100μmの細胞ストレーナーを通して濾過し、細胞塊を除去します。メーカーのプロトコルに従ってCD4磁気ビーズ分離によってCD4+細胞集団を豊かにします。
4. hiPSC由来CD8αβ+SP T細胞の抗原特異性の測定
注:このエクスペリメントに使用される APC のタイプは、hiPSC 派生 T セルの MHC 制限に依存します。ここで、T2細胞株が用いられ、これはTおよびBリンパ芽球細胞株のハイブリッドである。T2細胞はHLA-A*02:0123を発現し、これはMART1-iPSCが由来したJKF6細胞によって認識される18である。このT2細胞株はRPMI 1640 + 20% FBS + 1xペニシリン・ストレプトマイシンで拡張することができ、細胞が5 x 105細胞/mLの密度に達すると通過する。
- 生きたHLA-A*02:01+ T-Bハイブリッドリンパ芽球T2細胞をノイバウアー血球計とトリパンブルー色素を使用してカウントします。24ウェル組織培養プレートに1μg/mL MART-1ペプチドを含むAPCを37°Cで2時間インキュベートします。
注:最適なペプチド濃度は、細胞株および抗原特異性に応じて可変である。 - APCを収集し、余分なペプチドを除去するためにPBSの10 mLで2xを洗浄します。
- 100 IU IL-2 および 5 ng/mL IL-7 を使用して、OP9 メディアで APC をカウントし、2 x 5 x 105セル/mL で中断します。アリコート100μLの細胞懸濁液(2-5 x 104細胞)を超低着取り付けU底96ウェルプレートの各ウェルに、または事前にコーティングされたELISpotプレートに直接入れます。
- 細胞選別器を用いてhiPSC由来CD8αβ+SP T細胞(抗ヒトCD3/CD28抗体刺激の1週間後)を並べ替え、100 IU IL-2および5 ng/mL IL-7でOP9媒体で1 x 106細胞/mLで中断する。アリコート100μLの細胞懸濁液(1x105細胞)を各ウェルにAPC及び培養を37°Cで16〜20時間培養する。
- 16~20時間後、製造業者のプロトコルに従ってELISpotアッセイによるサイトカイン分泌プロファイルを分析する(図4)。
結果
OP9/DLL1と13日間のhiPSCの共培養後、CD34+CD43+造血前駆細胞が登場した(図1)。hSCF、hFLT3L、hIL-7の存在下で非ゼラチン化OP9/DLL1でさらに22日間培養した後、造血前駆体はCD3+CD7+CD4+ダブルポジティブ(DP)T系統細胞に分化し、その大部分はMART-1エピトープ(テトラマー)に特異的なTCRを発現した(図2)。
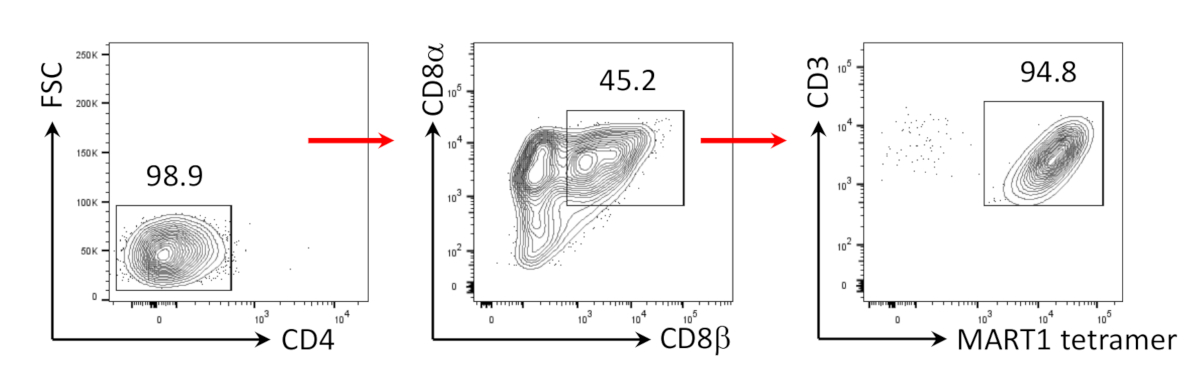
CD8+SP T細胞は、アゴニストペプチドまたは抗体駆動TCR刺激24、25を介したTCRシグナル伝達によってCD4+CD8+DP T細胞から誘導できることが以前から示されている。そこで、培養35日目に、hiPSC由来CD4+CD8+DP T細胞を、hIL-7およびhIL-2の存在下で抗ヒトCD3および抗ヒトCD28抗体で刺激した。 刺激の4日後、CD3+CD8αβ+SP細胞の数は劇的に増加し、MART-1エピトープに特異的なままであり、その遺伝性抗原特異性の保存を確認した(図3)。
hiPSC由来CD8αβ+SPT細胞の機能特性を決定するために、インターフェロンγ(IFN-γ)の抗原依存的活性化および分泌を解析した。抗ヒトCD3および抗ヒトCD28抗体による刺激後、hiPSC由来CD8αβ+SP T細胞を細胞選別剤を用いて単離し、16~20時間のCOGnate MART1ペプチドの有無にかかわらずHLA-A*02:01を発現するT2細胞株と共培養した。ELISpotアッセイは、hiPSC由来CD8αβ+SP T細胞が、MART-1ペプチドの存在下で培養した場合、CD8αα+SP T細胞と比較してより多量のIFN-γを分泌することを明らかにした。IFN-γ発現はT細胞およびAPC単独ではnullであり、ヒトT-iPSC由来T細胞が抗原特異的かつ機能的であることを実証した(図4)。

図1:hiPSC由来造血前駆細胞の生成(A) OP9/DLL1共培養を用いた造血系統へのhiPSCの差別化の概略図(B) 1 日目 (左上)、3 (右上)、7 (左下)、および 13 (右下) の hiPSC 派生構造体の外観。スケールバー = 100 μm. (C) 13日目のhiPSC由来CD34+CD43+造血前駆細胞のフローサイトメトリック分析。データは、6つの独立した実験(n=1〜2)を表す。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図2:マート1+CD4+CD8αβ+DP T細胞へのhiPSC分化。 (A) OP9/DLL1共培養を用いた未熟なT細胞に対するhiPSC由来造血系統の分化の概略的概要(B)35日目のhiPSC由来T細胞におけるCD4対CD8α、CD3対CD8β、およびMART-1四量体発現のフローサイトメトリクス分析。リンパ球、単細胞、PI陰性にゲート。データは、3つの独立した実験(n=3-8)の代表である。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図3:CD8αβ+SP T細胞表現型の誘導。CD4のフローサイトメトリクス分析-ヒト抗CD3およびヒト抗CD28抗体駆動刺激の4日後にhiPSC由来T細胞。リンパ球上にゲート、単一細胞、PI陰性(n=4)。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図4: hiPSC由来CD8αβ+SP T細胞の抗原特異性HIPSC由来CD8αβ+SP、CD8αα+SP、および20時間共培養後のバルクT細胞のELSpotアッセイによるIFN-γ分泌は、MART-1ペプチドとパルスされた(またはしない)T2細胞の有無にかかわらず。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
ディスカッション
OP9マウス間質細胞の共培養は、HSPCおよび多能性幹細胞からのリンパ球(すなわち、NK、B、およびT細胞)のインビトロ生成のための確立されたシステムである。ノッチシグナリングは、T系統のコミットメントを誘導するために必要とされ、T細胞生成1に対して同等の有効性を有するノッチリガンドDLL1またはDLL4の外来発現によって達成することができる。従って、OP9/DLL1共培養システムは、インビトロでT細胞を製造するための広く用いられている方法となっている。 さらに、この方法は臍帯血、骨髄HSPCおよびESCを含むヒト細胞のいくつかのタイプおよび源との使用のために適用可能である。 しかしながら、これらのソースからのT細胞の生成は、ソース細胞の検索が不十分であるか、またはT細胞1に対する非効率的な分化によって制限される。さらに、単一のTCR再結合を有するT細胞産物は、これらのオープンレパートリーソースから生成することはできません。再生医療技術、すなわち人工多能性幹細胞(iPSC)技術を用いることにより、細胞ベースの治療薬15に用いる抗原特異的T細胞を大量に製造することができるかもしれない。
hiPSCは、自己再生、無限の膨張、および体内の任意のタイプの体細胞に分化する可能性のための能力の多能ESCに似ています。しかし、彼らは臨床応用のための胚起源の製品の使用を取り巻く倫理的な懸念を欠いている。さらに、hiPSCは任意の体細胞から製造することができ、パーソナライズされた医療のための細胞製品の開発を可能にします。これまでの報告では、hiPSCは、全身末梢単核細胞、CD3+細胞、または単離された細胞傷害性Tリンパ球(CACL)をソース18、19、22、およびを用いてヒトT細胞から産生されてきた。 26.T細胞源(T-iPSC)からhiPSCが生成されると、元のTCR遺伝子再構成が継承されます。したがって、患者T-iPSC由来T細胞は、患者の明確な癌抗原を標的とすることによりパーソナライズされたACT治療のためのモデルを提供し得る。
ヒト多能性幹細胞をT系統細胞に分化させるのは、造血前駆細胞(HPC)27の生成とT系統細胞21へのさらなる分化の2つのステップに分けられる。どちらの手順も、OP9/DLL1 共培養システムを使用して実行できます。重要なことに、OP9/DLL1フィーダー細胞の品質は、T細胞分化の成功に不可欠です。OP9/DLL1細胞は不死化した均質な細胞株ではないため、FBSおよび培養条件の品質は、hiPSCの差別化をサポートする能力を失うことなく、その膨張を維持するために重要です。したがって、細胞間細胞質接触が起こり始めると、細胞分化や老化を防ぐために、FBSと通路の多くを一貫して事前評価することをお勧めします。考慮すべき点の1つは、顕微鏡の位相コントラストと倍率に応じて、細胞間接触が背景と区別できないように見えることがあるということです。私たちの経験では、ほとんどのOP9/DLL1料理は、通過する準備ができたときに80%のコンフルエントのように見えます。
OP9/DLL1共培養によりT-iPSCから生成された再分化T系統細胞は、刺激18、19時にCD8+SPT細胞を産生できることが示されている。しかしながら、再生されたCD8+SPT細胞は、先天様CD8ααホモダイマー22、28を取得し、これはTCRシグナル伝達29に対して効果のない共受容体である。 さらに、これらの再生CD8+SP T細胞は、強いTCR非依存性細胞傷害性を示しており、これらの細胞は臨床使用30に好ましくない。このプロトコルは、精製されたCD4+CD8+DP細胞の刺激を伴う最近の方法を記載し、より従来の表現型および改善された抗原特異的細胞毒性22を有するCD8αβ+SPT細胞を生成する。二次TCRαアレル再構成による抗原特異性の喪失は長期培養後のDP段階で起こるが、これはT-iPSC31におけるゲノム編集によって克服することができる。 私たちの経験では、hiPSC由来のDP細胞は培養30~35日目に現れ始め、新たに生成されたDP細胞はまだ二次TCRα転位を受けていない。したがって、35日目のほとんどのDP細胞は抗原特異性を保持し、抗原特異的CD8αβ+SP T細胞を生成するために使用することができる。
35日目にヒト抗CD3および抗CD28刺激の前に、CD4−CD8−DN細胞は、刺激22後にCD4+CD8+DP細胞の直接殺傷を引き起こすことが実証されているように、培養物から除去されなければならない。CD4磁気ビーズ濃縮(ステップ3.10)を用いて、DPおよびCD4+CD8-中間単一陽性(ISP)細胞1の両方に対して濃縮し、これは悪影響を及ぼさないことが示された22である。あるいは、フローサイトメトリーによる蛍光活性化細胞選別を行い、DP細胞を単離することができる。しかしながら、磁気ビーズ分離は、フローサイトメトリーによって誘発される機械的ストレスを回避するので好ましい。
活性化媒介性アゴニスト選択を伴わないヒト多能性幹細胞からのCD8αβ+SPT細胞の生成は、その後、3Dマウス間質細胞培養32の使用によって実証されている。しかしながら、生理学的陽性選択は、TCRと自己ペプチド-MHC複合体との相互作用に依存し、これは、胸腺皮質上皮細胞33によって独特に処理され、提示される。さらに、選択ペプチドに対するTCR親和性は、成熟CD8αβ+SP T細胞34のその後の機能能力を決定することが示されている。 現在、ノッチ間質細胞ベースの共培養システムが、生理学的陽性選択に必要な定義された選択ペプチドおよびMHC複合体を提供できることを示唆する証拠はない。
OP9/DLL1を単独で用いて腫瘍抗原特異的T細胞由来hiPSCから生成されたT系統細胞が従来の成熟を経験できないことが、マウスモデルで以前に報告されている。しかしながら、OP9/DLL1システムによって生成されたiPSC由来未熟T細胞は、3D培養システム28、35におけるさらなる生理学的胸腺教育によってナイーブ状のT細胞に成熟させることができる。したがって、OP9/DLL1システムによって生成されたiPSC由来未熟T細胞を産生するためにここに提示されるプロトコルは、生体内での長期持続性が可能な実際のヒト腫瘍抗原特異的胸腺後T細胞をさらに生成する試みに不可欠である。確立された血管化腫瘍を治療するための効率。
開示事項
著者は何の開示もない。
謝辞
アラン・B・フーフィングとエリナ・Hに感謝します。彼はグラフィカルな援助を求めていた。この研究は、国立がん研究所(ZIA BC010763)の壁内研究プログラムと、細胞ベースのがん免疫療法のための壁内NCIがんムーンショットイニシアチブによって支援されました。
資料
| Name | Company | Catalog Number | Comments |
| 10 cm dish | Corning, Inc. | 353003 | |
| Anti-CD3, human | BD Biosciences | Cat# 561812, RRID:AB_1089628 | |
| Anti-CD34, human | BD Biosciences | Cat# 348791, RRID:AB_400381 | |
| Anti-CD4, human | Biolegend | Cat# 344612, RRID:AB_2028479 | |
| Anti-CD43, human | BD Biosciences | Cat# 560198, RRID:AB_1645460 | |
| Anti-CD7, human | BD Biosciences | Cat# 555361, RRID:AB_395764 | |
| Anti-CD8a, human | BD Biosciences | Cat# 555369, RRID:AB_398595 | |
| Anti-CD8b, human | BD Biosciences | Cat# 641057, RRID:AB_1645747 | |
| Anti-TCRb, human | BD Biosciences | Cat# 555548, RRID:AB_395932 | |
| CD28 human monoclonal antibody (15E8), pure functional grade | Miltenyl Biotec | 130-093-375 | |
| CD3 human monoclonal antibody (OKT3), pure functional grade | Miltenyl Biotec | 130-093-387 | |
| CD4 Microbeads, human | Miltenyl Biotec | 130-045-101 | |
| Cell strainer 100 um | Fisher Scientific | 22-363-549 | |
| Fetal Bovine Serum (FBS) | Gemini | 100-500 | |
| Flt-3 ligand | R&D Systems | 427-FL | |
| Gelatin Solution 2% | SIGMA-Aldritch | G1393-100ML | |
| GlutaMAX (100X) | Thermo Fisher Scientific | 35050-061 | L-Glutamine supplement |
| HBSS Mg+Ca+ Phenol-Red Free | Gibco | 14025-092 | |
| Interleukin-2 | R&D Systems | 202-IL | |
| Interleukin-7 | R&D Systems | 407-ML | |
| iTAG MHC Tetramer HLA-A*0201 Mart1 Tetramer -ELAGIGILTV | MBL | Cat#TB-0009-2 | |
| Mart1-hiPSC | Vizcardo et al., Cell Stem Cell 2013 | RIKEN-IMS | |
| Melan-A, MART 1 (26-35) | InnoPep | 3146-0100 | |
| MEM Non-Essential Amino Acids Solution | Gibco | 11140050 | |
| αMEM powder | Gibco | 61100061 | |
| Mouse Embryonic Fibroblasts (MEF) | Thermo Fisher Scientific | C57BL/6 MEF MITC-TREATED 4M EACH; A34962 | |
| OP9/N-DLL1 | Riken Bioresource center | Cat# RCB2927; RRID:CVCL_B220 | OP9/DLL1 |
| Penicillin/streptomycin | Thermo Fisher Scientific | 15140-122 | |
| Phosphate buffered saline pH 7.4 (1x) | Thermo Fisher Scientific | 10010-023 | |
| Primate ES Cell Medium | Reprocell | RCHEMD001 | Human ESC Culture Media |
| Rhok inhibitor (Y-27632 dihydrochloride) | Tocris | 1254 | |
| RPMI 1640 | Gibco | 11875093 | |
| Stem Cell Factor (SCF) | R&D Systems | 455-MC | |
| StemPro | EZPassage | 23181-010 | |
| T2-tumor | ATCC | T2 (174 x CEM.T2) (ATCC® CRL-1992™) | |
| Trypsin-EDTA (0.05%), phenol red | Thermo Fisher Scientific | 25300-062 | |
| Trypsin-EDTA (0.25%), phenol red | Thermo Fisher Scientific | 25200-072 | |
| U Bottom 96 well plate | Corning, Inc. | 3799 |
参考文献
- Brauer, P. M., Singh, J., Xhiku, S., Zuniga-Pflucker, J. C. T Cell Genesis: In Vitro Veritas Est. Trends in Immunology. 37 (12), 889-901 (2016).
- Rosenberg, S. A., Restifo, N. P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 348 (6230), 62-68 (2015).
- Gattinoni, L., et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. Journal of Clinical Investigation. 115 (6), 1616-1626 (2005).
- Rosenberg, S. A., et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clinical Cancer Research. 17 (13), 4550-4557 (2011).
- Crompton, J. G., et al. Lineage relationship of CD8(+) T cell subsets is revealed by progressive changes in the epigenetic landscape. Cellular and Molecular Immunology. 13 (4), 502-513 (2016).
- Henning, A. N., Klebanoff, C. A., Restifo, N. P. Silencing stemness in T cell differentiation. Science. 359 (6372), 163-164 (2018).
- Henning, A. N., Roychoudhuri, R., Restifo, N. P. Epigenetic control of CD8(+) T cell differentiation. Nature Reviews Immunology. 18 (5), 340-356 (2018).
- Vodnala, S. K., et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science. 363 (6434), (2019).
- Restifo, N. P., Gattinoni, L. Lineage relationship of effector and memory T cells. Current Opinion in Immunology. 25 (5), 556-563 (2013).
- Tran, E., et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 344 (6184), 641-645 (2014).
- Gattinoni, L., et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nature Medicine. 15 (7), 808-813 (2009).
- Gautam, S., et al. The transcription factor c-Myb regulates CD8(+) T cell stemness and antitumor immunity. Nature Immunology. 20 (3), 337-349 (2019).
- Klebanoff, C. A., et al. Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clinical Cancer Research. 17 (16), 5343-5352 (2011).
- Klebanoff, C. A., Gattinoni, L., Restifo, N. P. Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy. Journal of Immunotherapy. 35 (9), 651-660 (2012).
- Crompton, J. G., Clever, D., Vizcardo, R., Rao, M., Restifo, N. P. Reprogramming antitumor immunity. Trends in Immunology. 35 (4), 178-185 (2014).
- Crompton, J. G., Rao, M., Restifo, N. P. Memoirs of a reincarnated T cell. Cell Stem Cell. 12 (1), 6-8 (2013).
- Kennedy, M., et al. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Reports. 2 (6), 1722-1735 (2012).
- Vizcardo, R., et al. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell. 12 (1), 31-36 (2013).
- Nishimura, T., et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 12 (1), 114-126 (2013).
- Lo, W., et al. Immunologic recognition of a shared p53 mutated neoantigen in a patient with metastatic colorectal cancer. Cancer Immunology Research. , (2019).
- Timmermans, F., et al. Generation of T cells from human embryonic stem cell-derived hematopoietic zones. Journal of Immunology. 182 (11), 6879-6888 (2009).
- Maeda, T., et al. Regeneration of CD8alphabeta T Cells from T-cell-Derived iPSC Imparts Potent Tumor Antigen-Specific Cytotoxicity. Cancer Research. 76 (23), 6839-6850 (2016).
- Salter, R. D., Howell, D. N., Cresswell, P. Genes regulating HLA class I antigen expression in T-B lymphoblast hybrids. Immunogenetics. 21 (3), 235-246 (1985).
- Snauwaert, S., et al. In vitro generation of mature, naive antigen-specific CD8(+) T cells with a single T-cell receptor by agonist selection. Leukemia. 28 (4), 830-841 (2014).
- Takahama, Y., Suzuki, H., Katz, K. S., Grusby, M. J., Singer, A. Positive selection of CD4+ T cells by TCR ligation without aggregation even in the absence of MHC. Nature. 371 (6492), 67-70 (1994).
- Seki, T., et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell. 7 (1), 11-14 (2010).
- Vodyanik, M. A., Slukvin, I. I. Hematoendothelial differentiation of human embryonic stem cells. Current Protocols in Cell Biology. , (2007).
- Vizcardo, R., et al. Generation of Tumor Antigen-Specific iPSC-Derived Thymic Emigrants Using a 3D Thymic Culture System. Cell Reports. 22 (12), 3175-3190 (2018).
- McNicol, A. M., et al. CD8alpha/alpha homodimers fail to function as co-receptor for a CD8-dependent TCR. European Journal of Immunology. 37 (6), 1634-1641 (2007).
- Themeli, M., Riviere, I., Sadelain, M. New cell sources for T cell engineering and adoptive immunotherapy. Cell Stem Cell. 16 (4), 357-366 (2015).
- Minagawa, A., et al. Enhancing T Cell Receptor Stability in Rejuvenated iPSC-Derived T Cells Improves Their Use in Cancer Immunotherapy. Cell Stem Cell. 23 (6), 850-858 (2018).
- Montel-Hagen, A., et al. Organoid-Induced Differentiation of Conventional T Cells from Human Pluripotent Stem Cells. Cell Stem Cell. 24 (3), 376-389 (2019).
- Takada, K., Kondo, K., Takahama, Y. Generation of Peptides That Promote Positive Selection in the Thymus. Journal of Immunology. 198 (6), 2215-2222 (2017).
- Takada, K., et al. TCR affinity for thymoproteasome-dependent positively selecting peptides conditions antigen responsiveness in CD8(+) T cells. Nature Immunology. 16 (10), 1069-1076 (2015).
- Vizcardo, R., et al. A Three-dimensional Thymic Culture System to Generate Murine Induced Pluripotent Stem Cell-derived Tumor Antigen-specific Thymic Emigrants. JoVE. , e58672 (2019).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved