
Method Article
次世代ウイルスRNA/DNA in situハイブリダイゼーションのヒト免疫不全ウイルス/サル免疫不全ウイルス研究における応用
要約
ここでは、ホルマリン固定パラフィン包埋(FFPE)組織中の特定のウイルスRNAまたはDNA配列を同定するための次世代のin situハイブリダイゼーションアッセイを紹介します。このアプローチにより、RNAおよびDNAの低コピーを24時間未満で、非常に高い感度と特異性で視覚化できます。
要約
In situハイブリダイゼーションは、組織切片の個々の細胞内の特定のRNAまたはDNA配列を同定する強力な技術であり、生理学的プロセスと疾患の病因に関する重要な洞察を提供します。In situハイブリダイゼーション(ISH)は、ウイルスに感染した細胞の位置を評価するために長年使用されてきましたが、最近、シグナル増幅とバックグラウンド抑制を同時に行うことができる独自のプローブ設計戦略により、組織の形態を維持しながら1分子の可視化を実現する次世代のISHアプローチが開発されました。この次世代ISHは、分岐型PCRのようなアプローチに基づいていますが、in situで実施され、従来のISH法やホルマリン固定パラフィン包埋(FFPE)組織中のRNAまたはDNAを日常的に検出するin situ PCRアプローチよりも、より容易で、感度が高く、再現性があります。ここ数年、当研究室では、このISHプラットフォームを、多数のFFPE組織内のヒト免疫不全(HIV)およびサル免疫不全(SIV)ウイルスRNA(vRNA)および/またはウイルスDNA(vDNA)陽性細胞の検出に応用してきました。この詳細な技術原稿により、次世代ISHを研究に活用することに関心のあるすべての方々と、私たちの知識とアドバイスを共有したいと思います。
概要
ISHは、細胞または組織内の特定のDNAまたはRNA配列に対する相補的なDNA、RNA、または修飾核酸鎖(プローブ)を標的化し、可視化するために使用される実験的アプローチです。ISHは、組織内の特定の核配列の特異的な局在化と可視化を可能にし、標的とその細胞環境との間の発現レベル、組織化、分布、相互作用を理解するために重要であり、これはqPCRなどの他の一般的な技術では得られない貴重な情報です。最近まで、ISHは標識された相補的DNAまたは相補的RNA(リボプローブ)のいずれかを使用して行われるのが一般的でした。これらのプローブは、ラジオ標識塩基、蛍光標識塩基、または抗原標識塩基( 35S、FITC、ジゴキシゲニンなど)と直接結合し、オートラジオグラフィー、蛍光顕微鏡法、または免疫組織化学検出アプローチのいずれかを使用して、それぞれ組織に局在化および定量しました。これらのin situテクノロジーは引き続き価値のあるアプローチですが、労働集約的でなく、よりシンプルで、より速く、感度が高く、特異なアプローチを開発するための改善の余地は十分にあります。
2012年に初めて報告された、ホストメッセンジャーRNA(mRNA)の検出のための代替の商用次世代ISHアプローチ(RNAscopeアッセイなど)は、分岐PCRに基づいています。mRNA検出はFFPE細胞および組織で行われ、個々の細胞で単一RNA分子の可視化に近づく感度で行われます1。このアプローチの特異性は、2つのdouble-Zターゲットプローブがそれぞれの相補的なRNA(またはDNA)配列に連続して結合し、シグナルプリアンプが1を連続的に結合するという独自の条件下で達成されます。これにより、分岐DNA(bDNA)1,2と同様に、その後のハイブリダイゼーションステップを介してシグナル増幅カスケードを開始することができます。さらに、このアプローチは非常に迅速かつ簡単で、わずか1日(<8時間)で結果が得られ、Radio-ISH 1,2などの代替技術で最大4週間かかるのに比べて大きな利点があります。この次世代のISHは、HIV/SIV研究に新たな視点と機会をもたらしました。HIV治療の主な障害は、病気の初期段階で確立される細胞および組織の貯蔵庫である3,4。この技術の全体的な目標は、ウイルスのリザーバーとして機能し、感染した宿主内で持続する主要な組織コンパートメントを特定し、位置を特定し、最終的に理解することです。これは、HIVに対する効果的な治療戦略の開発に役立ちます。
この原稿では、当社のデュプレックス次世代RNA/DNAマルチプレックスISHプロトコル(RNAscope/DNAscopeなど)について詳細に説明し、既存のRNA ISHプロトコルをどのように変更して、次世代ISHをサンプルと特定のターゲットに最適化したかを説明します。このプロトコルにより、5 μm組織切片内のHIV/SIVウイルスRNAおよびウイルスDNAの可視化、局在化、および定量が可能になります。vDNAとvDNAの両方の同時可視化は、2つのカスタムプローブセットを組み合わせることによって行われます:1つはvDNAコード鎖を標的とするセンス(C1 SIVmac239 Gag-Pol-Senseプローブ[416141-C1])、もう1つはウイルスゲノムの異なる領域をカバーするvRNA転写産物を標的とするアンチセンス(C2 SIVmac239 Vif-Env-Nef-Tar-Anti-Senseプローブ[416131-C2])です(表1) で、C1 と C2 の 2 つの異なる可視化チャネルを使用します。このプロトコルでは、チャネルC1とC2により、信号を異なる色(APは赤、HRPは茶色)で視覚化し、さまざまなアプローチでプローブを検出できます。組織固定処理と切断を除けば、このアッセイは2日間かかります。ここでは、細胞ペレットまたは組織切片で実施できる二本鎖vRNAおよびvDNAのin situハイブリダイゼーションプロトコルを示します。
プロトコル
1. 切片とスライドの準備
- パラフィンブロックをトリミングし、ミクロトームを使用して5 +/-1 μm切片を切断します。切片または細胞ペレットを、40-45 °C RNaseフリーウォーターバス内の荷電顕微鏡スライドにマウントします。エアドライスライドは37°CまたはRTで一晩スライドします。
注:スライドは、室温(RT)で最大3か月、4°Cで6か月間保存できます。 - FFPEスライドを脱パラフィンします。
- スライドをドライオーブンで60°Cで1時間焼きます。
- ヒュームフードに、スライド染色皿2枚に~200 mLの新鮮なキシレンを充填し、さらに2枚の染色皿に~200 mLの新鮮な100%エタノールを入れます。容器に蓋をします。
- スライドをラックに置き、最初のキシレン含有皿に浸します。攪拌しながらRTで5〜10分間インキュベートします。
- スライドを2番目のキシレン含有ディッシュに入れ、攪拌しながらRTで5〜10分間インキュベートします。
- すぐにスライドを100%エタノールが入った皿に入れます。スライドをRTで5〜10分間攪拌しながらインキュベートします。
- 直ちにスライドを100%エタノールを含む第2の皿に入れ、攪拌しながらRTで5〜10分間インキュベートする。
- ラックをエタノールから取り出し、ラックの側面を軽くたたいて余分なエタノールを取り除き、RNaseフリー水で5〜15分間すすぎます。
2.オーブンの準備
- ハイブリダイゼーションオーブンの電源を入れ、温度を40°Cに設定します。
- 布または丈夫な吸収性ペーパータオルをトレイに置き、湿度制御を可能にするために二重蒸留水で完全に濡らします。
- 蓋付きのトレイをオーブンに挿入し、オーブンのドアを閉じます。使用前に、トレイを40°Cで少なくとも30分間温めてください。使用しないときは、トレイをオーブンに入れてください。
3. 熱誘起エピトープ賦活化
- 0.5xクエン酸塩ベースのISHハイブリダイゼーションターゲット賦活化バッファー(10 nmol / L、pH = 6、 材料の表を参照)を調製します。加熱プレートのビーカーで沸騰させます。
- スライドを沸騰ターゲット回収バッファーに30分間入れることにより、熱誘起エピトープ回収を行います。
- スライドをターゲット回収バッファーから取り出し、すぐに再蒸留水で洗浄します。100%エタノールで5分間脱水してから、自然乾燥させます。
- スライドが風乾したら、疎水性バリアペンを貼ってスライドの組織切片を囲みます。疎水性バリアが完全に風乾するのを必ずお待ちください。
4. プロテアーゼ前処理
- 乾燥させたスライドをロック式スライドラックに置き、滅菌済みの冷たくしたPBSで1:5の比率で希釈して、プロテアーゼ前処理試薬(プロテアーゼ消化液、2.5 μg/mL)を調製します。よく混ぜます。
注:市販のキットには、濃度の異なる3種類のプロテアーゼ試薬が入っています。プロテアーゼIII(標準)、プロテアーゼIV(強)、およびプロテアーゼプラス(軽度)。最適な条件は組織の種類、固定、および厚さによって異なるため、研究に実施する前に、プロテアーゼ消化時間と希釈を経験的にテストしてください(「ディスカッション」を参照)。 - 希釈したプロテアーゼ溶液をスライド上に分注し、組織切片を完全に覆います。直ちにスライドを40°Cで20分間インキュベートし(ステップ1.4で調製)、スライドが湿ったハイブリダイゼーショントレイに密封されていることを確認します。プロトコルの残りの部分では、組織切片を乾燥させないでください。
- ロッキングスライドラックを二重蒸留水で満たされた洗浄トレイに沈めて、すぐに3回すすぎます。
- ペルオキシダーゼ溶液を各組織切片に滴下して完全に覆うことにより、内因性ペルオキシダーゼブロックを実行します。スライドを室温で10分間インキュベートします。完了したら、切片を二重蒸留水で3回すすぎます。
5. プローブハイブリダイゼーションとシグナル増幅
注:蒸発を防ぐために、インキュベーションステップ中に組織が乾燥しないように、湿度制御トレイが適切に密閉されていることを確認してください。洗浄ステップ中に湿度チャンバーをオーブンに戻し、40°Cに保たれるようにします。
- メーカーが推奨する通り、1容量のC2プローブから50容量のC1プローブをチューブ内にピペットで移し、C2プローブとC1プローブを1:50の比率で混合します。チューブを数回反転させます。ターゲットプローブ混合物を40°Cオーブンで~10分間予温し、使用前に沈殿物を溶解します。
注:混合ターゲットプローブは、4°Cで最大6か月間保存できます。 - スライドを水から取り出します。スライドをすすぎ、軽くたたいたりフリックしたりして、ティッシュ切片から余分な水分を取り除きます。すぐにプローブをスライド上に分注し、各組織切片が気泡なしで完全に覆われていることを確認します。プローブミックスを湿度チャンバー内で40°Cで一晩インキュベートします。
- 翌日、スライドをオーブンから取り出し、0.5x洗浄バッファーを入れた洗浄トレイに室温で5分間置き、洗浄手順をもう一度繰り返します。
- スライドを洗浄バッファーから取り出します。スライドをすすぎ、軽くたたくかフリックして、組織切片から余分な洗浄バッファーを取り除きます。
- 市販のAMP 1試薬(2 nmol / L)をすぐに使用できるAMP 1試薬(2 nmol / L)をハイブリダイゼーションバッファーB(20%ホルムアミド、5x SSC、0.3%リチウムドデシル硫酸、10%デキストラン硫酸、ブロッキング試薬)にスライド上に分注し、気泡のない組織切片を完全にカバーします。湿度チャンバー内で40°Cで30分間インキュベートします。手順1.6.3〜1.6.4を繰り返し、それぞれ2分間洗浄します。
- 市販のAMP 2を分注します。ティッシュ部分が気泡なしで完全に覆われていることを確認してください。スライドを湿度チャンバー内で40°Cで15分間インキュベートします。 手順5.3〜5.4を繰り返し、それぞれ2分間洗浄します。
- 市販のAMP 3を分注します。ティッシュ部分が気泡なしで完全に覆われていることを確認してください。スライドを湿度チャンバー内で40°Cで30分間インキュベートします。 手順5.3〜5.4を繰り返し、それぞれ2分間洗浄します。
- 市販のAMP4をディスペンスします。ティッシュ部分が気泡なしで完全に覆われていることを確認してください。スライドを湿度チャンバー内で40°Cで15分間インキュベートします。 手順5.3〜5.4を繰り返し、それぞれ2分間洗浄します。
- 市販のAMP 5を分注します。ティッシュ部分が気泡なしで完全に覆われていることを確認してください。スライドを湿度チャンバー内で40°Cで30分間インキュベートします。 手順5.3〜5.4を繰り返し、それぞれ2分間洗浄します。
- 市販のAMP 6を分注します。ティッシュ部分が気泡なしで完全に覆われていることを確認してください。スライドを湿度チャンバー内で40°Cで15分間インキュベートします。 手順5.3〜5.4を繰り返し、それぞれ2分間洗浄します。
- 検出する前に、スライドを1x TBS-Tween 20(0.05% v/v)で1回すすぎます。スライドを洗浄バッファーから取り出し、すすぎ、スライドを軽くたたいたりフリックしたりして、組織切片から余分な洗浄バッファーを取り除きます。すぐに1x TBS-Tweenバッファーを充填した洗浄トレイに入れます。
6. チャンネル1(C1)ターゲット信号検出
注:これは、アルカリホスファターゼ標識を含む2-プレックス検出キット( 材料表を参照)の赤色アルカリホスファターゼおよび高速赤色発色増幅6、および発色性検出を使用して実行されます。高速赤を基板として使用して、赤信号を生成します。
- Fast RED-BからFast RED-Aへの1:60希釈を使用して、Fast Red(FR)ワーキング溶液を調製します。よく混ぜます。沈殿物を減らし、よりクリーンなシグナルを得るには、シリンジを使用して0.45 μm MCEメンブレンで発色剤溶液をろ過します。
注意: FastRED-B溶液を5分以内に使用してください。直射日光や紫外線にさらさないでください。 - TBS-Tweenからスライドを取り出し、すすぎ、スライドを軽くたたいたりフリックしたりして、組織切片から余分なバッファーを取り除きます。
- 混合してろ過したFR溶液を各組織切片に分注し、各切片が完全に覆われていることを確認します。室温で6〜8分間インキュベートします。顕微鏡で観察します。
- スライドを0.5x洗浄バッファー2xですすいでください。スライドを洗浄バッファーから取り出し、すすぎ、スライドを軽くたたいたりフリックしたりして、組織切片から余分な洗浄バッファーを取り除きます。
- 市販のAMP 7をディスペンスします。ティッシュ部分が気泡なしで完全に覆われていることを確認してください。スライドを湿度チャンバー内で40°Cで10分間インキュベートします。 手順5.3〜5.4を繰り返し、それぞれ2分間洗浄します。
- 市販のAMP 8をディスペンスします。ティッシュ部分が気泡なしで完全に覆われていることを確認してください。スライドを湿度チャンバー内で40°Cで15分間インキュベートします。 手順5.3〜5.4を繰り返し、それぞれ2分間洗浄します。
- 市販のAMP 9をディスペンスします。ティッシュ部分が気泡なしで完全に覆われていることを確認してください。スライドを湿度チャンバー内で40°Cで30分間インキュベートします。 手順5.3〜5.4を繰り返し、それぞれ2分間洗浄します。
- 市販のAMP10をディスペンスします。ティッシュ部分が気泡なしで完全に覆われていることを確認してください。スライドを湿度チャンバー内で40°Cで30分間インキュベートします。 手順5.3〜5.4を繰り返し、それぞれ2分間洗浄します。
7. チャネル2(C2)ターゲット信号検出
注:これは、市販のBrown HRPおよびDAB Chromogen Kitsを使用して行います( 材料の表を参照)。2-プレックス検出からの増幅10は、西洋ワサビペルオキシダーゼ標識を含み、DABを用いて発色検出を行い、褐色シグナルを生成する。
- DAB信号を最適に検出するには、市販のキットを使用し、製造元の指示に従ってください( 資料の表を参照)。顕微鏡で観察します。
注意:DABは有毒です。この化学物質を取り扱い、廃棄する際には、適切な予防措置と安全ガイドラインに従ってください。
8. 対比染色と埋込み
- スライドをヘマトキシリンで対比染色します。
- スライドを攪拌しながら、50%新鮮なフィルター付きヘマトキシリンでスライドを30秒間対比染色します。スライドは紫色で表示されます。水が透明になるまでスライドを上下に攪拌しながら、すぐに流水ですすいでください。ティッシュ切片は紫色のままです。
- コントラストを良くするには、対向染色したスライドを炭酸リチウムで飽和させた蒸留水に1分間置きます。スライドを攪拌しながら、流水で少なくとも3回十分にすすいでください。最終すすぎには二重蒸留水を使用してください。
- FRは有機溶媒に弱いため、FRで染色したスライドは水性封入剤で覆い、RTで一晩乾燥させる必要があります。
- スライドを取り付けます。
- 水性封入剤で覆われた組織切片が乾燥していることを確認してください。
- キシレンにディップスライドさせてから、封入試薬を使用してカバーを滑らせます。カバーガラスとティッシュ部分の間の気泡を防ぐか取り除き、RTで16時間乾燥させてください。
9. CellProfiler5を使用したRNAscopeの定量的画像解析プロトコール
- 簡単に言うと、ソフトウェアがヘマトキシリンとFR染色を別々の画像に分離することを確認します。目的のオブジェクト(核、ビリオン、陽性細胞、凝集FR陽性染色)を特定し、測定します。測定値をCSVファイルに保存し、解析した画像を保存します。
- 「Unmix colors」のオプションを選択すると、汚れが分離され、元の関心領域(ROI)がヘマトキシリンとFRの画像に分割されます。
- ヘモシデリン、タトゥー、または同様の特徴が分析を妨げる場合は、ヘマトキシリン、FR、およびDABで2番目の「アンミックス」ステップを追加します。2 番目の FR 画像を使用して、最も強度の高い FR ピクセルを見つけます。この 2 番目の画像は、真の FR 染色のマスクとして使用されます。
- 必要に応じて、しきい値を設定する前に、染色された画像を滑らかにします。これは経験的な決定であり、必ずしも必要ではありません。「IdentifyPrimaryObjects」を使用して個々の染色画像にしきい値を設定し、正のピクセルを選択します。
- 3つの異なるタイプのオブジェクト(ビリオン、ビリオン凝集体、生産細胞)を特定します。
- 核がヘマトキシリンで染色された直径4〜100ピクセル(px)の物体であることを確認してください。しきい値処理後に穴をデクランプして埋めます。「IntenseFastRed」オブジェクトは直径4〜100 pxで、ビリオン、陽性細胞、およびB細胞卵胞(BCF)の濾胞性樹状細胞(FDC)に見られるような凝集陽性染色が含まれます。この画像は、偽陽性(ヘモシデリンなど)を除外するために使用されます。
- FRの小さなポジが直径2〜12ピクセルのオブジェクトであることを確認してください。この測定値には、ビリオンとvDNA+細胞が含まれます。この範囲外のオブジェクトは破棄します。しきい値処理後に穴をデクランプして埋めます。
- FRの大きなポジティブ:直径9〜100ピクセルのオブジェクトを確認します。この測定には、vRNA+陽性細胞と凝集陽性染色が含まれます。しきい値処理後にデクランプします。小さいFRポジティブオブジェクトと大きいFRポジティブオブジェクトのサイズは重なることがあります。これらは後のステップで分離されます。
注:ソフトウェア(Cellprofilerなど)はオブジェクトサイズにピクセルを使用し、ここで使用されるものは0.2510μm/ピクセル(40x)でスキャンされたスライドから導き出されます。
- 結果を定義して抽出します。
- オブジェクトが特定されたら、ビリオンの数、生産的な感染細胞、および核の結果を定義して抽出します。
- 偽陽性 (ヘモシデリンなど) を削除するように設定された IntenseFastRed オブジェクトで FastRedSmallPositives をマスクして、ビリオンを識別します。
- 次に、陽性細胞を同定し、FR陽性染色を凝集させます。 FastRedLargePositives を IntenseFastRed と virion オブジェクト セットでマスクすることで、誤検出とウイルス粒子を削除します。
- 精製した FastRedLargePositives から陽性細胞を抽出し、 Nucleiで再度マスキングします。接触しなくなったオブジェクトを分割し、オブジェクト領域で結果をフィルタリングして、小さなオブジェクトを削除します(≤6 px)。これにより、核の重なりをマスクすることによって作成された斑点が除去されます。その結果、陽性の細胞が得られます。
- 最後に、アグリゲート FR ポジティブを定義します。この手順では、 IdentifyTertiaryObjects を使用して、より大きな親オブジェクトに含まれるオブジェクトを検索します。この場合、調整された FastRedLargePositives オブジェクト セットが親であり、正のセルが減算されます。
- ビリオンと陽性細胞の数を数えます。
- 総正の面積を測定し、それをピクセルからmm2に変換します。オプションで、ビリオンと陽性細胞が占める領域をmm2 で記録します。分析で直接カウントの代わりに標準セルとビリオンサイズを使用する必要がある場合。
- 元の画像にポジオブジェクトを重ねて、結果を保存します。
注:ISHデータは、106 核(細胞)あたりのビリオン数と106 核(細胞)あたりの生産的に感染したvRNA+細胞の数によって報告され、理解を深め、qPCRデータとの比較を容易にしますが、結果はmm2の組織の面積によって報告することもできます。
結果
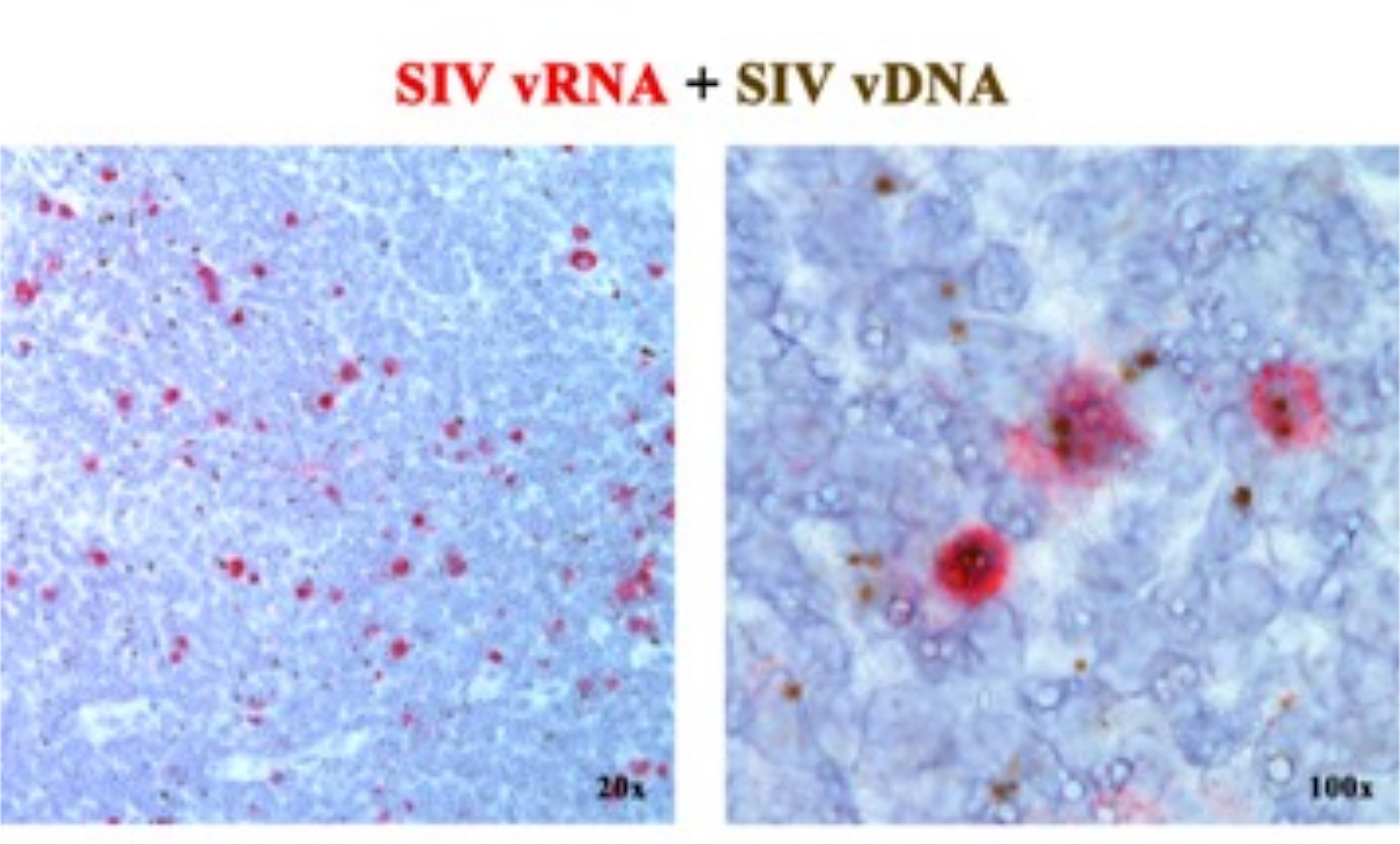
前回の論文2,6,7,8,9,10,11では、SIV/HIVゲノムの5'gag-pol部分を標的とするセンスプローブ(vDNA)と、ゲノムの3'半分の遺伝子を標的とするアンチセンスプローブ(vRNA)を用いて、vRNAまたはvDNAのいずれかを検出する次世代ISHプラットフォームを組み合わせることができることを報告した(vif、vpx、vpr、 tat、env、nef)、および5'ゲノムのTAR要素(表1)。このアプローチでは、転写活性細胞(vRNA+、vDNA+)を、転写活性細胞(潜在活性と推定される)感染細胞、または転写能力の低いプロウイルス(vRNA-、vDNA+)を同じ組織切片2に保有する細胞と区別します(図1)。

図1:同じ組織切片でのウイルスRNAとvDNAの検出。 急性SIV感染RMリンパ節におけるRNAハイブリダイゼーション(赤)とDNAハイブリダイゼーション(茶)の両方のアッセイの組み合わせにより、同じ組織切片でvRNAとvDNAを検出する能力を実証し、転写的にサイレントなvDNA+ vRNA-細胞をその場で同定するための強力なアプローチを提供します。この図は、Deleage C. et al.2から修正されています。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
| シングルプレックスRNA/DNAscopeプローブセット | |||
| 名前 | ACDカタログ# | ZZの数 | 形容 |
| SIVmac239(アンチセンス) | 312811 | 83 | D01065.1 の 1251-9420bp 以内をターゲットとするアンチセンスプローブ (gag、pol、vif、vpx、vpr、tat、env、および nef) |
| SIVmac239 (センス) | 314071 | 83 | D01065.1 の 1251-9420bp 以内の逆鎖を標的とするセンスプローブ (gag、pol、vif、vpx、vpr、tat、env、および nef) |
| V-HIV1-クレードA(アンチセンス) | 416101 | 80 | HIV-1 Clade A コンセンサス (gag、pol、vif、vpr、tat、rev、vpu、env、および nef) の 879-7629bp 以内を標的とするアンチセンスプローブ |
| V-HIV1-クレードA(センス) | 426341 | 80 | HIV-1クレードAコンセンサス(gag、pol、vif、vpr、tat、rev、vpu、env、およびnef)の879-7629bp内の逆鎖を標的とするセンスプローブ |
| V-HIV1-クレードB(アンチセンス) | 416111 | 78 | AF324493.2、HIV-1 Clade B NL4-3 (gag、pol、vif、vpr、tat、rev、vpu、env、および nef) の 854-8291bp 以内を標的とするアンチセンス プローブ |
| V-HIV1-クレードB(センス) | 425531 | 78 | AF324493.2、HIV-1 Clade B NL4-3 (gag、pol、vif、vpr、tat、rev、vpu、env、および nef) の 854-8291bp 以内の逆鎖を標的とするセンスプローブ |
| V-HIV1-クレードD(アンチセンス) | 416121 | 76 | HIV-1 Clade D コンセンサス (gag、pol、vif、vpr、tat、rev、vpu、env、nef) の 894-7697bp 以内を標的とするアンチセンスプローブ |
| V-HIV1-クレードD(センス) | 426351 | 76 | HIV-1クレードDコンセンサス(gag、pol、vif、vpr、tat、rev、vpu、env、nef)の894-7697bp以内の逆鎖を標的とするセンスプローブ |
| Multi-Plex RNA/DNAscopeプローブセット | |||
| 名前 | ACDカタログ# | ZZの数 | 形容 |
| V-SIVmac239-ギャグ-ポル-センス-C1 | 416141-C1 | 40 | D01065.1の1251-4093bp以内の逆鎖を標的とするセンスプローブ(ギャグおよびポール) |
| V-SIVmac239-vif-env-nef-tar-C2 (アンチセンス) | 416131-C2の | 47 | D01065.1 の 5381-10257bp 以内をターゲットとするアンチセンスプローブ (vif、vpx、vpr、tat、env、nef、および TAR 素子) |
| V-HIV1-Clade_B-ギャグ-ポル-センス-C1 | 444051からC1 | 40 | AF324493.2、HIV-1 Clade B NL4-3 (gag and pol) の 854-3940bp 以内の逆鎖を標的とするセンスプローブ |
| V-HIV1-Clade_B-vif-vpr-tat-rev-vpu-env-nef-tar-C2 (アンチセンス) | 444061-C2の | 40 | AF324493.2、HIV-1クレードB NL4-3(vif、vpr、tat、env、nef、およびTARエレメント)の5042-9673bp以内を標的とするアンチセンスプローブ |
| V-HIV1-Clade_C-ギャグ-ポル-センス-C1 | 444021-C1の | 48 | HIV-1 Clade Cコンセンサス配列(gagおよびpol)の888-5032bp以内の逆鎖を標的とするセンスプローブ |
| V-HIV1-Clade_C-vif-vpr- rev-vpu-env-nef-tar-C2 (アンチセンス) | 444041-C2の | 49 | HIV-1 Clade Cコンセンサス配列(vif、vpr、tat、env、nef、およびTARエレメント)の5078-9698bp以内を標的とするアンチセンスプローブ |
| V-HIV1-Clade_AE-ギャグ-ポル-センス-C1 | 444011からC1 | 55 | AF259954.1、HIV-1 Clade AE(ギャグおよびポール)の890-4812bp以内の逆鎖を標的とするセンスプローブ |
| V-HIV1-Clade_AE-vif-vpr-tat-rev-vpu-env-nef-tar-C2 (アンチセンス) | 444031からC2 | 57 | AF259954.1、HIV-1 Clade AE (vif、vpr、tat、env、nef、および TAR 要素) の 5052-9694bp 以内を標的とするアンチセンスプローブ |
表1:HIV-1およびSIVのvRNAおよびvDNAを標的とするプローブのリスト。
ディスカッション
In situハイブリダイゼーションは、核酸化学、細胞生物学、および組織学に関する厳密で基本的な知識を必要とする綿密なアッセイであり、十分に保存された環境でターゲットを局在化するための各重要なステップを適応させることができます。このディスカッションでは、正確で解釈可能な結果を得るためにトラブルシューティングが重要な重要なステップに焦点を当てたいと思います。
組織の固定と処理は重要であり、アッセイが最良の結果をもたらすことができるように、前もって対処する必要があります。中性緩衝PFA(4%調製したばかり)固定液は、デュプレックスアッセイに最適です。ただし、このアッセイは、凍結切片後の適切な固定条件で凍結組織(OCT)でも実行できます。
組織切片の前処理は重要なステップです。このアッセイには2つの前処理ステップがあり、1つ目は熱誘起エピトープ賦活化(HIER)です。このステップは、メチレン架橋の架橋の逆転や、固定組織に必要なタンパク質構造の修復に重要です。この処理の効率は、時間、温度、回収バッファーの種類、およびpHによって異なります。2つ目の前処理は、プロテアーゼ誘起エピトープ賦活化(PIER)です。このステップでは、ペプチドを切断し、抗原またはヌクレオチドを露出させ、プロテイナーゼK、トリプシン、ペプシンなどの酵素を使用します。これは非常に感度の高いステップであり、組織の形態と目的のターゲットの両方に損傷を与える可能性があります。このプロセスでは、酵素の濃度、およびインキュベーションの時間と温度が重要です。過剰消化は、核の境界が不十分になり、定量ステップが困難になります。RNA/DNAターゲットへの最適なアクセスと、目的の組織やターゲットに損傷を与えない前処理条件とのバランスを見つけることが重要です。各組織タイプは、これらの前処理のそれぞれに対して異なるレベルの感度を持っており、各パラメータ(酵素濃度、時間、温度)を経験的にテストする必要があります。
洗浄バッファーの厳密性は、温度、塩と洗剤の濃度、および時間の3つの主要なパラメーターに基づいています。洗浄バッファーは生理食塩水クエン酸ナトリウムバッファー(SSC)であり、バッファー内の塩濃度は洗浄ステップ中のストリンジェンシーを制御します。彼らのプロトコルでは、ACDは、0.1x SSC、0.03%ドデシル硫酸リチウムの最終濃度で洗浄バッファーを使用することを勧めています。DNAscopeとマルチプレックス最適化に取り組む中で、最終濃度0.05x SSCのウォッシュバッファーを使用すると、DNAシグナルの可視化結果が向上し、センスプローブの夜間インキュベーションに起因する非特異的なオフターゲットハイブリダイゼーションを大幅に減らすことができると判断しました。
検出アプローチ、発色剤(赤色または茶色)と蛍光性のどちらを選択するかは、アッセイを開始する前に、組織の種類と目標に基づいて検討する必要があります。赤は組織に自然に見られないため、赤の発色アプローチは素晴らしいコントラストを与えます。茶色の発色剤は、赤色の発色剤と同様の結果をもたらします。ただし、組織に存在する一部の血液分解生成物は同様の色をしており、タトゥーインクは定量化中に茶色の信号から分離するのが難しいことに留意することが重要です。蛍光検出アプローチにより、さまざまな細胞マーカーを明確に区別でき、マルチプレックス化により、vRNAおよび/またはvDNAを保有する細胞の表現型を完璧にアッセイできます。
プローブの特異性とアッセイの品質を確保するためには、複数のコントロールが必要です。新しく設計された各プローブは、既知の陽性および陰性のコントロール組織または細胞ペレットでテストする必要があります。私たちはしばしば、標的配列を含むプラスミドを作製し、細胞株へのトランスフェクションを行ってポジティブコントロールを生成します。各分析では、既知の陰性組織(HIVまたはSIV陰性)、プローブ希釈剤のみを含むプローブなしコントロール、およびRNase処理コントロールを追加して、アッセイの品質と特異性を確保します。
定量化は非常に重要なステップであり、尋ねられた質問に基づいて適切なツールとアルゴリズムを使用して実行する必要があります。この原稿では、複数のオプションを評価した後に選択した画像解析ソフトウェア(Cellprofilerなど)を紹介しました。このソフトウェアが私たちのニーズに最適なソフトウェアであると推定しましたが、使用できる画像分析ソフトウェアプログラムは多数あります。
開示事項
著者は何も開示していません。
謝辞
このプロジェクトは、契約番号の下で、国立がん研究所、国立衛生研究所からの連邦資金で全額資金提供されています。HHSN261200800001Eおよびオレゴン国立霊長類研究センターNIH助成金P51OD011092(JDE)によって授与されました。この出版物の内容は、必ずしも保健福祉省の見解や政策を反映しているわけではなく、商号、商品、または組織についての言及は、米国政府による承認を意味するものではありません。デュプレックスは、Advanced Cell Diagnosticsの助けを借りて開発されました。
資料
| Name | Company | Catalog Number | Comments |
| ACD HybEZII Hybridization system (110V) with ACD EZ-Batch Slides system | ACD | 321710 | Hybridization oven |
| CAT Hematoxylin | Biocare medical | CATHE-GAL | colorstain |
| Clear-Mount | ELECTRON MICROSCOPY SCIENCES | 17985-15 | mounting reagent for red chromogen |
| Immpact DAB Peroxidase Kit | Vector | SK-4105 | Used to reveal HRP - DAB (Brown) to replace the DAB coming in the ACD kit |
| lithium carbonate | Fisher chemical | L119-500 | bluing solution |
| paraformaldehyde | ELECTRON MICROSCOPY SCIENCES | 15714-S | for tissue fixation (4%) |
| PBS | life technology | 14190-136 | |
| Permount Mounting Medium | ThermoFisher Scientific | SP15-100 | mounting regaent for brown chromogen |
| Prolong Gold | ThermoFisher Scientific | P36930 | mounting regaent for fluorescence |
| ribonucleases A | ThermoFisher Scientific | 12091039 | for RNAse treatment in DNAscope protocol |
| ribonucleases T1 | Roche | R1003 | for RNAse treatment in DNAscope protocol |
| RNAscope 2.5, 2-plex detection reagent | ACD | 322430 | Brown and red kit chromogen detection |
| RNAscope Target Retrieval Reagents | ACD | 322000 | retrieval buffer |
| SuperFrost Plus Glass Slides | ThermoFisher Scientific | 12-550-17 | |
| TBS | BOSTON BIOPRODUCTS | BM-301-4L | for washes |
| TSA Plus Fluorescence palette kit (Cy3, Cy5, TMR, Fluorescein) | Perkin elmer | NEL760001KT | HRP Fluorescence detection |
| Tween 20 | SIGMA | P1379-1L | for washes |
| XYLENE 20LT | ThermoFisher Scientific | AC422680200 |
参考文献
- Wang, F., et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. Journal of Molecular Diagnosis. 14 (1), 22-29 (2012).
- Deleage, C., et al. Defining HIV and SIV Reservoirs in Lymphoid Tissues. Pathogens and Immunity. 1 (1), 68-106 (2016).
- Sengupta, S., Siliciano, R. F. Targeting the Latent Reservoir for HIV-1. Immunity. 48 (5), 872-895 (2018).
- Churchill, M. J., Deeks, S. G., Margolis, D. M., Siliciano, R. F., Swanstrom, R. HIV reservoirs: what, where and how to target them. Nature Reviews Microbiology. 14 (1), 55-60 (2016).
- McQuin, C., et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biology. 16 (7), e2005970(2018).
- Deleage, C., Chan, C. N., Busman-Sahay, K., Estes, J. D. Next-generation in situ hybridization approaches to define and quantify HIV and SIV reservoirs in tissue microenvironments. Retrovirology. 15 (1), 4(2018).
- Deleage, C., Turkbey, B., Estes, J. D. Imaging lymphoid tissues in nonhuman primates to understand SIV pathogenesis and persistence. Current Opinion in Virology. 19, 77-84 (2016).
- Estes, J. D., et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nature Medicine. 23 (11), 1271-1276 (2017).
- Mavigner, M., et al. Simian Immunodeficiency Virus Persistence in Cellular and Anatomic Reservoirs in Antiretroviral Therapy-Suppressed Infant Rhesus Macaques. Journal of Virology. 92 (18), (2018).
- Peterson, C. W., et al. Differential impact of transplantation on peripheral and tissue-associated viral reservoirs: Implications for HIV gene therapy. PLoS Pathogen. 14 (4), e1006956(2018).
- Deleage, C., et al. Impact of early cART in the gut during acute HIV infection. Journal of Clinical Investigation Insight. 1 (10), e87065(2016).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved