
Method Article
細胞外マトリックスタンパク質に対する細胞応答を研究するためのマイクロパターニングのためのPRIMOシステムを用いてタンパク質の光誘導分子吸着
* これらの著者は同等に貢献しました
要約
私たちの全体的な目的は、細胞が指示された軸方向の成長につながる細胞外の手がかりを感知する方法を理解することです。ここでは、軸ゾンの成長と経路検査を支配する特定の事象を研究するために、細胞外マトリックス成分の定義されたマイクロパターンを生成するために使用されるタンパク質の光誘導分子吸着の方法論について説明する。
要約
細胞は、細胞外マトリックスの組成と幾何学を含む様々な細胞外手がかりを感知し、細胞自体によって合成され、改造される。ここでは、単一または組み合わせのタンパク質を用いてマイクロパターン化細胞外マトリックス(ECM)基質を作製するパターニング技術としてPRIMOシステムを用いたタンパク質(LIMAP)の光誘導分子吸着法を紹介する。この方法は、優れた再現性を持つミクロン分解能でECMパターンの印刷を可能にします。我々はステップバイステップのプロトコルを提供し、これをニューロン経路発見のプロセスを研究するためにどのように適用できるかを示す。LIMAP には、複数のコンポーネントのパターン化の容易さと、ジオメトリやグラデーションを使用してパターンを生成する機能という点で、既存のマイクロ印刷方法に比べて大きな利点があります。プロトコルは、細胞の運命と細胞の行動に対するほぼすべての化学成分の寄与を研究するために容易に適応することができる。最後に、発生する可能性のある一般的な問題とその回避方法について説明します。
概要
近年、生物科学は物質科学の進歩を生かしてますます進歩を遂行しています。顕著な例の一つは、細胞増殖などの細胞応答を研究するために使用することができる基板のマイクロパターニングです1,2分化3,4,5,6、セルの移行7,8,9とパスファインディング10,11.多光子励起光化学など、基板のマイクロパターニングを可能にする技術は数多く用意されています。12, AFM ディップペンナノリソグラフィー13、ピンおよびインクジェット直接印刷14、電子ビームリソグラフィ15またはマイクロ流体16.しかし、生物分野で広く使用されている2つの技術は、マイクロコンタクト印刷です。17,18,19またはレーザー支援パターニング3(図 1).レーザーアシストパターニングは、マイクロコンタクト印刷と比較して、タンパク質およびPEG安定性およびパターン上の細胞閉じ込めの点でより信頼性の高い結果を提供すると考えられています。20.ここで説明するマイクロパターニングのためのより新しいアプローチは、タンパク質の光誘発分子吸着の使用です21(リマップ、図 1D)を使用して、市販のシステム(PRIMO、材料の表).各方法には、以下で簡単に説明する利点と制限があります。マイクロコンタクト印刷では、リソグラフ付きマスタから生成される必要なマイクロフィーチャーを使用して、PDMS 金型 (スタンプ) を使用します。切手は選択したタンパク質でインキュベートされ、細胞培養基板に移される(スタンプ)。18(図 1A).レーザーアシストパターニングは、UV光を使用して防汚フィルムをクチクします22,23,24,25、その後、目的のタンパク質でコーティングすることができる領域を公開する(図 1B).フォトパターニングアプローチで達成される解像度はミクロンの範囲内にありますが、25,26これらの技術のほとんどは、サンプルと接触するか、顕微鏡目的の物体面に位置するフォトマスクを必要とします。23,27,28.マイクロコンタクト印刷とフォトパターニングの両方におけるマスクの要件は、制限される場合があります。特定のマスクは、すべての幾何学的パターンとサイズに必要であり、発生にコストと時間がかかる場合があります。これらの手法とは対照的に、LIMAP はマスクを必要としません(図 1D).LIMAP に PRIMO システムを使用すると、機器の購入が必要なため、最初はコストが高くなる可能性があります。しかし、オープンソースソフトウェアは、任意のジオメトリのパターンを設計するために使用され、はるかに自由を与え、タンパク質濃度勾配の使用を含むより複雑な実験を可能にします。PRIMOレーザーはデジタル制御されたマイクロミラー装置(DMD)によって制御され、ユーザー定義の幾何学の任意の数のパターンを作成するために指示される。LIMAPは、培養面に細胞の付着を防ぐ分子でコーティングする必要があります。ポリエチレングリコール(PEG)は、このような「防汚」試薬として最も一般的に使用されます。それはガラスまたはプラスチック表面に密な抗粘着性フィルムを形成する29.その後、光化機構を介してPEGフィルムを高精度に除去できるフォトイニシエータが追加されます。30DMDの制御下で紫外線への局所的な露出によって。これらのPEGフリー領域は、レーザーエッチング表面に吸着するタンパク質でコーティングすることができ、マイクロパターンを生成します。レーザーパワーを変化させることにより、異なる量のPEGを表面から除去し、ユーザーがタンパク質勾配を生成できるようにします。PEG除去とコーティング手順を繰り返して、同じマイクロウェル内に2つ以上の異なるタンパク質を含むパターンを作成することができます21.生成されたマイクロパターンは、細胞の接着面を提供し、細胞の挙動の研究を可能にします。我々の研究では、神経細胞株(CAD(カテコラミン作動性-分化)細胞のニューライトまたは軸線の経路を研究するためにマイクロパターニングを使用しています。31)または原発ラット後根神経節(DRG)ニューロンは、それぞれ。ここでは、LIMAP のステップ バイ ステップ プロトコルの概要を説明します。図 2)市販のPRIMOシステムと付属のレオナルドソフトウェアを使用しています。定義された幾何学と複数のタンパク質を用いたパターンの生成にどのように使用できるかを示し、軸道の経路発見を研究するために使用する。発生する可能性のある一般的な問題とその回避方法について説明します。
プロトコル
1. パターンテンプレートのデザイン
注:パターニング用のテンプレートは、デジタル描画ソフトウェア(材料の表)で生成されます。異なる灰色のレベルで描画すると、レーザー強度が決定されます。ソフトウェアを使用してパターン テンプレートを設計すると、任意のジオメトリとグラデーションを使用してパターンを迅速に生成できます (図 3)。
- 描画ソフトウェアを使用して、目的のパターンテンプレートをデジタルで描画します。1824 ピクセルの長さと 1140 ピクセルの幅のイメージ サイズを選択します (このスタディでは、長さ 415 μm と幅 260 μm に対応します)。パターン テンプレートを 8 ビット Tiff ファイルとして保存します。
注:テンプレートを生成するためのステップバイステッププロトコルは、マイクロパターニング装置(材料のテーブル)を商品化するブランドへの要求に応じて利用可能です。
2. プラズマ洗浄
注:最適な結果は、すべての有機物を除去し、表面を活性化するパターニングの前に表面のプラズマ洗浄を必要とします。この場合、周囲空気は表面活性化のために十分である。プラズマクリーナー(材料の表)は、1000-1300 mTorrのプロセス圧力と1-5分間29.6 Wの電力で使用されました。
- 20mmの内側の井戸サイズと0.16-0.19 mmのガラス厚のガラス底皿/esを使用してください。複数の条件をテストするには、6ウェルガラス底皿を使用します。それ以外の場合は、単一のウェルガラス底皿(材料のテーブル)を使用してください。
注:プラズマ洗浄装置を製品化するブランドへの要望に応じて、プラズマ洗浄のためのステップバイステッププロトコル(材料の表)を利用できます。
3. パッシベーション
注:このステップは、ガラス表面へのタンパク質吸着を防ぐ防汚フィルムを生成します。PEGは、防汚剤としてタンパク質吸着29に対して高い耐性を提供する。LIMAP は、フォト イニシエータを使用して、UV フォトシジョンを通じて PEG をローカルに除去します。目的のタンパク質/sは、これらのPEGフリー表面21に吸化し、マイクロパターンを生成する。
- PLL-PEG を使用したパッシベーション
- 無菌条件下では、PDMSステンシル(図4A、B、材料表を参照)を切断して、内側のガラス底部によく収まるようにし、生殖不能鉗子で内側のマイクロウェル充填物を取り除きます。 鉗子を使用してよくガラスにステンシルを貼ります。
- ステンシルがガラスによく固く付き、パッシベーションプロセス中に漏れを引き起こす可能性のある気泡の形成を防ぎます。
注: PDMS ステンシルは、公開されたプロトコル18,32を使用して社内で製造することもできます。 - リン酸緩衝生理食べ物(PBS)にPLL-PEG溶液(材料表、0.1mg/mL)を調製します。各マイクロウェルに20μLのPLL-PEG溶液を加え、室温で1時間インキュベートします。
- マイクロウェルから15μLのPLL-PEGを取り出し、乾燥させることなく20μLのPBS(材料の表)で5回洗います。
注:常に洗い出しの間に約5 μLのPBSを残してください。その小さな容積を考えると、マイクロウェルは特に乾燥の影響を受けやすい。乾燥すると、品質の悪いマイクロパターンになります。 - 培養皿をPBS(井戸当たり3mL)で最大3日間4°Cに保つか、次のステップ(ステップ3.1.5)を続けます。
- 1つのマイクロウェルから18 μLのPBSを取り外します(例えば、左上のマイクロウェルを参照)、5 μLの光イニシエータ(PLPP、材料の表)を追加し、残りのマイクロウェルに20 μLのPBSを残します。 PLPPを使用したこのマイクロウェルは、システムキャリブレーションステップ(ステップ4)中に参照パターンを作成するために使用されます(図4D、Eを参照)。PLPPを光から保護してください。
- 長続きがするPEG-SVAとのパッシベーション
注:1)ポリL-リジンにリンクされたPEG(PLL-PEG、ステップ3.1)または2)PEG-コハク酸N-ヒドロキシシニミド(PEG-SVA)を使用することができます。どちらか一方または他方のパッシベーション オプションを選択する決定は、ストレージ・オプションによって異なります (ステップ 10 を参照)。PEG-SVAでインキュベートされた培養皿は、2倍のパッシベーションとフォトパターニング時間を必要とします。- 手順 3.1.1 で説明するようにステンシルを準備します。
- 各マイクロウェルに0.01%ポリL-リジン(PLL、材料表)の20 μLを加え、室温で30分間インキュベートし、PLLでプレコートします。
- マイクロウェルから15μLのPLLを取り出し、ウェルを乾燥させることなく、1M HEPESバッファー(材料のテーブル)の20 μLで3回洗浄します。
注:常に洗い出しの間に約5 μLのHEPESまたはPBSを残してください。その小さな容積を考えると、マイクロウェルは特に乾燥の影響を受けやすい。乾燥すると、品質の悪いマイクロパターンになります。 - PEG-SVA ソリューションを準備します。PEG-SVA溶液(HEPESバッファ1Mの50mg/mL)は、使用直前に毎回新鮮に調製する必要があります。マイクロウェルあたり20 μLのPEG-SVAを準備します。
- HEPES バッファは 8 ~ 8.5 の間の pH を持つ必要があります。pH紙でPEG-SVA溶液を調製する前にHEPES pHをテストします。精密スケールを使用して遠心管のPEG-SVAの重量を量る。溶かされるまでHEPESバッファーと渦30sを追加します。PEG-SVAは、溶液が透明な場合に完全に溶解します。
注:SVAは、以前にコーティングされたPLLへのPEGの結合を可能にするエステルです。HEPES バッファが PEG-SVA に追加されると、SVA の半減期は 15 分で、すぐに使用する必要があります。 - 各マイクロウェルに20 μLのPEG-SVA溶液を加え、室温でインキュベートし、1時間でマイクロウェルから15μLのPEG-SVAを取り出し、ウェルを乾燥させることなく20 μLのPBS(材料のテーブル)で5回洗浄します。
- 培養皿を長期保存用に準備するか(最大1ヶ月、ステップ10.2を参照)、または次のステップ(ステップ3.2.8)に進みます。
- 1つのマイクロウェルから18 μLのPBSを取り出します(例えば、左上のマイクロウェルを参照)、図4Dを参照してください、PLPP(材料の表)の5 μLを追加し、残りのマイクロウェルに20 μLのPBSを残します。このマイクロウェルは、参照パターンを作成するために使用されます(Fi gure 4D、Eを参照)。マイクロウェルの全面にPLPPが均質であることを確認します。PLPPを光から保護してください。
4. システムキャリブレーション
注:これらのステップでは、レーザーの焦点は、培養皿の特定のタイプに調整されます(ステップ4.1)。基準パターンは1つのマイクロウェル(ステップ4.2)のみで生成され、その後タンパク質溶液(ステップ4.3)でインキュベーションを行い、レーザーの最適な焦点条件を確保し(ステップ4.4)、鋭く定義されたパターンを得るために必要です。
- レーザーキャリブレーション
注:キャリブレーションプロセス中に、キャリブレーションレーザー画像は蛍光ハイライトガラス表面(キャリブレーションウェル、蛍光蛍光蛍光光機でマークされた図4C)に投影され、顕微鏡。- 蛍光蛍光灯(材料の表)を使用して、空の内側のガラスをよくマークします。
注:キャリブレーションウェルは、マイクロパターンが生成される培養皿と同じガラス厚さ(0.16~0.19mm)でなければなりません。6ウェルガラス底皿を使用している場合は、空の井戸をキャリブレーションに使用することができ、無菌条件下で蛍光蛍光灯でマークする必要があります。 - 顕微鏡、ステージ、コンピュータのスイッチを入れます。PRIMOマイクロパターニング装置のスイッチ、マイクロマネージャとレオナルドソフトウェアを開きます。レオナルドソフトウェアは、プラグインの下でマイクロマネージャを介して動作します.材料の表の機器やソフトウェアのブランド/カタログ番号を確認してください。
- レオナルドの初期メニューで、キャリブレーションを選択します。専用の PRIMO フィルタ キューブがフィルタ タレット内の正しい位置 (光パス) にあることを確認します。顕微鏡とレオナルドソフトウェアの両方で20Xの目的(0.75 DIC Sプランフルー、フェーズリングなし)を選択します。
注: このプロトコルは、レオナルドバージョン4.4に調整されています。プロトコルは、他のバージョンの調整が必要な場合があります。 - 以前に強調表示されたキャリブレーション (図 4C)を目標の上に配置します。カメラパスを選択します。PRIMOロゴとタグラインの両方のレーザー投影が焦点を合わせるまで、目的の焦点を調整します。
- デフォルトのカメラの露出時間を 25 ミリ秒のままにして、レーザー強度を調整して、PRIMOロゴ プロジェクションの文字Iを灰色で表示し、残りの文字を白で表示します。
- 焦点面のZ位置を記録し、後でキャリブレーションのZ位置と呼ばれる(試料に対する目的の高さ)。これは、参照パターンの生成後に得られる最適なフォーカスに近似されます(ステップ 4.2-4.4 を参照)。
- 蛍光蛍光灯(材料の表)を使用して、空の内側のガラスをよくマークします。
- 参照パターン
- フォトイニシエータ(参照パターンマイクロウェル、図4D)を含むマイクロウェルで培養皿を目的の上に配置し、レオナルドソフトウェアでパターンを選択します。
- カメラを透過した光でマイクロウェルのエッジを視覚化し、右側のメニューからROIシンボルを選択します。ROI 円の直径を 4000 μm に設定し、デジタル ROI のエッジを現在のマイクロウェルのエッジに合わせます。
- マイクロウェルのエッジの周りにステージを移動することにより、デジタルROIと現在のマイクロウェルの間に正確な重複があることを確認します。ROI位置はステージの動きに結合されます。
- 目的の位置にROIをロックするには、[レオナルドソフトウェアでロック]を選択します。透過光をオフにします。
- 設計単位として ROI に投影される目的のパターン テンプレートをアップロードするには、PRIMOを選択します (図 5を参照)。パターンは、アクションと呼ばれるドロップダウン リストに表示され、ソフトウェアの[アクション] メニューに表示されます。
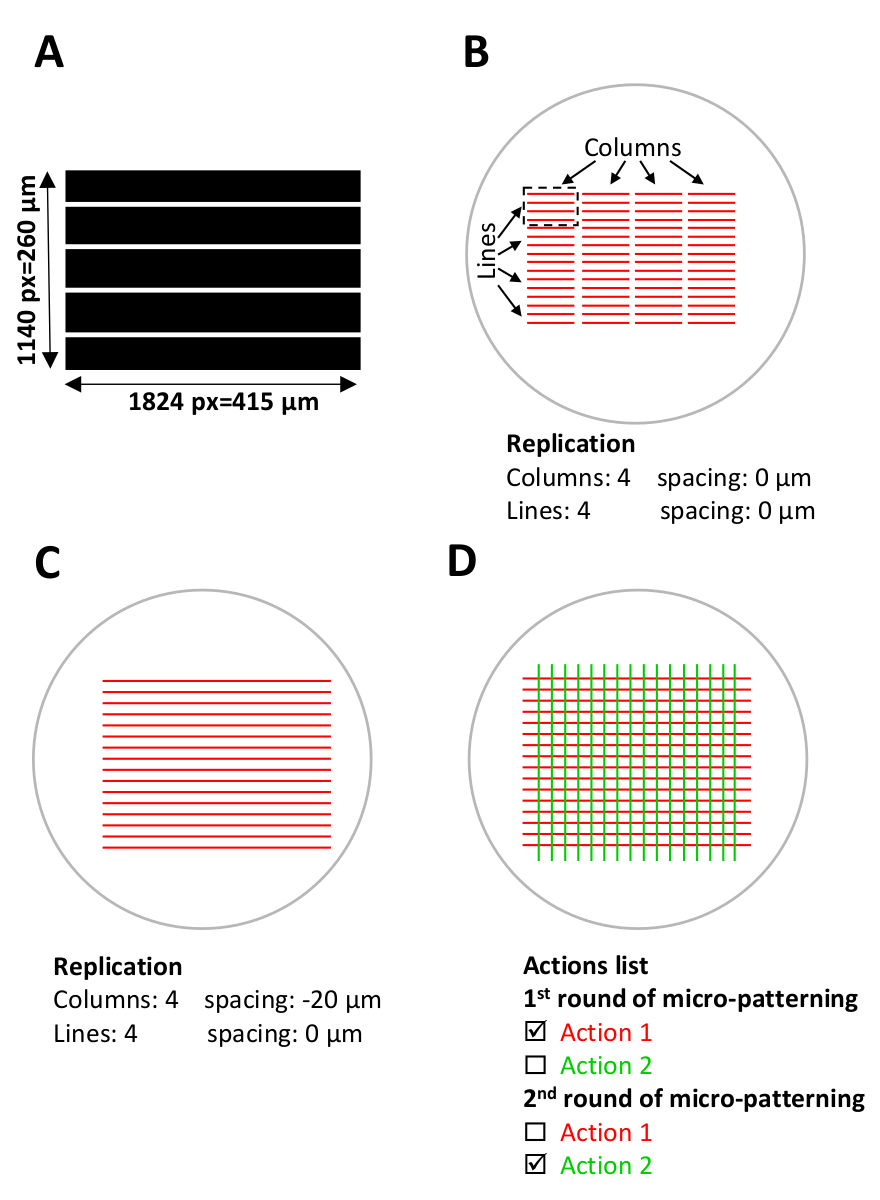
注: パターン テンプレートは、ソフトウェアにテンプレートを読み込む前に、事前に設計し (手順 1 を参照)、8 ビット Tiff ファイルとして保存する必要があります。 - 参照パターンには小さなパターンのみが必要です。たとえば、3 行、1 列(図 4Eおよび図 5Bを参照)。[レプリケーション]メニューで、必要な列数と行数 (Leonardo ソフトウェアの行)を設定します。[更新]をクリックして、パターンデザインのデジタル プレビューを確認します。
- [複製]メニューでレーザー線量を設定します。この設定で最適なレーザー線量とPLL-PEGを使用すると、1390 mJ/mm2です。
注:レーザーパワーは5-7.5 mW/mm2の間で変化することができます。この場合、1390 mJ/mm2のレーザー線量を使用して、各設計ユニットをパターン化するのに約30sかかります7.5 mW/mm2です。培養皿表面がPEG-SVA(PLL-PEGと比較して約2倍のレーザー線量)でパッシベートされている場合は、より高いレーザー用量が必要な場合があります。これは事前にテストする必要があります。 - マイクロウェルの周辺領域(例えば、上部)をマイクロウェルのパターン生成(中央領域)の主領域から離して(図4Eを参照)、[ロック]を選択します。パターンが表示されるまで待ちます。
- キャリブレーションの Z 位置にフォーカスを調整します(ステップ 4.1.6 を参照)。
注:別のユーザーがフォトパターニングラウンド間で同じ顕微鏡を使用している場合は、追加のシステムキャリブレーションステップを行うことをお勧めします。 - パターニングを開始するには、再生シンボルを選択します。ソフトウェアでレーザーがオンであることを確認します。パターニングプロセスが完了するまで待ちます。パターニング期間は[推定時間]パネルに表示されます。レオナルドソフトウェアバージョン4.4でパターニングは、すべてのアクションがビジュアライゼーションメニューで青色に表示されたときに完了します。
- 参照パターン上のタンパク質インキュベーション
- 無菌条件下で、基準パターンマイクロウェルを20μLのPBSで3回洗浄し、PLPPを除去します。
- 蛍光標識ECMタンパク質(PBSで10μg/mLラミニン、フィブロネクチンまたはフィブリノーゲン、材料表およびステップ6)を参照パターンマイクロウェルに20 μLを添加する。光から保護された(タンパク質に応じて)10〜20分間室温でインキュベートする(アルミ箔で皿を包む)。
- インキュベーション後、18 μLのタンパク質溶液を取り出し、20μLのPBSで3回洗浄する。PBSの20 μLで参照パターンを作成するために使用されないマイクロウェルを保持します(図4D,Eを参照)。
- 最適なレーザーフォーカスの可視化と設定
- エピ蛍光顕微鏡、20X目的および適切なソフトウェアを用いて参照パターンを可視化する(材料の表でこの研究で使用されるソフトウェアをチェックする)。参照パターンは、参照パターンが生成された周辺領域(例えば、上部ウェル領域)に表示される必要があります(図4Eを参照)。
- カメラ パスを通じてパターン エッジにフォーカスを調整します。参照パターンに最適なフォーカスに従って Z 位置を記録します。この調整されたZ位置は、後続のパターニングに使用される最適なレーザーフォーカスになります。
5. ソフトウェアのセットアップとフォトパターニング
注:システムのキャリブレーションが達成されると(ステップ4)、ユーザーは、それぞれに1つまたは複数のタンパク質のパターンを生成するオプションを使用して、フォトパターニング用に必要なパターンテンプレート(テンプレート構成、図5)をアップロードします。マイクロウェル。マイクロパターニングプロセスには、フォトパターニングおよびタンパク質インキュベーションステップが含まれます(図2参照)。
- 無菌条件下で、すべてのマイクロウェルから18μLのPBSを取り出し、各マイクロウェルに5μLのPLPPを加えます。PLPPがマイクロウェルの全面に均質であることを確認します。
- カルチャ皿を参照パターンマイクロウェル(図4D,E)を目標の上に配置し、レオナルドソフトウェアでパターンを選択します。
- カメラパスを透過した光でマイクロウェルを視覚化し、ROIシンボルを選択します。ROIの直径を4,000 μmに設定し、デジタルROIのエッジを現在のマイクロウェルのエッジに重ね合います。[ロック]を選択します。
注: ROI の形状と直径は、使用する PDMS ステンシルの設計とサイズによって異なります。たとえば、5,000 x 5,000 μm PDMS 二乗ステンシルを使用する場合は、5,000 x 5,000 μm 平方 ROI を使用します。 - 皿のマイクロウェルごとに重なり合うステップ(ステップ5.3)を繰り返します。完了後、透過光をオフにします。
- 参照パターン領域から離れた参照パターンマイクロウェルの中心に移動し、PRIMOを選択して、設計単位として ROI に投影される目的のパターン テンプレートをアップロードします (図 5参照)。パターンは、アクションと呼ばれるドロップダウン リストに表示され、ソフトウェアの[アクション] メニューに表示されます。
注: パターン テンプレートは、テストの前に設計し、ソフトウェアにテンプレートをロードする前に 8 ビット Tiff ファイルとして保存する必要があります。 - マイクロウェル全体にパターンを作成するには、設計ユニットを複製する必要があります。設計単位はマイクロウェル区域の約0.1 mm2をカバーする。[レプリケーション]メニューで、必要な列数と行数 (ソフトウェアの行)を設定します (図 5を参照)。
- 連続パターンを生成するには、列と線の間隔を調整します。本研究では、連続ストライプのパターンは、列間の-20 ~-35 μm 間隔(負の間隔)を使用して得られます。この負の間隔は、設計単位間の重複を作成します (図 5B,C)。
- [複製]メニューでレーザー線量を設定します。この設定で最適なレーザー線量とPLL-PEGを使用すると、1390 mJ/mm2です。パターニング期間は[推定時間]パネルに表示されます。
注:この場合、レーザーパワーは7.5 mW/mm2であり、1390 mJ/mm2の用量を使用して、各設計ユニットをパターン化するために約30sを取ります。たとえば、4 列と 4 行 (約 1.6 mm2)でレプリケートされた設計単位 (0.1 mm2)は、パターン化するのに 8 分かかります。培養皿表面がPEG-SVA(PLL-PEGと比較して約2倍のレーザー線量)でパッシベートされている場合は、より高いレーザー用量が必要な場合があります。 - [ロック]を選択し、パターンが仮想的に表示されるまで待ちます。
- パターンのパラメータを更新するには、関連するアクションをクリックし、パラメータのロックを解除して更新します。パターンの更新が完了したら、[もう一度ロック]を選択します。
- 同じマイクロウェル内の複数のタンパク質のパターニング(シーケンシャルマイクロパターニングラウンド)は、パターンの正確な位置合わせを必要とします。このようなアライメントを実現するには、必要なパターンテンプレートのすべてのセットを同時にアップロードします(第1および第2のフォトパターニングラウンドのパターン)。
- パターン テンプレートの複製パラメータと線量パラメータを設定します。パラメータを設定すると、パターンは[アクション]リストにアクションとして表示されます。このテンプレート構成をソフトウェアのファイルとして保存します (図 5Dを参照)。
- [アクション]リストで、最初のパターニング ラウンド中にパターン化する特定のアクションのみを選択し、2 番目のパターニング ラウンド中にパターン化されるアクションの選択を解除します (図 5Dを参照)。
- ステップ 4.2 で参照パターンが生成された領域に移動します (たとえば、参照パターンマイクロウェルの上部領域は、図 4E を参照してください)。フォーカスを最適な Z 位置に調整します(ステップ 4.4 で取得)。
注:使用されている顕微鏡に完璧なフォーカスシステムを選択することを強くお勧めします(可能な場合)。 - パターニングを開始するには、再生シンボルを選択します。パターニング期間は[推定時間]パネルに表示されます。レオナルドソフトウェアバージョン4.4で、すべてのアクションがビジュアライゼーションメニューで青色に表示されると、パターニングが完了します。
6. タンパク質インキュベーション
注:マイクロウェルは、ECMタンパク質(好ましくは蛍光標識)でインキュベートされます。これらは、手順 5 で説明するフォト パターニング プロセスを通じて PEG が切り分けされた領域にのみバインドされます。各ウェルには4つのマイクロウェルを備えたPDMSステンシルが含まれており、各マイクロウェルで異なるタンパク質のインキュベーションなど、4つの異なる条件を同時にテストすることができます(図4D参照)。
- マイクロパターンを視覚化するために、蛍光標識タンパク質(例えば、ラミニン、フィブロネクチン、またはフィブリノーゲンを赤色または緑色蛍光色素に結合)を使用して、マイクロパターンを視覚化します(ステップ6.7および9.4を参照)。あるいは、標識されていないタンパク質を吸着し、免疫蛍光を用いて後の段階で可視化することができる。
注:ECMタンパク質(例えば、フィブロネクチン)は、既存のプロトコル33および市販の蛍光標識キット(すなわち、Alexa 488標識キット)を使用して標識することができ、または容易に標識(例えば、共役フィブリノゲン-488または)を購入することができます。ラミニンレッド蛍光ローダミン、材料の表を参照してください) - 無菌条件下で、ECMタンパク質の所望の濃度(PBSで10μg/mLラミニン、フィブロネクチンまたはフィブリノーゲン、材料の表およびステップ4.3を参照)を調記します。
注:蛍光標識されたECMタンパク質は光に敏感であり、光(アルミ箔で皿をラップ)から保護する必要があり、常に氷の上に保管する必要があります。 - 無菌条件下で、マイクロウェルを20μLのPBSで3回洗浄し、PLPPを除去します。
- すべてのマイクロウェルから18μLのPBSを取り出し、各マイクロウェルに20 μLのECMタンパク質溶液を加えます。室温で20〜30分間インキュベートし、光から保護します。
注:最適なコーティングインキュベーション時間は、タンパク質の種類と濃度によって異なる場合があります。 - インキュベーション後、ECMタンパク質溶液の15μLを取り出し、20μLのPBSでマイクロウェルを3回洗浄します。
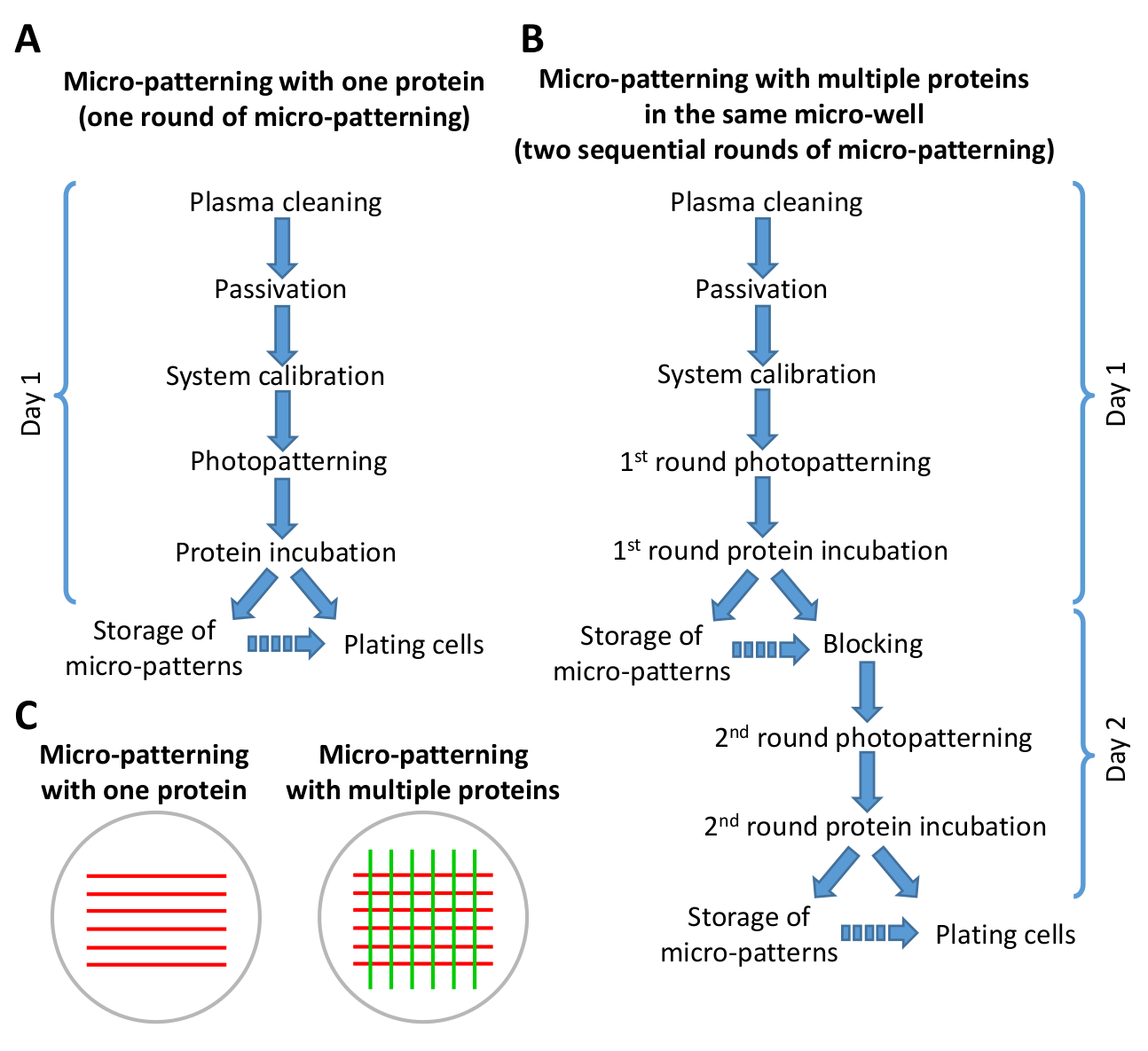
注:常に洗い出しの間に約5 μLのPBSを残してください。 - たんぱく質を1つ(マイクロパターニングの1ラウンド)でパターニングする場合は、培養皿の貯蔵(ステップ10)または細胞めっき(ステップ11、図2参照)のいずれかに進みます。同じマイクロウェル内で異なるタンパク質を用いて第2ラウンドのマイクロパターニングを行う場合は、培養皿の貯蔵(ステップ10)または非特異的結合部位の遮断のいずれかに進む(ステップ7、図2を参照)。
- オプションの品質管理ステップ:めっき細胞の前にエピ蛍光顕微鏡を使用して印刷パターンを視覚化し、画像化します。適切な蛍光チャネルを選択し、それに応じて露光時間を調整します。
注:実験間のパターンの蛍光強度を比較するには、同じタンパク質に対して同じ露光時間を使用することが不可欠です。
7. 非特異的結合部位の遮断(複数のタンパク質パターンのみ)
注:同じマイクロウェル内の複数のタンパク質を用くマイクロパターニングには、順次パターニング工程が含まれます(図2参照)。ブロッキング剤(PLL-PEGまたはBSA)をマイクロウェルに添加してクロス結合を防ぎ、2番目のインキュベートされたタンパク質(ステップ9)が最初のインキュベートされたタンパク質(ステップ6)に結合する際に起こり、パターン内のタンパク質の混合物を回避します。
- 無菌条件下では、クロスバインディングを防ぐためのブロッキングステップとして、PLL-PEG(PBSでは0.1mg/mL)またはBSA(PBSでは1%BSA)を追加します。
注:ブロッキング効率は、使用されるタンパク質の性質およびそれらの間の親和性によって異なる場合があります。PLL-PEG と BSA の両方をブロッキング剤として事前にテストすることをお勧めします (図 10D-I)。 - ブロッキング剤を室温で1時間インキュベートし、光から保護します。ブロッキング剤の15 μLを取り外し、PBSの20 μLですべてのマイクロウェルを3回洗浄します。常に洗い出しの間に約5 μLのPBSを残します。
- すべてのマイクロウェルから18μLのPBSを取り出し、各マイクロウェルに5μLのPLPPを加え、PLPPがマイクロウェルの全面に均質であることを保証します。
8. フォトパターニングの第2ラウンド(複数のタンパク質パターンのみ)
注:フォトパターニングとタンパク質インキュベーションの最初のラウンドの後、マイクロパターンが生成されます。フォトパターニングの第2ラウンドでは、2番目のタンパク質のパターンが同じマイクロウェルで生成されます(図2Cおよび図5Dを参照)。ソフトウェアで、このラウンド中にパターン化される正しいパターン テンプレート (アクション) を選択します (図 5Dを参照)。
- 最初のマイクロウェル(図4D)に移動します。デジタルROIとマイクロウェルの間に適切な重複を確認します。
- 以前に保存したテンプレート構成を読み込み(ステップ5.12)、2回目のフォトパターニングラウンド中にパターン化するアクションを選択します(最初のラウンドからアクションの選択を解除し、図5Dを参照)。
- パターニングを開始するには、再生シンボルを選択します。ソフトウェアでレーザーがオンであることを確認します。
9. タンパク質インキュベーションの第2ラウンド(複数のタンパク質パターンのみ)
注:プロトコルのこの部分では、蛍光標識タンパク質/sは、フォトパターニングの第2ラウンドの後に培養皿上でインキュベートされます。
- 無菌条件下で、PBSの20 μLでマイクロウェルを3回洗浄し、PLPPを除去します。18 μL の PBS を取り出し、各マイクロウェルに 20 μL の ECM タンパク質溶液を追加します。室温で20~30分間インキュベートし、光から保護します。
注:最適なコーティングインキュベーション時間は、タンパク質の種類と濃度によって異なる場合があります。 - インキュベーション後、ECMタンパク質溶液の15μLを取り出し、20μLのPBSでマイクロウェルを3回洗浄します。常に洗い出しの間に約5 μLのPBSを残します。
- 培養皿の貯蔵(ステップ10)または細胞めっき(ステップ11、図2参照)のいずれかに進みます。
- オプションの品質管理ステップ:エピ蛍光顕微鏡を使用して、めっき細胞の前に印刷パターンを視覚化し、画像化します。適切な蛍光チャネルを選択し、露光時間を調整します。
注:実験間のパターンの蛍光強度を比較するには、同じタンパク質に対して同じ露光時間を使用することが不可欠です。
10. マイクロパターンの保存
注:吸着タンパク質を含むマイクロパターンは、プロトコルの異なるステップ中に保存することができます(図2参照)。複数のタンパク質を用いたマイクロパターニングの場合、マイクロパターンは、マイクロパターニングの最初のラウンドの後、またはマイクロパターニングの2つの順次ラウンドが完了した後に保存することができます(図2B参照)。
- PLL-PEGでパッシベートした場合は、マイクロパターンをPBS(ウェル当たり3mL)で最大3日間4°Cに保存します。
- 培養皿をPEG-SVAでパッシブした場合、マイクロパターンを最大1ヶ月間保存できます。そのために、二重蒸留脱イオン水で集中的にマイクロパターンをすすいで、アルゴンまたは窒素の無菌エアガンで乾燥させますが、通常の空気も使用できます。乾燥後、マイクロパターンは最大1ヶ月間4°Cで保存できます(PRIMOシステムサポートチーム、パーソナルコミュニケーション)。
11. めっき細胞
注:次のステップの間に、細胞は、調製されたマイクロパターン培養皿/esにめっきされます。これらの研究では、ニューロン細胞株(CAD細胞)が31を使用する。しかし、このプロトコルは、他の細胞タイプの目的を研究するように調整することができる(必要に応じて細胞めっきプロトコルを調整する)。
- 分化培地を用いて48時間のCAD細胞を分化する(DMEMは1%グルタミンを補充し、血清なしで、1%のペン/ストレップで、材料の表を参照)。
- 内側ガラスに細胞を含む培地のプレート1 mLを、すべてのマイクロウェルを覆い、培養皿を37°C、5%CO2インキュベーターに入れます。
結果
上記のプロトコルに続いて、目的のECMタンパク質/sでコーティングされたマイクロパターン表面をもたらす。これらのパターンを使用して、ニューロンの経路を追跡しています。
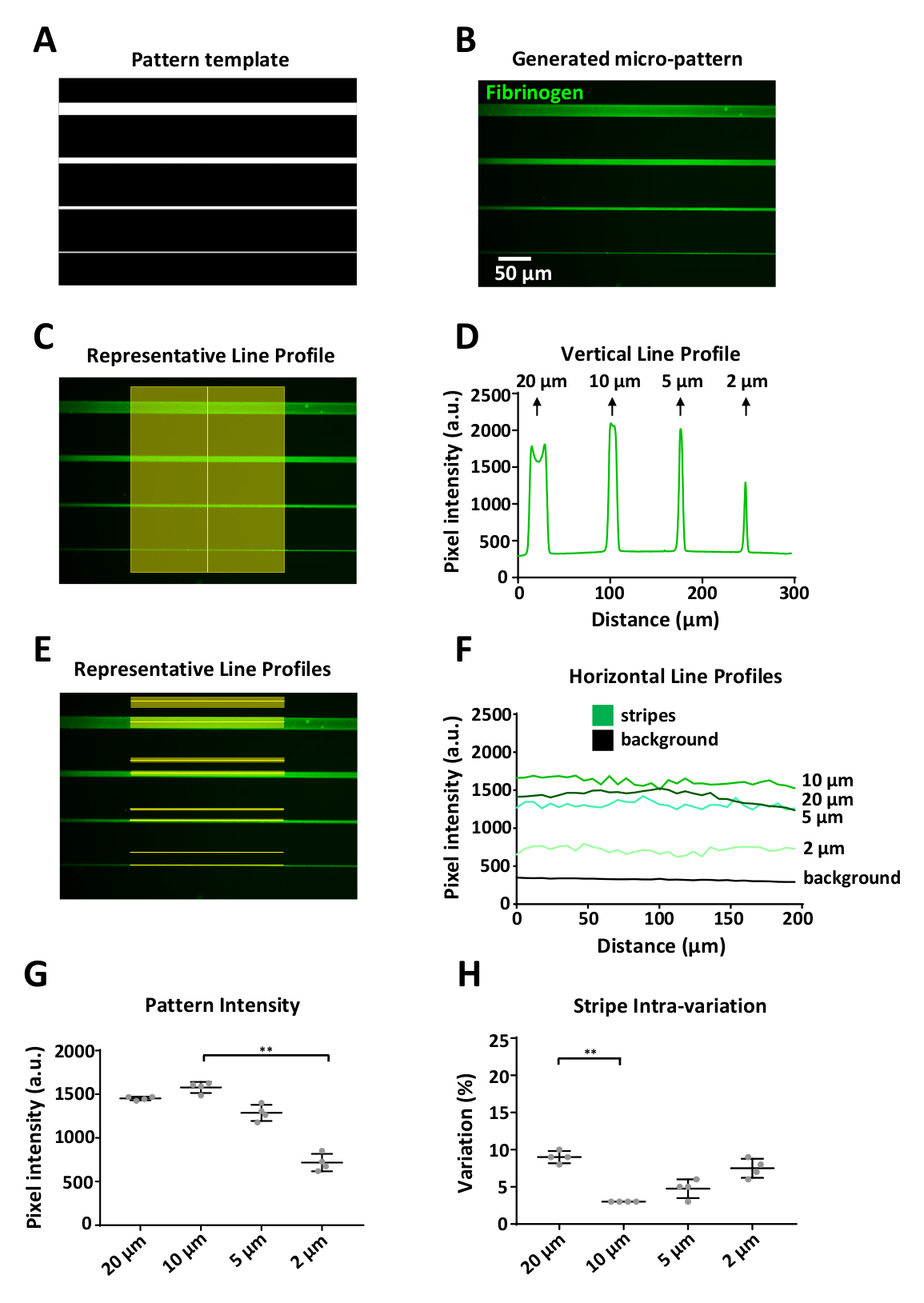
生成されたパターンは、テンプレートを正確に表現する必要があります。図 6に示す例は、1 つの設計単位 (図 5B)を表すデジタル パターン テンプレート (図6A)で、20 ~ 2 μm 幅の定義されたマイクロ パターンがラベル付きでコーティングされた場合に示されています。フィブリノーゲン (図 6B)ImageJを用いて、蛍光強度測定は、ストライプに沿って垂直方向(図6C)と水平方向(図6E)の両方を、各ストライプの上の対応するバックグラウンド領域15μmから得た。背景測定値は、各ストライプ幅のパターン測定値から差し引かれました。
システムの1つの制限は、印刷機能≥20 μmを印刷する際にエッジ効果が観察され、中心と比較してパターンエッジでより高い強度信号を持つ(図6D、最初のピーク)蛍光強度プロファイル)。我々の実験では、分解能限界は約2μmであった。この幅では、大幅な減少(約50%)を観測しましたより広い縞の強度と比較してフィブリノーゲン蛍光強度で(図6F,G)。PRIMOシステムとここで概説したプロトコルを用いてパターニングを行い、4つの個別の複製設計単位から2μm幅で測定された平均蛍光強度の最高標準偏差で再現可能なパターンを生成しました(図6)G)パターンストライプ内のバリエーションも低いことがわかった。変動係数は3~10%の範囲で、20μmと2μmのストライプが最大の内部変動を有する。これは、それぞれエッジ効果とシステムの解像度制限の結果である可能性があります。これらの測定では、これらの画像の取得に使用される目的から生じる不均一な照明を避けるために、ストライプの中心の強度のみを測定しました(ケラレ、図6E)。
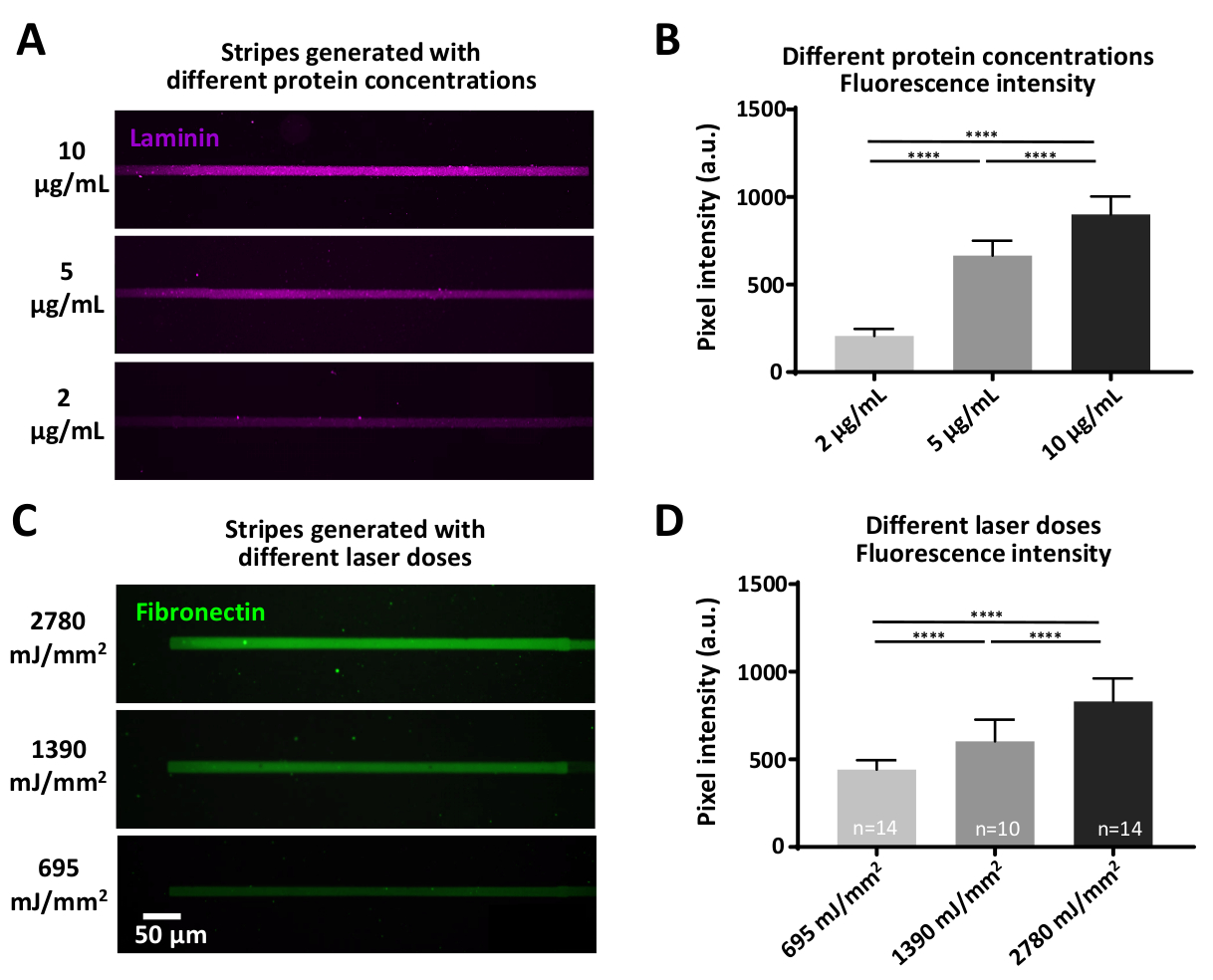
特定の実験には、定義されたタンパク質濃度を必要とする質問が含まれる場合があり、これは2つの方法で達成することができる:1)タンパク質濃度の変化(図7A、B)。ラミニンの異なる濃度を有するインキュベーションは、有意に異なる蛍光強度をもたらす、より高いタンパク質濃度で増加する(図7B)。2)抗粘着フィルム(PEG)をクチクするのに用いられるレーザー線量は変化させることができる。より高いレーザー用量は、より大きな程度に防汚フィルムを除去し、目的のタンパク質のためのより多くの結合部位を生成します(図7C,D)、有意に異なる蛍光強度をもたらし、より高いレーザーで増加します用量 (図 7D)
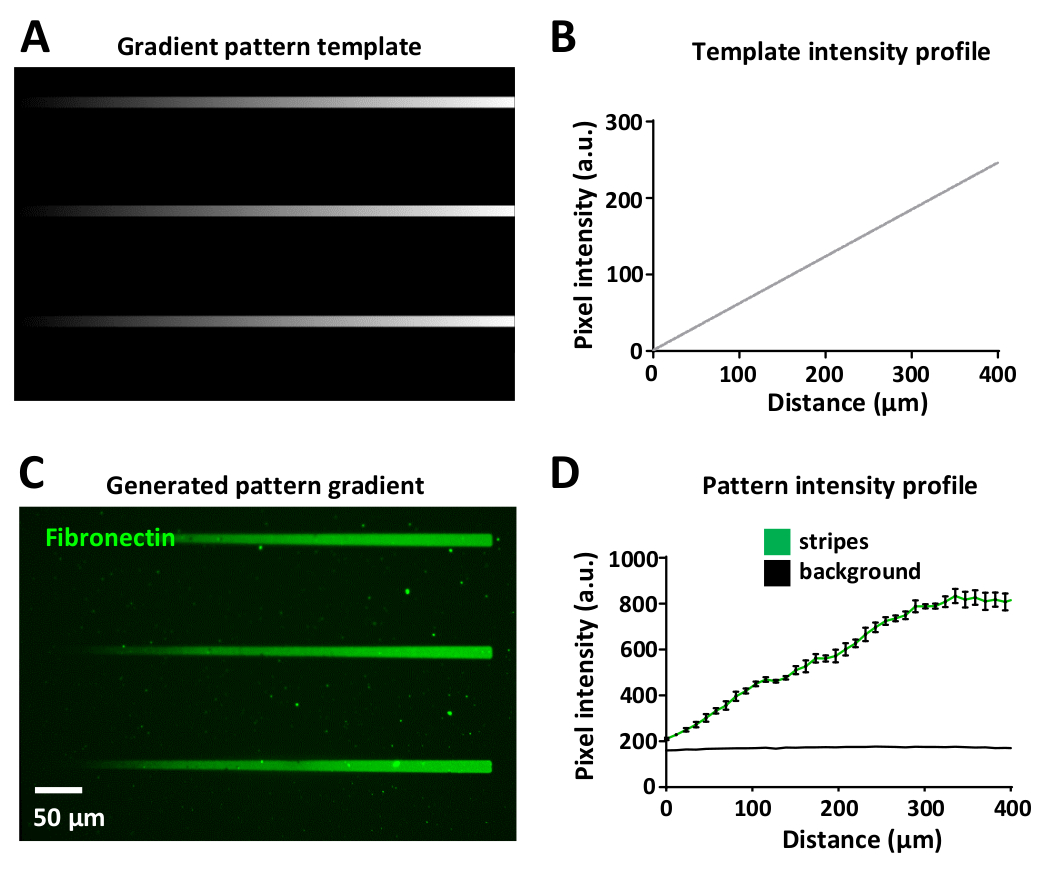
レーザー線量を変化させることで、同じパターン内でタンパク質勾配を生成できます。これは図 8Aに表示され、グラデーション テンプレートは、黒 (レーザーパワーなし) から白 (最大レーザーパワー) まで、さまざまなグレースケール レベルを使用して設計されています。
レーザー強度は、テンプレートのグレースケール レベル(8 ビット 画像では 0 ~ 255 の範囲)に比例し、UV イルミネーションのグラデーションを生成します。グラデーションストライプに沿った蛍光強度プロファイルの測定は、パターンテンプレート(図8B)および生成された勾配パターン(図8C,D)において線形である。これは、同じテンプレートとグラデーションパターン内のすべてのグラデーションストライプの中で再現可能です(図8B,D)。このような勾配の生成は非常に有用であり、細胞がしばしば生理活性タンパク質34、35、36、37の勾配に反応する生体内環境で模倣するのに役立つ。
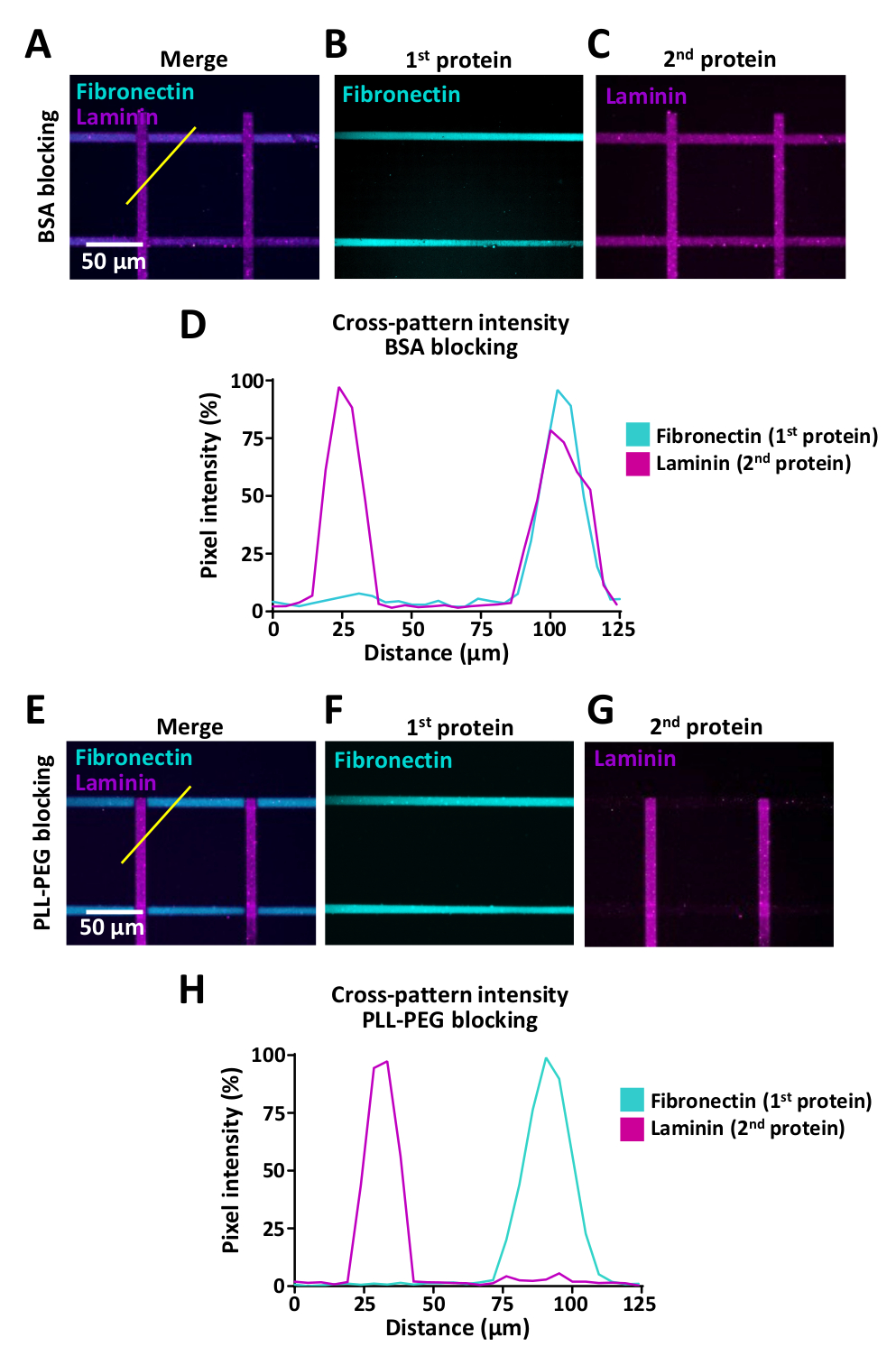
細胞は細胞外環境の変化を感知しますが、細胞がそのような変化に遭遇した場合の細胞行動の研究を可能にするアッセイは限られています。LIMAPは、同じマイクロウェル内の複数のタンパク質を用いるために使用することができる。図9に例を示し、フィブロネクチン(水平)とラミニン(垂直)のストライプでクロスパターンが生成された図9に示す。複数のタンパク質を持つパターンを作成する場合、タンパク質のクロス結合を防ぐために、第1タンパク質インキュベーションと第2タンパク質インキュベーションの間にブロッキングステップを使用することが重要です(ステップ7参照)。ブロッキング効率は、コーティングに使用されるタンパク質の生化学的特性によって異なる場合があり、PLL-PEG(0.1 mg/mL)およびBSA(1%)を含むいくつかのブロッキングバッファーを試験することをお勧めします。このクロスバインディング効果を評価するために、ImageJ(図9)を用いて蛍光強度測定を行い、フィブロネクチンおよびラミニンに対するPLL-PEGバッファー(0.1mg/mL)を用いてクロス結合を劇的に低減できることを示した。クロスパターン(図9D,H)。
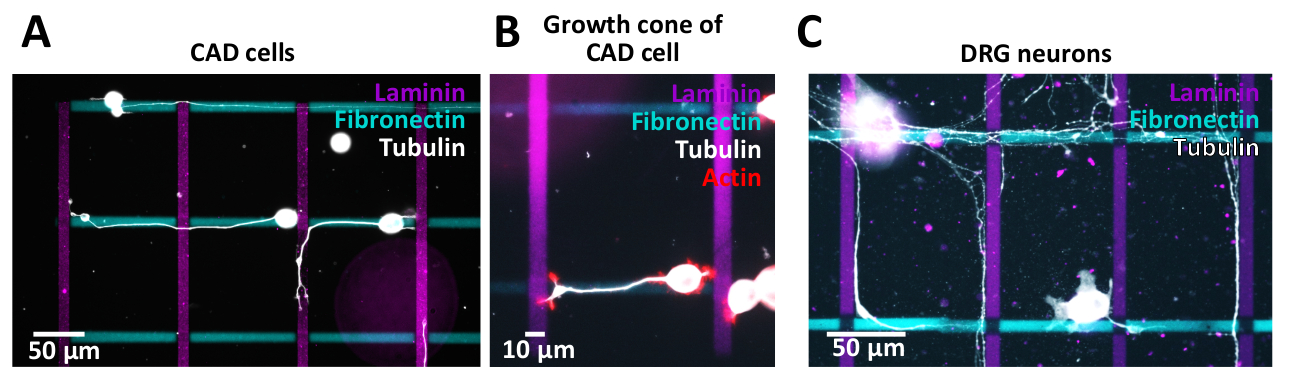
生成されたクロスパターンは、CAD細胞(図10A,B)またはラット後根神経節(DRG)ニューロン(図10C)を用いた細胞アッセイに用いた。彼らのニューライト(CAD)と軸(DRG)は、異なるラインに沿って成長します。CAD細胞は、一次ニューロンと比較して同様のインテグリン発現プロファイルを示し、培養中48時間後にアクチンが豊富な成長コーンを表示するため、ニューロンモデルとして使用され(図10B)、パスファインディングに適しています。研究。
一次ニューロンに対して発生したマイクロパターンの細胞傷害性の影響を調べるために、DRGニューロンは、以前に公開されたプロトコル38に続いてマイクロパターン上で単離および培養された。結果は、一次ニューロンがマイクロパターン環境に耐える(図10C)ことを示している。現在、様々なECMタンパク質が軸質(ニューライト)経路発見にどのように影響するかを研究しています。CAD細胞で見つかった概念の予備的証拠は、DRGニューロンを使用してさらに調査されます。生成されたマイクロパターンの品質を検証するためには、細胞めっきに進む前にパターンエッジが明確に定義されていることを確認するために、蛍光顕微鏡による画像パターンが望ましい。イメージングプロセスでは、後部分析とデータの解釈に影響を与える周辺の黒ずみ効果(ケラネット)を避けるために、顕微鏡とカメラの間の光学的調整を確実にすることが重要です。さらに、パターンイメージをイメージし、パターンイメージからこのイメージを減算するために使用されるのと同じ露出時間を使用して、パターンのない領域のイメージを取得します。
要約すると、良好なマイクロパターン生成のために、タンパク質濃度(図7A,B)、レーザー用量(図7C,D)、タンパク質の背景レベル(図6 E、F、H)および複数のタンパク質を使用する場合の効率的なブロッキングステップ(図9)。最終的には、細胞アッセイから信頼性が高く再現可能なデータを得るためには、LIMAPで生成されるマイクロパターンの品質が不可欠です。

図1:マイクロパターニング技術のスキーム:マイクロコンタクト印刷とレーザー支援パターニング。(A)マイクロコンタクト印刷は、定義されたマイクロ機能を備えたリソグラフ化されたマスターを使用して、目的のタンパク質でインキュベートされたPDMSスタンプを生成します。このタンパク質はガラス表面に移され(スタンプ)され、タンパク質のマイクロパターンが生成されます。(B)レーザー支援パターニング技術には、フォトパターニングおよび直接レーザーパターニングが含まれる。(C)ほとんどのフォトパターニングアプローチでは、特定の位置でPEG防汚面をクリートするために、所望の形状を持つUV光源とフォトマスク(基板表面に接触するか、目的の焦点面内のいずれか)を使用し、定義されたパターンを作成します。その後のタンパク質インキュベーションステップは、レーザー切断領域にのみタンパク質吸着をもたらす。(D)LIMAPは、基板に接触するフォトマスク(すなわち、マスクレスおよび非接触アプローチ)を必要としないフォトパターニング技術である。LIMAPは、光露出領域を切断するレーザーの低用量によって活性化される光イニシエータを使用しています。これにより、順次タンパク質吸着のためのアタッチメント部位が作成されます。(E)ダイレクトレーザーパターニングは、高エネルギー光を使用してPEGフィルムを直接エッチングし、エッチング領域でタンパク質結合を可能にします。この図のより大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図2:マイクロパターニングプロトコルにおけるステップの概要を示すスキーム。(A)1つのタンパク質を用いたマイクロパターニングは、マイクロパターニング(フォトパターニングおよびタンパク質インキュベーション)の1ラウンドのみを含み、8時間以下(B)複数のタンパク質を用いたマイクロパターニングを2回の連続的なラウンドを必要とする。マイクロパターンの数に応じて、1-2日で完了することができます。作業の1日でプロトコルのBバージョンを通過することが可能です。連続矢印は、プロトコル内のステップの直接流れを示します。不連続矢印は、1 つのステップともう一方のステップの間に大きな時間差があることを示します(ステップ 6.6 および 9.3 を参照)。(C)マイクロパターニング(赤いストライプ)の1ラウンドまたはマイクロパターニングの2つの順次ラウンド(赤と緑のストライプ)の後に得られた例パターンの概略図。この図のより大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図 3: パターン テンプレートの生成は LIMAP で汎用性があります。(A, B)ImageJ (クロスボウ、B 文字) で設計されたパターン テンプレートの例。白で描かれた形状は最大レーザーパワーで投影され、黒で描かれた形状は投影されませんでした。(C, D)10 μg/mLフィブリノーゲン(緑)をインキュベーションした後のテンプレートからLIMAPで得られたマイクロパターン。(C)クロスボウは幅50μm、高さ50μm、水平方向75μm、垂直方向50μmです。(D)文字は幅 80 μm、高さ 85 μm です。C と D のスケール バーは 50 μm を表します。
{kind=link}

図 4: LIMAP プロトコルに不可欠な材料。(A)このプロトコルで使用されるステンシルは、直径20mm、薄い円形PDMSピース(厚さ250μm)で、4マイクロウェル(直径4mm)を含む。マイクロウェルで使用される体積は5~20μLの範囲であり、各実験に必要な試薬およびタンパク質の量を大幅に削減します。(B)ステンシルが各井戸に既に配置されている6ウェルガラス底皿。マイクロウェルには20μLのPBSが含まれており、それらを可視化します。(C)内側のガラスによく緑色の蛍光光体が付いているキャリブレーション皿は、レーザーフォーカスをキャリブレーションするために使用されます。(D)Bの6ウェルガラス底皿(赤い丸で輪郭を描いた)から左上の模式図。内側のガラス底部は白で表示され、ステンシルは灰色で表示されます。ステンシルには、4つのマイクロウェル(番号1〜4)が含まれており、4つの異なる実験条件(例えば、異なるタンパク質濃度、パターン形状、タンパク質の組み合わせなど)の試験用です。アスタリスクは、参照パターンを含むマイクロウェルを表します。(E)上部部分(矢印が塗りつぶされた矢印)で参照パターンが生成されたマイクロウェルの概略図。このリファレンスパターンは、パターニングに最適なレーザーフォーカスを得るために必要です(ステップ4参照)。空の矢印を持つ矢印は、システムキャリブレーション後の後続のパターニングに使用されるマイクロウェルの中央領域を示します。この図のより大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図 5: マイクロパターニング用のソフトウェアセットアップ。(A) ImageJ で設計され、8 ビット Tiff ファイルとして保存された並列ストライプを持つパターン テンプレート。(B-D)マイクロパターンが生成される現在のマイクロウェルと重複するデジタルRO(対象地域)のスケマティックビュー。(B) 使用するパターン テンプレート (長さ 1824 ピクセル =415 μm、幅 1140 ピクセル =260 μm) が Leonardo で選択され、設計ユニット (黒い破線の長方形の赤いストライプ) として ROI に投影され、約 0.1 mm2をカバーします。マイクロウェルエリア。設計単位は、レプリケーションメニュー (テンプレート構成) の 4 列と 4 行でレプリケートされ、マイクロウェル全体にパターンが作成されます。列間のスペースをメモします。(C)連続ストライプをパターン化するには、列間の間隔を調整する必要があります。この場合、設計単位間の重なり合いを達成するために、列間の間隔は、レプリケーションメニューで負の間隔として設定され、-20 μm.(D)同じマイクロウェルで複数のタンパク質をパターン化するために、パターンの正確な位置合わせが行われます。必須。ソフトウェアのセットアップステップ (手順 5) 中に、必要なすべてのパターン テンプレートを同時にアップロードします。[アクション]リストで、各パターニング ラウンド中にパターン化する特定のアクションのみを選択し、残りのアクションの選択を解除します (ステップ 5.12、5.13、および 8.2)。この図のより大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図6:LIMAPを用いてパターン変動性の解析(A)Width(上から下に 20、10、5、2 μm)の 4 つのストライプをマイクロパターン化するために使用される ImageJ で設計されたパターン テンプレート。(B)蛍光標識フィブリノーゲン(緑色)の10μg/mLをインキュベーションした後に得られたマイクロパターン。(C)マイクロパターンのストライプを横切る垂直線に沿った強度測定。(D)(C)での測定から得られた垂直蛍光強度プロファイル。より大きな幅(20 μm)では、ストライプのエッジにタンパク質の蓄積によって引き起こされる垂直プロファイルにばらつきがあり、2つの異なる蛍光強度ピーク(エッジ効果)が生じることに注意してください。この効果は、描かれた水平線(蛍光および背景)に沿ったストライプ幅≥20μm(E)強度測定でのみ見られます。(F)(E)での測定から得られた水平蛍光強度プロファイル。(G)各ストライプ幅の平均強度を示すグラフは、4つの個々の複製された設計単位(パターン間変動)から測定した。2 μm ストライプ幅のパターンに対するタンパク質吸着の減少に注意してください。(H)パターンストライプ内の変動(変動係数)は、3~10%の範囲で、すべてのストライプ幅に対して低かった。平均±SDとして示されたG及びHのデータは、GおよびHにおける一方通行分散分析(Kruskal-Wallis)非パラメトリック検定を用いて複数の比較を用いて行った。P 値は **の有意値は <0.001 です。この図のより大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図7:タンパク質吸着効率に対するレーザー力およびタンパク質濃度の変動の影響(A)PLL-PEG表面を一定のレーザー用量(1390mJ/mm2)でレーザー切り離し、蛍光標識ラミニン(マゼンタ)の示された濃度でインキュベートした。(B)ラミニンストライプの蛍光強度(A)の定量。(C)異なる示されたレーザー用量を適用し、その後、蛍光標識フィブロネクチン(緑色)の同じ濃度(10μg/mL)を有するインキュベーションを行った。(D)(C)におけるフィブロネクチンストライプの蛍光強度の定量化は、より高いレーザー用量が高レベルの吸着タンパク質と相関することを示す。すべての測定値は背景を減算されます。サンプル番号は列の下部に表示されます。データは平均±SEMとして示され、両尾計算を用いて非パラメトリックマン・ホイットニー検定を用いて統計分析を行った。P 値は 、**** の有意性に対して <0.0001 です。この図のより大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図8:マイクロパターン内のタンパク質濃度勾配の生成。(A) グレースケールのグラデーション パターン テンプレート。(B)(A)から測定した蛍光強度プロファイル。(C)蛍光標識フィブロネクチン(緑色)の10μg/mLをインキュベーションした後(A)におけるパターンテンプレートからLIMAPで得られたパターン。(D)n=3ストライプおよび背景の蛍光強度プロファイルは、平均±SEMとして表され、タンパク質勾配の線形増加を示す。この図のより大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図9:複数のタンパク質を順次パターン化する場合のクロスバインディング効果。(A-C, E-G)蛍光標識フィブロネクチン(シアン、水平)および蛍光標識ラミニン(マゼンタ、垂直)の10μmストライプを有するクロスパターン。(A-C)BSAブロッキングバッファで処理されたサンプル。(E-F)非特異的結合部位を遮断するためのPLL-PEGで処理されたサンプル(ステップ7)。(A, E)フィブロネクチンとラミニンの両方を示す結合蛍光チャネル。(B,F)フィブロネクチンのみを示す画像。(C, G)ラミニンのみを示す画像。Cの場合、BSAとの非占有結合部位の非効果的なブロッキングに起因する水平フィブロネクチン陽性ストライプ上にもラミニンの存在に注意してください。Gの場合、PLL-PEGによるブロッキングは、ラミニンとフィブロネクチンストライプへの効率的な結合を防ぐことに注意してください。(D,H)AおよびEにおける示された測定値(対角黄色線)から得られた蛍光強度プロファイルは、それぞれ。この図のより大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図10:ニューライト/軸子経路を調べるためのクロスパターン。(A-C)蛍光標識フィブロネクチン(シアン、水平)および蛍光標識ラミニン(マゼンタ、垂直)の10μmストライプを有するクロスパターン。(A, B)マイクロパターンに沿って成長する神経ライトを持つCAD細胞の蛍光画像。ニューライトを可視化するために、細胞を48時間培養し、4%PFAで固定し、チューブリン(A)またはチューブリンおよびアクチン(B)用に染色した。(C)ラットの後根神経節(DRG)ニューロンは、微小パターンに沿って成長する軸を有する。軸を可視化するために、DRGニューロンを72時間培養し、4%のPFAで固定し、チューブリン用に染色した。この図のより大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図11:LIMAPを使用してマイクロパターンを生成する際に得られる一般的な負の結果の例。(A-C)異なる状況下で得られる10 μg/mL蛍光標識フィブリノーゲン(緑)のサブ最適パターンストライプ。(A)パターン生成時にマイクロウェルが乾燥した。背景(矢印)の蛍光レベルが高く、PBS結晶(アスタリスク)の存在に注意してください。(B)ソフトウェアのセットアップ中にストライプ間のステッチが適切に調整されなかったため、設計単位間にギャップ(矢印)が付いている不連続なストライプが発生しました(ステップ 3.4.7 を参照)。(C)レーザーフォーカスは、パターンテンプレートの実際のストライプ幅を表さない拡散ストライプ(矢印)を引き起こすのが最適で、上から下に20、10、5、2μmである必要があります(B)。この図のより大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
ディスカッション
LIMAP(PRIMO)マイクロパターニングの利点とマイクロコンタクト印刷との比較
マイクロコンタクト印刷は、おそらく生物学的分野で最も一般的に使用されるマイクロパターニング技術である一方で、LIMAP技術40、41、42を使用する研究者が増加しているようです。 、43、44。ここでは、LIMAPの市販システムであるPRIMOを用いてプロトコルを紹介した。以下では、マイクロコンタクト印刷とLIMAPフォトパターニングの潜在的な利点と限界について簡単に説明します。
マイクロコンタクト印刷では、フォトマスク(一般にSU-8)をガラスまたはシリコンウエハにスピンコーティングして作成したリソグラフマスターが必要です。これらのマスタは、PDMS スタンプ45を作成するためのテンプレートとして使用されます。スタンプは、選択したタンパク質でインビキュベートされ、細胞培養皿に移されます。PDMSスタンプにタンパク質を吸着するプロセスは、タンパク質濃度、緩衝液およびインキュベーション時間に依存する。これらのパラメータは、最適な結果46を事前にテストする必要があります。
マスターは、正しく保存されている場合、数ヶ月または数年続く実験のかなりの数で使用することができます。しかし、この技術の制限要因は、すべての所望の変更のための新しいリソグラフマスターを再設計する必要性です。実験計画の変更により、新しいマスターの生産に時間がかかる(数週間まで)ため、実験が遅れる可能性があります。これに対し、LIMAP フォトパターニングでは物理マスターは必要ありません。ソフトウェア生成のパターンテンプレートを使用して、マイクロパターンの所望のジオメトリを変化する研究の質問に柔軟に適応させることができます。LIMAPは、同じマイクロパターン内でタンパク質勾配を生成するためにも使用できます(図8)。
さらに、LIMAPで達成されるマイクロパターン分解能は、我々の場合、2 μm(図6B)である。
この解像度に近づくと、パターン内およびパターン間の可変性が増加しました。幅10μm以上のパターンを生成する場合は、非常に再現性が高かった(図6G,H)。逆に、マイクロコンタクト印刷では、10 μm未満の解像度を一貫して得ることは困難であり、小さな特徴をスタンプする際にアーティファクトを見つけるのが一般的です(データは示されていません)。
LIMAPは、同じマイクロウェル内で複数のタンパク質(図9)をマイクロパターン化するために使用できることを示し、実験にさらに複雑さのレベルを追加することができます。これはマイクロコンタクト印刷で実現できますが、異なるタンパク質を高い精度で整列させることは技術的にかなり厳しい場合があります。LIMAPを用いて複数のタンパク質をパターン化することは簡単に見えますが、シーケンシャルコーティング手順を通じてタンパク質のクロスバインディングはブロッキング試薬を通じて減少することができますが、完全に排除されるわけではありません(図9)。
一方または他の技術のコストに関しては、ここで説明するLIMAPは、異なる蛍光顕微鏡に設置できるマイクロパターニング装置(PRIMO)の購入を必要とし、電動段階を必要とする。この投資は最初はコストが高いですが、LIMAP に関連する長期的には消耗品(ステンシル、PEG、PLPP)以外の追加購入はありません。あるいは、PDMSステンシルは、公開されたプロトコル18、32に続く独自の実験者によってラボで製造することもできる。マイクロコンタクト印刷の最大のコストは、新しいマスターの生産に関連する可能性があり、実験に新しいパターンが必要な場合は相当な額になる可能性があります。
LIMAP の欠点の 1 つは、この手法の比較的低いスループット アプローチです。マイクロコンタクト印刷は、LIMAPで必要なシーケンシャルレーザーマイクロパターニングと比較して、同時プレスステップで多数のマイクロパターンを迅速かつ効率的に生成できます。例えば、PDMSスタンプ(スタンプ調製を除く)を用いてマイクロコンタクト印刷を行い、約2時間で6枚のスタンプガラスカバースリップを製造することが可能です。LIMAPで同様の領域(6ウェル皿)をパターン化すると、表面のパッシベーションの手順を除いて約4時間かかります(ステップ5.12で説明するパターンテンプレート構成を考慮すると、図5Bを参照)。
LIMAP 技術のもう 1 つのレート制限要因は、大きな領域をパターン化するために必要な長い照明時間 (7.5 mW/mm2レーザーを使用した設計単位あたり 30 s) です。このような場合は、マイクロコンタクト印刷が推奨される場合があります。新しく利用可能なフォトイニシエータ(PLPPゲル、材料のテーブル)は、パターニングに要する時間を大幅に短縮し、わずか数分で大きな領域(最大8mm2)で数百のマイクロパターンを生成できるようにする必要があります。
細胞培養用のマイクロパターニング表面を考慮に入れるもう一つの重要な要因は、マイクロコンタクト印刷で得られた変動性と比較して、異なる実験繰り返し間のマイクロパターンの再現性である。例えば、図7B、Dに示すグラフは、非常に類似した結果を持つ3つの独立した実験繰り返しの代表的なデータである(図は示さない)。私たちの経験と以前の出版物に基づいて、再現性のこのレベルは、マイクロコンタクト印刷48、49、50、51、52で達成することは困難です。
光感受性材料を設計するための専用の化学または一般的にあまり生体適合性のない光感度3の使用を必要とする他の光パターニング技術とは対照的に、LIMAP(PLPP))は、細胞21によって生体適合性と十分に許容される。私たちの手の中で、CAD、DRGニューロン(図10)、線維芽細胞、上皮細胞、黒色腫細胞を含む様々な細胞間で細胞毒性を経験していません(データは示されていません)。他のフォトパターニング技術と比較してPRIMOを使用するLIMAPのもう一つの利点は、フォトマスクが必要ではないことです。マイクロコンタクト印刷と同様に、新しいフォトマスクは、必要なパターンごとに設計および生成する必要があります。
マイクロコンタクト印刷に関して上記のすべての制限事項は、技術の手動アプローチを参照してください。しかしながら、スタンプ負荷及び圧力制御53を備えた自動化装置を用いてマイクロコンタクト印刷のスループットと再現性を高めることができる。
PRIMO を使用した LIMAP のプロトコルと問題解決の主な手順
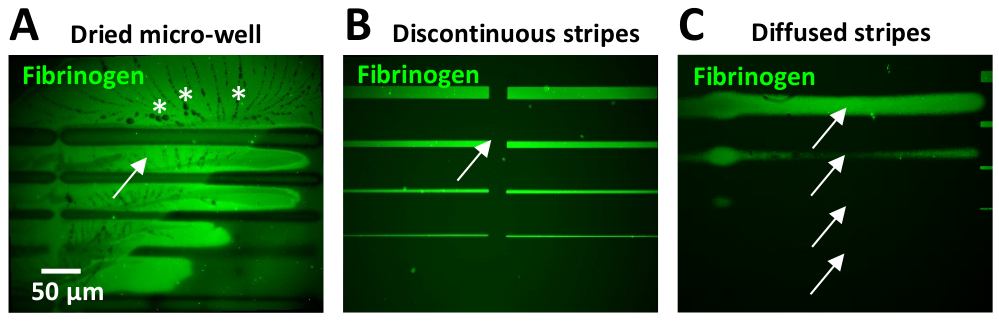
このプロトコル中に見つかった最も一般的な問題の1つは、マイクロパターン内のバックグラウンド蛍光の高レベルを有することです。これは、多くの場合、その小さな体積のために発生するマイクロウェルの乾燥に起因することができます。これが起こると、PBS結晶はしばしばECMパターンの周囲に現れる(図11A)。
タンパク質インキュベーション後の不十分または非効率的な洗浄ステップはまた、バックグラウンド蛍光の高レベルをもたらすことができます。これは、特に10μg/mL(図11B)以上のタンパク質濃度を用いた場合に観察することができる。バックグラウンドでのタンパク質の過剰は、PBSと追加の洗浄ステップを含むことで低減することができます。
タンパク質の背景の存在を測定し、各実験で特徴付け、背景蛍光強度(図6E)を計算し、マイクロパターン強度から差し引く(図6F-H)図7B,D)高タンパク質の背景は、CAD細胞の付着および発芽に影響を与え、結果の解釈を損なう可能性があります。
設計単位間にギャップを持つことは、ユーザーの経験が限られている場合に一般的な問題です (図 11B)。レオナルドソフトウェアの2つのパラメータは、これを克服するために調整することができます:1)パターンの設計に応じて、列間の負の間隔が必要な場合があります(ステップ5.7と図5B、Cを参照)。または、2) [エキスパート]メニューのグラデーション オプションを使用して列をステッチします。最適な間隔パラメータを決定するためのクイックテストは、UV接着剤(材料の表)を使用して行うことができます。この接着剤の小さな滴は、ガラススライドに適用され、その後、フィルムを作る、ガラスカバースリップで覆われています。埋め込まれたUV接着剤は、低レーザー線量(30 mJ/mm2)を使用して目的のパターンテンプレートでフォトパターン化されています。埋め込まれた接着剤のUV露出領域は硬化し、明視野顕微鏡下で見えるようになります。テスト結果は、パターン内で得られた間隔を評価するために視覚化されます。私たちのニューロン実験では、ストライプ間のギャップが細胞の挙動に悪影響を及ぼし、成長力学の変動(速度の低下または経路の放棄)を生み出す可能性があります。
レオナルドソフトウェアの最新アップデート(公開時点、レオナルド4.11)では、マイクロウェル表面のはるかに大きな領域(20X目的を使用して最大8ミリメートル2)をカバーする以前に設計された大きなパターンテンプレートをアップロードすることが可能です。設計単位ごとの現在の0.1 mm2と比較して、より小さい設計単位を一緒にステッチする必要性を除去する。未定義のエッジは、パターン生成中にレーザーフォーカス調整が行われなことから生じる可能性があります(図 11C)。したがって、レーザーを校正し、パターニングの前に参照パターンステップ(ステップ4を参照)を実行することが重要です。ストライプの定義が不十分な場合、ストライプ幅のばらつきが生じ、軸が成長ダイナミクスとストライプ幅の相関関係を困難にします。アクソンはまた、エッジが拡散したストライプを放棄する傾向があります。さらに、幅10~20μm以上のストライプを印刷するとエッジのばらつき性も見出され、パターンの中央領域と比較してエッジでタンパク質含有量が高くなります(図6B,D)。このエッジ効果は、フォトパターニングプロセス中にフォトイニシエータの非均質な拡散によって生成されます。光化反応は酸素依存性であり、エッジでより拡散します。このエッジ効果は、フォトパターニングプロセス中にマイクロウェルにピペットを使用してフォトイニシエータを均質化することを最小限に抑えることができます。さらに、新たに製品化されたフォトイニシエータ(PLPPゲル)は、エッジ効果を低減することもできる(PRIMOシステムサポートチーム、パーソナルコミュニケーション)。
複数のタンパク質のマイクロプリンティングは、クロスバインディングをもたらす可能性があります(図9A-D)。これは、2つの異なるタンパク質のインキュベーションステップ間の非特異的結合部位を占めるために使用されるブロッキング効率を高めることによって最小化することができる。タンパク質のクロスバインディングは、実験結果の再現性を乱し、各タンパク質が軸次成長力学やその他の細胞挙動に対する寄与を決定することが困難であるため、データの誤解釈につながる可能性があります。
結論
LIMAPを用いて提供されるプロトコルが、PRIMOシステムを用いてタンパク質マイクロパターンの生成を容易にすることを期待する。我々のプロトコルは、2Dガラス表面でマイクロパターンを確実に生成する方法に焦点を当てていますが、他の人は、柔らかい基板54のマイクロパターニングにLIMAPを使用することが可能であることを示しています42.これらのマイクロパターンは、マイクロ環境の変化に対する細胞応答を研究するための汎用性の高いツールです。
開示事項
著者は何も開示していない。
謝辞
この作業は、BBSRC、EPSRC、MRC、ウェルカムトラストによってサポートされています。C.B.ラボは、ウェルカム・トラスト(助成番号088785/Z/09/Z)からのコア資金によって支えられているマンチェスター大学のウェルカム・トラスト・センターの一部です。著者らは、バイオテクノロジー・生物科学研究評議会(BBSRC)からC.M.J.(BB/M020630/1)、およびP.A.(BB/P000681/1)および工学・物理科学研究協議会(EPSRC)および医学によって提供された資金を承認したいと考えています。研究評議会(MRC)再生医療博士後期訓練センター(EP/L014904/1)著者は、アルベオールの対応とアフターサポートチームに感謝しています。著者らは、マンチェスター大学バイオイメージング施設のピーター・マーチとロジャー・メドウズに、顕微鏡検査の助けに感謝している。この研究で使用されるバイオイメージング施設顕微鏡は、BBSRC、ウェルカムトラスト、マンチェスター大学戦略基金からの助成金で購入されました。
資料
| Name | Company | Catalog Number | Comments |
| Alexa 488 protein labeling kit | Invitrogen | A10235 | Working concentration: N.A. |
| Alexa 647 protein labeling kit | Invitrogen | A20173 | Working concentration: N.A. |
| CAD cells | ECACC | 8100805 | Working concentration: N.A. |
| Conjugated fibrinogen-488 | Molecular Probes | F13191 | Working concentration: 10 μg/ml |
| DMEM culture medium | Gibco | 11320033 | Working concentration: N.A. |
| Epifluorescence Microscope** | Nikon | Eclipse Ti inverted | Working concentration: N.A. |
| Fibronectin | Sigma | F4759 | Working concentration: 10 μg/ml (after labelling with Alexa 488 protein labeling kit, see above) (diluted in PBS) |
| Fiji-Image J | www.imagej.nih.gov | Version 2.0.0-rc-54/1.51f | Working concentration: N.A. |
| Fluorescent highlighter | Stabilo | Stabilo Boss Original | Working concentration: N.A. |
| HEPES | Gibco | 15630080 | Working concentration: 1M |
| Inkscape software | Inkscape | Check last update | Working concentration: N.A. |
| Laminin-red fluorescent rhodamine | Cytoskeleton, Inc. | LMN01 | Working concentration: 10 μg/ml (diluted in PBS) |
| Leonardo software | Alvéole | version 4.11 | Working concentration: N.A. |
| L-Glutamine | Sigma | G7513 | Working concentration: 1% |
| Micro-manager software | Open imaging | Check last update | Working concentration: N.A. |
| Motorized x/y stage | PRIOR Scientific | Proscan II | Working concentration: N.A. |
| NIS Elements Software | Nikon | NIS Elements AR 4.60.00 64-bit (With Nikon jobs) | Working concentration: N.A. |
| PBS (without Ca2+, Mg2+) | Sigma | D8537 | Working concentration: 1X |
| PDMS Stencils | Alvéole | visit www.alveolelab.com | Working concentration: N.A. |
| PEG-SVA | Laysan bio, Inc. | MPEG-SVA-5000-1g | Working concentration: 50 mg/ml |
| Phalloidin 405 | Abcam | ab176752 | Working concentration: 1:1000 |
| Photo-initiator (PLPP) | Alvéole | Classic PLPP | Working concentration: 14.5 mg/ml |
| Photo-initiator (PLPP gel) | Alvéole | PLPP gel | Working concentration: 4.76% diluted in ethanol |
| Plasma cleaner | Harrick Plasma | PDC-32G (115V) | PDC-32G-2 (230V) | Working concentration: N.A. |
| PLL-PEG | SuSoS (also distributed by Alvéole) | www.alveolelab.com | Working concentration: 0.1 mg/ml (diluted in PBS) |
| Poly-L-Lysine | Sigma | P4707 | Working concentration: 0.01% |
| Primo equipment | Alvéole | www.alveolelab.com | Working concentration: N.A. |
| Pen/Strep | Thermo Fisher | 15140122 | Working concentration: 1% |
| Tubulin anti-alpha antibody | Abcam | DM1A | Working concentration: 1:1000 CAD cells |
| Tubulin anti-beta 3 antibody | Sigma | T8660 | Working concentration: 1:500 DRG neurons |
| UV adhesive | Norland Products | NOA81 | Working concentration: N.A. |
| 1 well glass bottom dish | Cellvis | D35-20-1.5-N | Working concentration: N.A. |
| 6 well glass bottom dish | Cellvis | P06-20-1.5-N | Working concentration: N.A. |
| 20x objective** | Nikon | no phase ring (check updated catalogue) | Working concentration: N.A. **Epifluorescence microscope: images were acquired and patterns were generated on an Eclipse Ti inverted microscope (Nikon), coupled to PRIMO micro-patterning equipment (Alvéole), using a 20x objective (0.75 S Plan Fluor (nophasering, Nikon). Nikon specific filter sets for GFP, mCherry and Cy5 were used and fluorescent light source was LED (Lumencor) although other fluorescence sources and filter sets can be used. The microscope has an automated x/y stage (PRIOR Scientific) for the printing of multi-field patterning and Nikon Perfect Focus to prevent focus drift. The images were collected using a Retiga R6 (Q-Imaging) camera. |
参考文献
- Alamdari, O. G., Seyedjafari, E., Soleimani, M., Ghaemi, N. Micropatterning of ECM Proteins on Glass Substrates to Regulate Cell Attachment and Proliferation. Avicenna Journal of Medical Biotechnology. 5 (4), 234-240 (2013).
- Sunami, H., Yokota, I., Igarashi, Y. Influence of the pattern size of micropatterned scaffolds on cell morphology, proliferation, migration and F-actin expression. Biomaterials Science. 2 (3), 399-409 (2014).
- Thery, M. Micropatterning as a tool to decipher cell morphogenesis and functions. Journal of Cell Science. 123 (Pt 24), 4201-4213 (2010).
- Marino, A., et al. Two-photon polymerization of sub-micrometric patterned surfaces: investigation of cell-substrate interactions and improved differentiation of neuron-like cells. ACS Applied Materials & Interfaces. 5 (24), 13012-13021 (2013).
- Joo, S., et al. Effects of ECM protein micropatterns on the migration and differentiation of adult neural stem cells. Scientific Reports. 5, (2015).
- Morgani, S. M., Metzger, J. J., Nichols, J., Siggia, E. D., Hadjantonakis, A. K. Micropattern differentiation of mouse pluripotent stem cells recapitulates embryo regionalized cell fate patterning. Elife. 7, (2018).
- Javaherian, S., O'Donnell, K. A., McGuigan, A. P. A Fast and Accessible Methodology for Micro-Patterning Cells on Standard Culture Substrates Using Parafilm (TM) Inserts. Plos One. 6 (6), (2011).
- Smirnov, M. S., Cabral, K. A., Geller, H. M., Urbach, J. S. The effects of confinement on neuronal growth cone morphology and velocity. Biomaterials. 35 (25), 6750-6757 (2014).
- Albert, P. J., Schwarz, U. S. Dynamics of Cell Ensembles on Adhesive Micropatterns: Bridging the Gap between Single Cell Spreading and Collective Cell Migration. PLOS Computational Biology. 12 (4), (2016).
- Evans, A. R., et al. Laminin and fibronectin modulate inner ear spiral ganglion neurite outgrowth in an in vitro alternate choice assay. Developmental Neurobiology. 67 (13), 1721-1730 (2007).
- Nichol, R. H., Hagen, K. M., Lumbard, D. C., Dent, E. W., Gomez, T. M. Guidance of Axons by Local Coupling of Retrograde Flow to Point Contact Adhesions. Journal of Neuroscience. 36 (7), 2267-2282 (2016).
- Burdick, J. A., Khademhosseini, A., Langer, R. Fabrication of gradient hydrogels using a microfluidics/photopolymerization process. Langmuir. 20 (13), 5153-5156 (2004).
- Schwartz, P. V. Molecular transport from an atomic force microscope tip: A comparative study of dip-pen nanolithography. Langmuir. 18 (10), 4041-4046 (2002).
- Barbulovic-Nad, I., et al. Bio-microarray fabrication techniques--a review. Critical Reviews in Biotechnology. 26 (4), 237-259 (2006).
- Shafagh, R. Z., Vastesson, A., Guo, W. J., van der Wijngaart, W., Haraldsson, T. E-Beam Nanostructuring and Direct Click Biofunctionalization of Thiol-Ene Resist. Acs Nano. 12 (10), 9940-9946 (2018).
- Kobayashi, J., Yamato, M., Itoga, K., Kikuchi, A., Okano, T. Preparation of microfluidic devices using micropatterning of a photosensitive material by a maskless, liquid-crystal-display projection method. Advanced Materials. 16 (22), (2004).
- Bernard, A., et al. Printing patterns of proteins. Langmuir. 14 (9), 2225-2229 (1998).
- Ruiz, S. A., Chen, C. S. Microcontact printing: A tool to pattern. Soft Matter. 3 (2), 168-177 (2007).
- Qin, D., Xia, Y., Whitesides, G. M. Soft lithography for micro- and nanoscale patterning. Nature Protocols. 5 (3), 491-502 (2010).
- Fink, J., et al. Comparative study and improvement of current cell micro-patterning techniques. Lab Chip. 7 (6), 672-680 (2007).
- Strale, P. O., et al. Multiprotein Printing by Light-Induced Molecular Adsorption. Advanced Materials. 28 (10), 2024-2029 (2016).
- Belisle, J. M., Correia, J. P., Wiseman, P. W., Kennedy, T. E., Costantino, S. Patterning protein concentration using laser-assisted adsorption by photobleaching, LAPAP. Lab Chip. 8 (12), 2164-2167 (2008).
- Belisle, J. M., Kunik, D., Costantino, S. Rapid multicomponent optical protein patterning. Lab Chip. 9 (24), 3580-3585 (2009).
- Heinz, W. F., Hoh, M., Hoh, J. H. Laser inactivation protein patterning of cell culture microenvironments. Lab Chip. 11 (19), 3336-3346 (2011).
- Azioune, A., Carpi, N., Tseng, Q., Thery, M., Piel, M. Protein Micropatterns: A Direct Printing Protocol Using Deep UVs. Microtubules: In Vivo. 97, 133-146 (2010).
- Vignaud, T., Ennomani, H., Thery, M. Polyacrylamide hydrogel micropatterning. Methods in Cell Biology. 120, 93-116 (2014).
- Waldbaur, A., Waterkotte, B., Schmitz, K., Rapp, B. E. Maskless projection lithography for the fast and flexible generation of grayscale protein patterns. Small. 8 (10), 1570-1578 (2012).
- Kang, J., Choi, J. C., Kim, M., Jung, H. R., Doh, J. Photopatterning with a printed transparency mask and a protein-friendly photoresist. Methods in Cell Biology. 119, 55-72 (2014).
- Falconnet, D., Csucs, G., Grandin, H. M., Textor, M. Surface engineering approaches to micropattern surfaces for cell-based assays. Biomaterials. 27 (16), 3044-3063 (2006).
- Morlat, S., Gardette, J. L. Phototransformation of water-soluble polymers. Part II: photooxidation of poly(ethylene oxide) in aqueous solution. Polymer. 44 (26), 7891-7897 (2003).
- Qi, Y., Wang, J. K., McMillian, M., Chikaraishi, D. M. Characterization of a CNS cell line, CAD, in which morphological differentiation is initiated by serum deprivation. Journal of Neuroscience. 17 (4), 1217-1225 (1997).
- Shrirao, A. B., et al. A Versatile Method of Patterning Proteins and Cells. Journal of Visualized Experiments. (120), (2017).
- Pankov, R., Momchilova, A. Fluorescent labeling techniques for investigation of fibronectin fibrillogenesis (labeling fibronectin fibrillogenesis). Methods in Molecular Biology. 522, 261-274 (2009).
- Dertinger, S. K., Jiang, X., Li, Z., Murthy, V. N., Whitesides, G. M. Gradients of substrate-bound laminin orient axonal specification of neurons. Proceedings of the National Academy of Sciences of the United States of America. 99 (20), 12542-12547 (2002).
- Chelli, B., et al. Neural cell alignment by patterning gradients of the extracellular matrix protein laminin. Interface Focus. 4 (1), (2014).
- Tang, Y., Qiu, Q. F., Zhang, F. L., Xie, M., Huang, W. H. Quantifying orientational regeneration of injured neurons by natural product concentration gradients in a 3D microfluidic device. Lab Chip. 18 (6), 971-978 (2018).
- Srinivasan, P., Zervantonakis, I. K., Kothapalli, C. R. Synergistic effects of 3D ECM and chemogradients on neurite outgrowth and guidance: a simple modeling and microfluidic framework. PLoS One. 9 (6), (2014).
- de Luca, A. C., Faroni, A., Reid, A. J. Dorsal root ganglia neurons and differentiated adipose-derived stem cells: an in vitro co-culture model to study peripheral nerve regeneration. Journal of Visualized Experiments. (96), (2015).
- Khadpekar, A. J., Khan, M., Sose, A., Majumder, A. Low Cost and Lithography-free Stamp fabrication for Microcontact Printing. Scientific Reports. 9 (1), (2019).
- Delepine, C., et al. Altered microtubule dynamics and vesicular transport in mouse and human MeCP2-deficient astrocytes. Human Molecular Genetics. 25 (1), 146-157 (2016).
- Decock, J., Schlenk, M., Salmon, J. B. In situ photo-patterning of pressure-resistant hydrogel membranes with controlled permeabilities in PEGDA microfluidic channels. Lab Chip. 18 (7), 1075-1083 (2018).
- Stoecklin, C., et al. A New Approach to Design Artificial 3D Microniches with Combined Chemical, Topographical, and Rheological Cues. Advanced Biosystems. 2 (7), (2018).
- Toraille, L., et al. Optical Magnetometry of Single Biocompatible Micromagnets for Quantitative Magnetogenetic and Magnetomechanical Assays. Nano Letters. , (2018).
- Theodoly, O., et al. Live nanoscopic to mesoscopic topography reconstruction with an optical microscope for chemical and biological samples. PLoS One. 13 (12), (2018).
- Ermis, M., Antmen, E., Hasirci, V. Micro and Nanofabrication methods to control cell-substrate interactions and cell behavior: A review from the tissue engineering perspective. Bioactive Materials. 3 (3), 355-369 (2018).
- von Philipsborn, A. C., et al. Microcontact printing of axon guidance molecules for generation of graded patterns. Nature Protocols. 1 (3), 1322-1328 (2006).
- Ricoult, S. G., Kennedy, T. E., Juncker, D. Substrate-bound protein gradients to study haptotaxis. Frontiers in Bioengineering and Biotechnology. 3, (2015).
- Bietsch, A., Michel, B. Conformal contact and pattern stability of stamps used for soft lithography. Journal of Applied Physics. 88 (7), 4310-4318 (2000).
- Hui, C. Y., Jagota, A., Lin, Y. Y., Kramer, E. J. Constraints on microcontact printing imposed by stamp deformation. Langmuir. 18 (4), 1394-1407 (2002).
- Sharp, K. G., Blackman, G. S., Glassmaker, N. J., Jagota, A., Hui, C. Y. Effect of stamp deformation on the quality of microcontact printing: theory and experiment. Langmuir. 20 (15), 6430-6438 (2004).
- Delamarche, E., Schmid, H., Michel, B., Biebuyck, H. Stability of molded polydimethylsiloxane microstructures. Advanced Materials. 9 (9), 741-746 (1997).
- Perl, A., Reinhoudt, D. N., Huskens, J. Microcontact Printing: Limitations and Achievements. Advanced Materials. 21 (22), 2257-2268 (2009).
- Chakra, E. B., Hannes, B., Dilosquer, G., Mansfield, C. D., Cabrera, M. A new instrument for automated microcontact printing with stamp load adjustment. Review of Scientific Instruments. 79 (6), (2008).
- Pasturel, A., Strale, P., Studer, V. Tailoring 3D cell culture templates with common hydrogels. bioRxiv. , (2019).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved