
Method Article
連続結晶構造解析実験のためのエンドチアペプシン結晶の成長の最適化
要約
この記事の目的は、大きな単一タンパク質結晶を成長させるための少量の蒸気拡散プロトコルを、シリアル結晶学用の大量のバッチマイクロ結晶化法に変換する方法を視聴者に確実に理解させることです。
要約
ここでは、シンクロトロンとXFELの両方での連続結晶学実験に適した大量(> 100 μL)の微結晶スラリーの作成を容易にするためのプロトコルが提示されています。この方法は、タンパク質結晶相図の理解と、その知識をどのように活用できるかに基づいています。この方法は、(1)結晶形態の最適化、(2)バッチへの移行、(3)スケーリングの3つの段階に分けられます。ステージ1では、うまくいけば、必ずしもそうとは限りませんが、立方体のような形態で提示される、よく回折する単結晶を見つけます。ステージ2では、ステージ1の条件は結晶成長時間によって最適化されます。この戦略は、蒸気拡散によって成長した結晶をバッチに変換できます。約24時間以内に結晶成長が起こると、タンパク質と沈殿剤混合物のモルフォグラムをプロットし、スケーリング戦略の基礎として使用できます(ステージ3)。結晶をバッチで成長させることができる場合、スケーリングを試みることができ、体積が増加するにつれて結晶サイズと濃度が最適化されます。エンドチアペプシンは、このプロトコルの実証タンパク質として使用されています。提示された決定のいくつかはエンドチアペプシンに特有のものです。しかし、それらが適用された方法が、他の人が自分のプロジェクトに適応できるこの手順についての考え方を刺激することが期待されています。
概要
室温(RT)高分子結晶構造解析は、構造生物学のコミュニティで再び人気があります。X線自由電子レーザー(XFEL)光源の開発は、RTサンプル送達アプローチ1,2,3,4の開発に拍車をかけ、これらの方法はシンクロトロン5,6,7,8に適用されています。RT法は、ポンププローブ実験の可能性を開くだけでなく、タンパク質13,14,15,16,17内の代替立体配座状態を促進するという証拠も増えています。
しかし、1990年代後半にクライオ法がRTアプローチよりも勢いを増した主な理由は、氷点下の結晶温度による放射線損傷の減速でした18。クライオ法19は、単一のタンパク質結晶から完全なデータセットの収集を可能にし始めました。XFELおよびシンクロトロンにおける最新のRT法は、高速(> 100Hz)結晶送達戦略の開発によって単結晶放射損傷の問題を解決した1,2,3,4。これらの方法により、個別に露出した何千もの結晶から完全なデータセットを収集できます。したがって、これらのRT送達アプローチでは、均質な微結晶(>100 μLの< 50 μm結晶)を含む大量の溶液を製造する必要があります。しかし、クライオ法は単結晶のみを必要とする傾向があるため、このような微結晶スラリーを作成する方法は、現在、タンパク質結晶構造解析ラボ全体で遍在していません。
文献には、連続結晶構造解析サンプルの微小結晶化最適化手順の一部を実行する方法の例があります。ここでは、膜タンパク質と可溶性タンパク質を区別する必要があります。モノオレイン(または他の何らかの脂質)中で成長させた微小膜タンパク質結晶の成長を最適化するためのプロトコルは、脂質立方相(LCP)について、十分に記載されている20、21、22。しかしながら、非LCP条件下で増殖させた膜タンパク質を含む可溶性タンパク質の微小結晶化のための方法は一般に欠けている。以前の研究では、微結晶スクリーニング23,24、核生成の増強24、フリー界面拡散25を使用したスケーリングなど、プロセスの特定の部分に焦点を当ててきましたが、完全な方法ではありませんでした。
しかしながら、完全なプロトコルを提供しようとする方法が最近記載された26 。タンパク質結晶学の多くの側面と同様に、それは新しいものではありません。提案されたアイデアの多くは、Rayment(2002)27によってすでに説明されています。この方法は、蒸気拡散を使用して成長させた単一の結晶から、数千の微結晶を成長させるバッチ手法への変換を実行する方法を結晶学者に示すことを目的としています。この方法は、すべてのタンパク質データバンク(PDB)堆積物の95%が蒸気拡散プレート26内で成長した結晶に由来するため、共通の出発点として蒸気拡散に焦点を当てる。しかしながら、蒸気拡散は微小結晶化のための理想的な方法ではない26ので、蒸気拡散をバッチ結晶化に変換する方法論が説明される。結晶をバッチで成長させることができれば、より大きなボリュームへのスケーリングルートがより実用的になります。タンパク質の結晶化の気まぐれさを考えると、著者らは、この方法がフェイルセーフではないことを強調するだろう。ただし、プロトコルは、少なくとも、タンパク質の「結晶化空間」への洞察を提供する必要があります。
この方法は、タンパク質結晶化相図と、その図を理解することが微小結晶化の最適化中のガイドとしてどのように機能するかに依存しています。タンパク質の状態図は、通常、x軸とy軸にそれぞれ沈殿剤とタンパク質濃度を持つx/yプロットとして表されます(図1A)。純水点(左下隅-図1A)から、タンパク質と沈殿剤の両方の濃度が溶解度線に達するまで増加します。溶解度線は過飽和点を示します(紫色の線-図1A)。タンパク質が過飽和になると、溶液は熱力学的に不安定になり、「タンパク質が豊富な」溶液と安定した飽和溶液の2つの相に分離し始めます。この分離は溶解度線を超えた場所で発生する可能性があり、その速度論はタンパク質の特性と溶液の成分に依存します。
タンパク質と沈殿剤の濃度が高すぎると、タンパク質は溶液から不安定に分解し、非晶質の沈殿物を生成します(ピンク色の領域- 図1A)。しかし、規則的な相分離は核生成領域で発生する可能性があり[詳細な説明についてはGarcia-Ruiz(2003)28 を参照]、結晶核は形成される傾向があります(緑色領域- 図1A)。核生成と成長は溶液からタンパク質を除去し、溶解度線に達するまで成長を続けることができる準安定領域に液滴を移動させます[詳細な議論についてはMcPherson and Kuznetsov (2014)29 を参照]。この図は、結晶化条件の大部分について、大部分が過度に単純化されている30。ただし、これに関係なく、ダイアグラムのマッピングにより溶解度線と核生成の速度論を決定できるため、このダイアグラムはマイクロ結晶学者にとって依然として非常に有用です。
微結晶の生成に関して、最適化する必要がある結晶化中の2つの要因は、結晶の数(Xn)とそれらの平均最長寸法(Xs)です。X nは核生成イベントの数(n )に比例します(式1)。
 式 1
式 1
Xsは 、溶解度線(Ps)をXn で割った値(式2)より上の遊離タンパク質の濃度に比例する。
 式 2
式 2
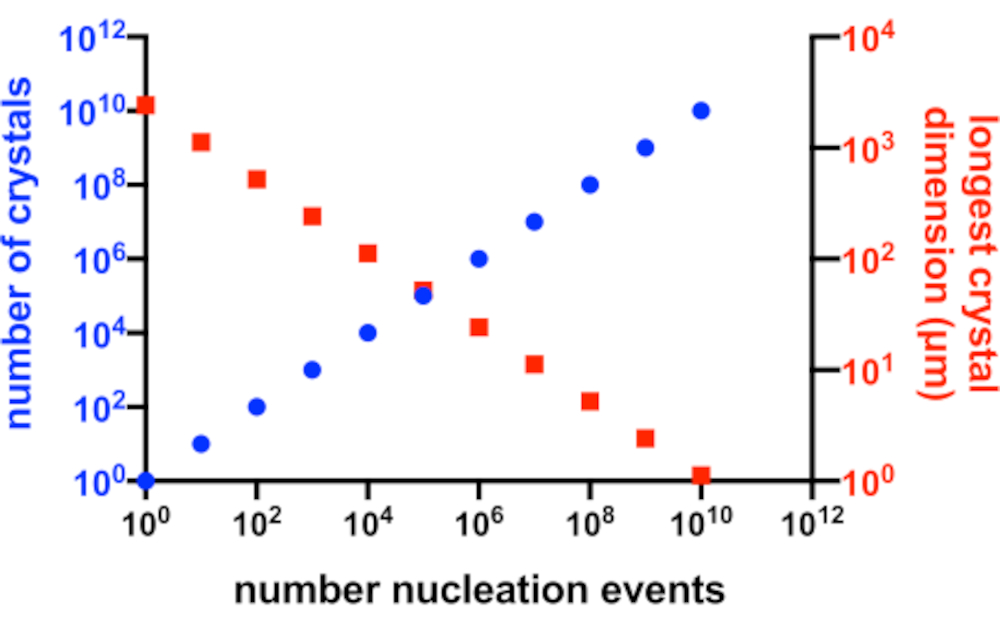
完璧な状況では、すべての核生成イベントで可能な結晶が生成され、これらの結晶のすべてが溶液中の利用可能なタンパク質に等しくアクセスできます。 図2 は、Xn とXsとの関係の理想的なシナリオからのグラフ表現である。実際には、結晶学者がXn およびXs に対して有する主要な制御は、核生成の量に影響を与えること、または種結晶の添加によるものである。マイクロ結晶学者は、適切な結晶濃度と結晶サイズの両方を作成できるように、Xn を増加させる方法を判断する必要があります。
結晶化技術の大部分は「移行期間」を必要とします(図1B)。例えば、蒸気拡散実験では、タンパク質溶液と沈殿剤溶液を混合すると、液滴がウェル溶液と平衡化するにつれて、それぞれの濃度が変化します。これらの変化が、結晶化の傾向が増加する核生成ゾーンへのドロップを徐々に移行させることが期待されます。結晶が核形成して成長し始めると、溶液中のタンパク質の量が減少し始め、さらなる核形成の可能性が減少します。核生成の最終的な量は、タンパク質と条件に固有であり、核生成ゾーンへの浸透の深さにも依存します。移行段階を必要とする方法の核生成ゾーンの浸透が制限されていることを考えると、核生成のレベルは、最終的には準安定核生成領域の境界での核生成速度に制限されます。
微結晶学者にとって核生成レベルを高めることができることが重要であるため、バッチ結晶化方法論に移行することが重要です。バッチは、核生成領域全体をより有効に活用できます(図1C)。バッチ法では、タンパク質と沈殿剤を混合して、成分濃度を変更することなく過飽和溶液を作成するという考え方です。核生成は混合後すぐに可能であるはずです。したがって、バッチ法では、理論的に核生成ゾーン全体に到達することができます。準安定核生成境界を超えた核生成速度論の増加を利用することができます。
結晶核生成の基礎レベルが大きなXnを生成するのに十分でない場合は、マイクロシード法を使用することができる。マイクロシーディングでは、予め成長した結晶を分解して、新鮮な結晶成長のための足場として作用することができる結晶断片のスラリーを作成する31、32。マイクロシーディングは、結晶核生成を増加させることなくXn を増加させる方法として、連続結晶学的サンプル調製において広く使用されています(図1C)。
蒸気拡散からバッチへの遷移は、実験の開始点を非過飽和領域または準安定領域から核生成ゾーンに移動するものとして、相図上で視覚化できます。これは、タンパク質および/または沈殿剤の濃度、および/または液滴内の2つの比率を増加させ(図1D)、どの条件が急速に現れる結晶を生成するかを観察することによって行うことができます(<24時間)26。完全な蒸気拡散液滴平衡化には数日または数週間かかることがあります33。したがって、急速に現れる結晶を示す条件を探すことによって、マイクロバッチ34、35、36、37などの代替結晶化スクリーニングフォーマットに移行することなく、バッチ条件を見出すことができる。
核生成ゾーンが見つかると、バッチ条件が見つかり、モルフォグラム(ここでは大まかな状態図)を作成できます。モルフォグラムは、シードバッチプロトコルとストレートバッチプロトコルのどちらを使用するかを検討するときに非常に役立ちます。Xnをタンパク質および沈殿剤濃度の関数としてプロットすることによって、核生成速度論の評価を行うことができる26。Xnが核生成領域全体にわたって低いままである場合、結晶成長を制限するのに十分な大きさのXnを作るためにシードバッチが必要となるかもしれない。この評価は、大容量(> 100 μL)へのスケーリングプロセスの最初のステップです。
この方法は、標準的な蒸気拡散結晶化装置を使用して、ほとんどの結晶化実験室で実施できるように設計されています。機器が利用可能である場合、このプロセスの多くの部分を容易にする技術を説明する多くの研究も実施されています。これらには、動的光散乱(DLS)25,27、非線形イメージング20,24,25、粉末回折20,24,27、および電子顕微鏡26が含まれますが、これらに限定されません[素晴らしいレビューについては、Cheng et al. (2020)40を参照してください]。
この研究の目的は、少量(< 500 nL)の蒸気拡散結晶化から大容量(> 100 μL)のバッチ結晶化に移行する方法の視覚的なデモンストレーションを提供することです。 Cryphonectria parasitica 由来のエンドチアペプシンは、この翻訳を実証するためのシステム例として使用されています。微結晶が必要とされる実験のタイプおよび試料送達方法は、理想的なXs 出力26に影響を与えるであろう。ミリ秒の時間分解能41 または気体力学仮想ノズル42を必要とする混合実験のためには、5μm<の最終Xsが 望ましい場合がある。この場合の目標は、光子活性化ポンププローブ実験のために、固定ターゲット送達アプローチを使用して、約1.5 Åまで回折するタンパク質結晶を生成することでした。
エンドチアペプシンを用いたこのような連続結晶学実験のサンプル要件の説明を与えるために、 表1 は、仮想実験の実験パラメータを示す。サンプル情報は、以下に説明するプロトコルに基づいていました。ヒット率とデータ収集要件に関する控えめな推定値を考えると、50 mgは実験全体の総サンプル消費量の推定値です。
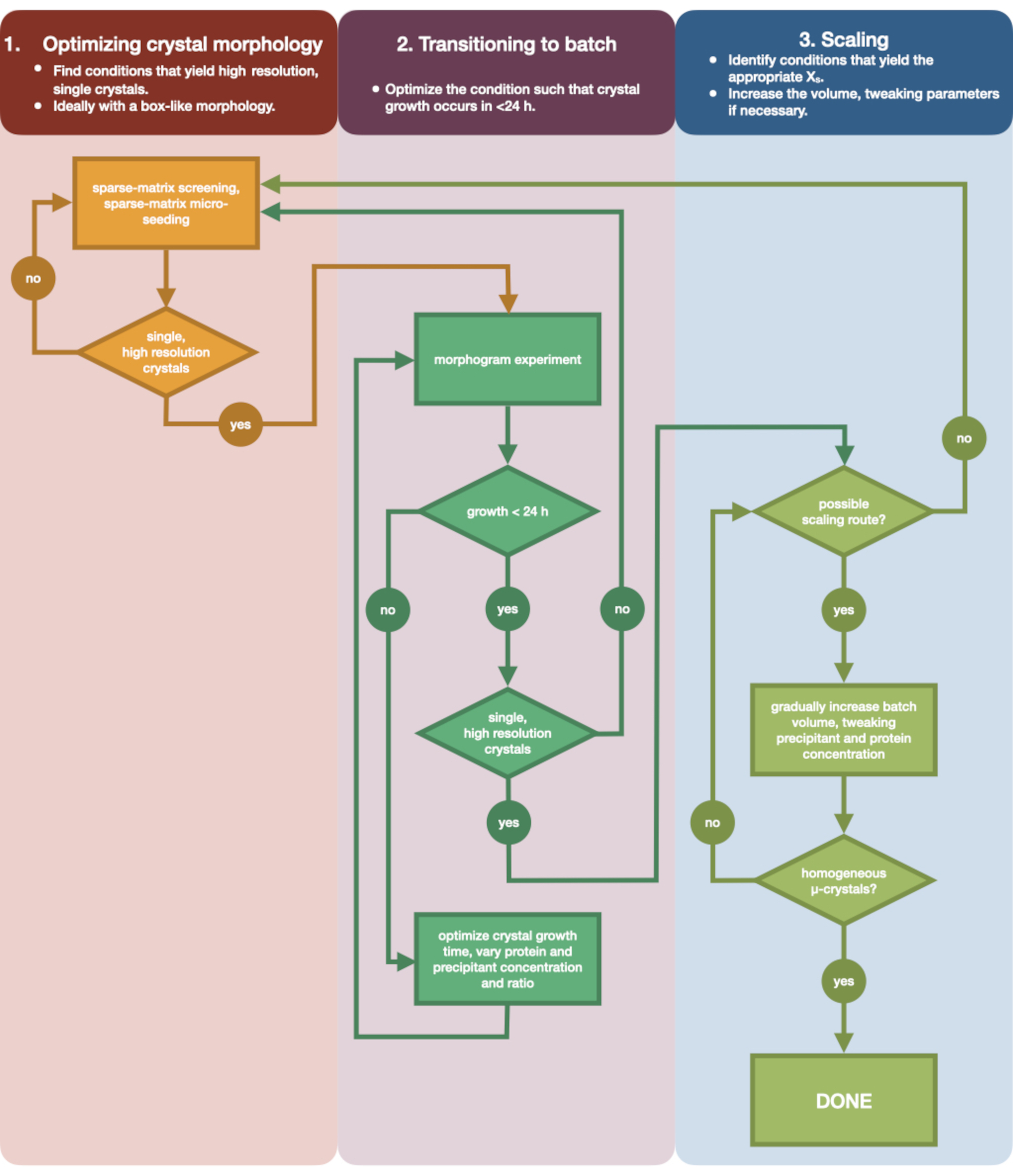
図3 は、初期の小容量蒸気拡散結晶化から大規模バッチまでの完全な最適化プロセスのフローチャートを示しています。ほとんどのシリアル結晶構造解析プロジェクトでは、ターゲットタンパク質はすでに結晶化されているため、このプロトコルはステップ2「バッチへの移行」から始まります。ただし、ステップ1は、完全を期し、読者にその重要性を思い出させるために含まれています。よく回折する単一の大きな結晶を生じる条件を見つけることは、微小結晶最適化の最良の出発点です。ステップ2では、この条件を蒸気拡散からバッチまで最適化し、核生成領域と準安定領域のモルフォグラムをプロットできます。これが完了したら、バッチ条件をより大きなボリュームにスケーリングすることをステップ3で実行できます。フローチャートの終わりまでに、結晶学者はエンドチアペプシンの再現性のある大容量(> 100 μL)の微量結晶化バッチプロトコルを作成します。この方法は、次いで、目的の特定のタンパク質に適用することができる。
プロトコル
注:すべての96ウェルシッティングドロップ結晶化実験は、2または3ドロッププレートのいずれかを使用してセットアップされました。リキッドハンドリングロボットと結晶化イメージャー/ホテルを使用して、すべての96ウェルスクリーンの準備とモニタリングを容易にしました。結晶化実験のためのすべての試薬濃度は、混合前の開始濃度で与えられます。
1. 結晶形態の最適化
注: 手順 1.1.1.および 1.1.6.エンドチアペプシンの結晶化条件がどのように見つかったか、およびこれらの条件を最適化して、単一のよく回折する結晶を生成する単一の条件を見つける方法を説明する。
- スパース行列の最適化
- 新鮮なエンドチアペプシン溶液を調製する。
注:エンドチアペプシンは、Superan 600として調達する場合、保存溶液からバッファーを移して濃縮する必要があります。- 3 Lの0.1 M Na酢酸塩pH 4.6を4°Cで調製します。
- 透析チューブを20 cm切断し、バッファーで短時間洗浄します。クリップを使用してチューブの一方の端をシールし、50 mLのエンドチアペプシン溶液をチューブに入れてから、もう一方の端をシールします。
- 溶液を1 Lの酢酸Na緩衝液中で4°Cで少なくとも4時間(または一晩)透析するために放置します。保存バッファーの成分により、透析バッグ内の溶液は約100 mLになります。
- エンドチアペプシンを入れた透析バッグを、4°C、0.1 M Na 酢酸pH 4.6の新鮮なリットルに移します。元のバッファーが酢酸Naに対して2000倍に希釈されるように、この手順をもう一度繰り返します。
- エンドチアペプシンは約10 mg / mLになります。10 kDa遠心濃縮機と遠心分離機を使用して100 mg/mLまで濃縮します。
- エンドチアペプシン溶液を液体窒素中の50 μLアリコートで瞬間冷却し、-80°Cで保存します。
- PACTプレミア96ウェルスパースマトリックススクリーンを準備します。
- リキッドハンドリングロボットを使用して、100 nL の 70 mg/mL エンドチアペプシンと 100 nL のウェル溶液を 1 つのサブウェルに分注します。タンパク質とウェル溶液を結晶化バッファーの添加時に3回混合する。
- プレートを密封し、20°Cで28日間放置し、最初の1週間は毎日、その後は4週間毎週画像を撮影します。
- スパース行列解析
- 単一のエンドチアペプシン結晶を生成するヒットを特定します。PACTスクリーニングから、MgCl2 を含む条件は、針クラスターではなくシングルトンとして成長した。
- スパース行列の最適化
- ステップ1.1.3.1で特定したMgCl2 含有条件から、異なるウェル成分をランダムに組み合わせて変化させる96ウェルスクリーンを作成します。
- リキッドハンドリングロボットを使用して、100 nL の 70 mg/mL エンドチアペプシンと 100 nL のウェル溶液を 1 つのサブウェルに分注します。タンパク質とウェル溶液を結晶化バッファーの添加時に3回混合する。
- プレートを密封し、20°Cで28日間放置し、最初の1週間は毎日、その後は4週間毎週画像を撮影します。
- 最適化分析
- 適切な表計算ソフトを用いて、結晶品質および沈殿レベル、結晶なし(0)〜理想(5)および低(0)〜高(5)に基づいて、結晶を生じる結晶化条件をそれぞれランク付けする。結晶品質に関しては、幅広い基準は箱状の形態を有する単結晶である。
- 晶析条件の内容と結晶量および析出準位とのピアソンの相関分析を行います。
- これらのデータをヒート マップとしてプロットします。好ましい結果と相関した成分と条件を探します。
- 回折分析。
- ステップ1.1.5で特定した条件から成長した結晶が連続結晶構造解析に適していることをX線回折実験により確認する。
- 同定された各条件からのエンドチアペプシン結晶のサンプルを、100または293 Kのいずれかでデータ収集を可能にする支持体にロードし、X線回折実験を実行します。クライオで作業する場合は、クライオ保護剤として25%エチレングリコールを使用してください。
- 適切なソフトウェアスイート を介して これらのデータを処理します。エンドチアペプシン結晶は1.5 Åを超えて回折するはずです。双晶は連続結晶データ処理を著しく複雑にする可能性があるため、双晶を確認してください。
- 結晶がシングルトンで1.5 Åまで回折する場合は、ステップ2に進みます。そうでない場合は、ステップ1.1.2に戻り、よりスパースマトリックス画面を試して、有望な条件を特定します。ステップ1.1.5で行った分析の後。1.1.6.、25%(w / v)PEG 6,000、0.1 M Tris-HCl pH 7.0および0.15 M MgCl2 の結晶化条件がおおよその理想として見出されるはずであった。
- 新鮮なエンドチアペプシン溶液を調製する。
2. バッチへの移行
- モルフォグラム実験
- マイクロクリスタルシードストックを作成します。
注:シードストックを作るときは、タスクのために特別に成長させた結晶からシードを作るのがベストプラクティスです。これは再現性に大いに役立ちます。ステップ2.1.1.1から2.1.1.11で提示された他のアイデアは、常に標準数のウェル(ここでは5)から成長した結晶を使用し、凍結融解サイクルを無効にするためにストックを分注することです。- 結晶化バッファーを含むウェルを含む96ウェル結晶化プレートを準備します:25%(w / v)PEG 6,000、0.1 M Tris-HCl pH 7.0および0.15 M MgCl2。
- リキッドハンドリングロボットを使用して、解凍した70 mg/mLエンドチアペプシン200 nLとウェル溶液200 nLをウェルあたり1つのサブウェルに分注します。タンパク質とウェル溶液を結晶化バッファーの添加時に3回混合する。
- プレートを密封し、24時間放置します。
- 1.5 mLの遠沈管に250 μLの結晶化バッファーと10-15個の1 mmガラスビーズを入れます。遠沈管を氷の上に置いて5〜10分間冷却します。
- 結晶を含む5つのウェルを選択し、メスでウェルを開き、ピペットチップを使用してウェル内の結晶を粉砕します。
- 氷結した遠沈管から1 μLのバッファーを吸引し、破砕した結晶スラリーをホモジナイズします。均質になったら、スラリー全体を吸引し、冷却した遠沈管に集めます。
- 5つのサブウェルのそれぞれについてステップ2.1.2.6を繰り返します。
- バッファー、プールされたスラリー、ビーズを入れた遠沈管を1000rpmで30秒間ボルテックスします。
- 遠沈管を氷に30秒間戻します。
- 手順 2.1.2.8 と 2.1.2.9 をさらに 2 回繰り返します。
- これでシードストックの準備が整い、10 μLのバッチに分注し、-20°Cで保存できます。
- モルフォグラム実験を行います。
- 2ドロップ96ウェルグリッドスクリーンを準備します。プレートカラムに沿ってPEG 6,000の濃度を5〜40%(w / v)の範囲で変化させ、バッファーと塩をそれぞれ0.1 M Tris-HCl pH 7.0および0.15 M MgCl2に保ちます。
- エンドチアペプシンを0.1 M Na 酢酸塩pH 4.6で100から12.5 mg/mLまで8ステップで順次希釈します。プレートの各列に異なる濃度のエンドチアペプシンが使用されます。
- リキッドハンドリングロボットを使用して、150 nLのエンドチアペプシンをサブウェル1と2の両方に分注します。サブウェル1に、150 nLのウェル溶液を分注します。サブウェル2で、50 nLの解凍したシードストックと100 nLのウェル溶液を多重吸引し、両方をタンパク質溶液に分注します。結晶化バッファーの添加時に溶液を3回混合する。
- プレートを密封し、20°Cで放置して、0、3、6、12、18、24時間ごとに画像を撮影し、最初の1週間は毎日、次の4週間は毎週画像を撮影します。自動イメージングが不可能な場合は、1日目の1時間ごとのイメージングについて心配する必要はありません。
- マイクロクリスタルシードストックを作成します。
- モルフォグラム解析
- 24時間後に撮影された画像を見て、各ウェルに存在する結晶の数を推定し、これらの推定値を提供された「モルフォグラムジェネレーター」ワークシートに記録します。これらの見積もりは正確である必要はありません。何千もの微結晶が存在する場合、個別にカウントすることは実用的でも必要でもありません。主に、推定値がプレート全体で一貫していることを確認するようにしてください。
注:24時間ルールは、Beale et al. (2019)26で行われた観察に基づいています。蒸気拡散結晶化条件は、平衡化するのに数日または数週間かかる場合があります。急速に現れる結晶は、液滴成分の段階的な平衡化ではなく、バッチプロセス によって 成長した可能性が高くなります。したがって、24時間の基準はやや恣意的であり、バッチ実験と蒸気拡散実験の間の正確なカットオフ時間は、条件の特定の混合物に依存します[Beale らを参照。 (2019)詳細については26 ]。 - エンドチアペプシンとPEG 6,000の開始濃度を示されたボックスに入力します。
- ワークシートは、沈殿剤とタンパク質の濃度をそれぞれ x 軸と y 軸に含む従来の状態図形式で結果を自動的にプロットします。播種液滴で結晶のみを生じるウェル条件は、図の準安定領域(透明な青色)を示し、播種液滴と非播種液滴の両方に結晶がある条件は核生成帯(緑色)を示します。
注:理想的には、核生成ゾーンの大部分が図上に存在する必要があります(つまり、図の下部にいくつかの透明なウェルがあり、いくつかの沈殿物が高タンパク質および沈殿剤濃度で見えるはずです)。そうでない場合は、おそらく実験を繰り返しますが、タンパク質および/または沈殿剤の濃度を上げます(可能な場合)。 - クリスタルが24時間以内に現れた場合は、ステップ2.3.1に進みます。そうでない場合は、ステップ2.4に進み、バッチに向けて最適化を続行します。
- 24時間後に撮影された画像を見て、各ウェルに存在する結晶の数を推定し、これらの推定値を提供された「モルフォグラムジェネレーター」ワークシートに記録します。これらの見積もりは正確である必要はありません。何千もの微結晶が存在する場合、個別にカウントすることは実用的でも必要でもありません。主に、推定値がプレート全体で一貫していることを確認するようにしてください。
- 結晶解析
- ステップ1の最後に述べたように、次のステップに進む前に、これらの結晶が望ましい形態と回折品質を持っていることを確認してください。形態に関しては、結晶は針球や扇のような構造ではなく、観察的に絡み合っておらず、シングルトンとして形成されていますか?回折に関しては、可能であれば結晶から回折データを収集してください。これらの結晶が回折しない場合、より大きな体積で成長した結晶が回折する可能性は低いです。
- モルフォグラム実験からのエンドチアペプシン結晶のサンプルを、100または293 Kのいずれかでデータ収集できる支持体にロードし、X線回折実験を実行します。クライオで作業する場合は、クライオ保護剤として25%エチレングリコールを使用してください。
- 適切なソフトウェアスイート を介して これらのデータを処理します。エンドチアペプシン結晶は1.5 Åを超えて回折するはずです。結晶のサンプル全体で、細胞サイズ、観測の総数、およびモザイク性を観察します。これらの測定値は、回折結晶の均質性に関する指標を与えるであろう。
- 結晶形態と回折品質が十分であれば、ステップ3に進みます。
- 結晶成長時間を最適化します。
注:モルフォグラム分析(ステップ2.2)では、結晶化の開始点(つまり、 沈殿剤とタンパク質溶液を混合したときに液滴が位置する相図の領域)が示されます。準安定領域の低下ですか、それとも溶解度線を下回っていますか?バッチ結晶化は核生成ゾーンで始まります(図1C)。このステップの目標は、この開始点を溶解度線または準安定領域の下から核生成ゾーンに移動することです(図1D)。シードがステップ2.2からドロップした場合。急速に結晶を生じさせている場合、これは液滴混合物がすでに準安定領域にあることを示しており、そうでない場合は、液滴が過飽和ではない可能性が高い。- 結晶成長時間の最適化。
- ステップ2.1.3と同じ画面を使用して、3滴プレートで96ウェル蒸気拡散結晶化実験を準備します。
- y軸上のエンドチアペプシンの開始タンパク質濃度を上げます(つまり、エンドチアペプシンの場合はおそらく120 mg / mLのタンパク質をさらに濃縮します)。
- ステップ2.1.3.2のように、プレートの各行に順次低いタンパク質濃度が含まれるように、段階希釈を実行します。
- プレート上の3つの滴のそれぞれに異なる滴率を使用します:1:1、1:2、および2:1、タンパク質:沈殿剤。
- プレートを最初の日の0、3、6、12、18、24時間に表示または画像化し、最初の週は毎日、次の4週間は毎週プレートを表示します。自動イメージングが不可能な場合は、1日目の1時間ごとのイメージングについて心配する必要はありません。
- 最も急速に現れる結晶を生成する液滴を特定し、24時間以内に結晶成長が起こるまで、これらを反復最適化の出発点にします。
- 急速に現れる結晶状態が特定されたら、ステップ2.1に戻り、スケーリングを開始するための前奏曲としてモルフォグラムを再プロットします。
- 結晶成長時間の最適化。
3. スケーリング
- スケーリング ルートをランク付けします。この段階では、単一のスケーリングルートを決定する必要はなく、オプションを特定してランク付けするだけで、順番に探索できます。スケーリング手順中にバッチ混合物の体積が増加すると、核生成速度と結晶サイズの範囲に変化が生じます。ただし、これらは、スケーリングされた体積が増加するにつれて成分濃度を注意深く調整することで克服できます。
注: 手順 3.1.1 および 3.1.2 では、モルフォグラムから、バッチプロトコルとシードバッチプロトコルのどちらがより適切かを識別する方法について説明します。- ストレートバッチプロトコル
- 核生成帯のXnは、タンパク質および/または沈殿剤の濃度に比例しますか?すなわち、Xnは沈殿剤および/またはタンパク質濃度のいずれかの関数として増加するか?-はい。ステップ 3.1.1.2 に進みます。いいえ。ステップ 3.1.2 に進みます。
- 必要なサイズの結晶を生成する条件を見つけて、ステップ3.2に進みます。
- シードバッチプロトコル
- Xn は核生成帯を横切って平らですか? すなわち、Xn は、沈殿剤および/またはタンパク質濃度のいずれかの関数として増加しない。
- 必要なサイズの結晶を生成するシード条件を見つけて、ステップ3.2に進みます。すべてのクリスタルが大きすぎる場合は、ステップ3.1.2.3に進みます。
- モルフォグラム実験(ステップ2.1)を繰り返しますが、今回は播種ウェルで使用する種子ストックの濃度を上げます。シードストックは、その作成により多くの結晶を使用することで増やすことができます。たとえば、ステップ 2.1.1.5 の 5 つのウェルの代わりに、10 個のウェルを使用します。
- 最初の0、3、6、12、18、24時間にわたってプレートを表示または画像化します。
- Xn はシードドロップで増加し、Xsは 減少したはずです。より小さな結晶が必要な場合はこのサイクルを繰り返し、シードバッチプロトコルに従ってください。
- ストレートバッチプロトコル
- 段階的なスケーリング
- 96ウェルプレートでのスケーリング。エンドチアペプシンモルフォグラムから、結晶化条件0.1 M Tris-HCl pH 7.0、0.15 M MgCl2、および30%(w / v)PEG 6,000を使用したストレートバッチ法をスケーリングに最初に選択しました。100 mg/mL エンドチアペプシンを結晶化バッファーと 1:1 の比率で混合しました。
- 100 μLの0.1 M Tris-HCl pH 7.0、0.15 M MgCl 2、および30%(w / v)PEG 6,000を含む2ウェル96ウェルシッティングドロッププレートに2〜3ウェルを調製します。
- 解凍したばかりの100 mg/mLエンドチアペプシン溶液を使用し、ウェルあたり0.5 μLのタンパク質と0.5 μLの沈殿剤を分注し、密封して20°Cで保存します。
- 最初の0、3、6、12、18、24時間にわたってプレートを表示または画像化します。Xs とXnの範囲の変化に注意してください。
- 変更が発生した場合は、手順3.2.1.1から3.2.1.2を繰り返しますが、タンパク質、沈殿剤、および/または種子の濃度を増減して、Xs およびXnの範囲への変更を復元します。
- Xs とXn の範囲が許容できる場合は、手順3.2.2に進みます。
- 24ウェルハンギングドロッププレートでのスケーリング
- 24ウェルのハンギングドロッププレートの端に真空グリースを塗って、1つのウェルを準備します。
- 0.5 mLの0.1 Mトリス塩酸pH 7.0、0.15 M MgCl2、および30%(w / v)PEG 6,000を調製し、グリースを塗ったウェルを満たします。
- 解凍したばかりのエンドチアペプシン溶液を使用して、1 μLのタンパク質をガラスカバーガラスの表面にピペットで入れます。1 μLの結晶化バッファーをタンパク質滴上にピペットで入れ、ピペットを使用して混合します。
- 最初の0、3、6、12、24時間にわたってプレートを表示または画像化します。Xs とXnの範囲の変化に注意してください。
- 変更が発生した場合は、ステップ3.2.2.1から3.2.2.4を繰り返しますが、タンパク質、沈殿剤、および/または種子の濃度を増減して、Xs およびXnの範囲への変更を復元します。
- Xs とXn の範囲が許容できる場合は、手順3.2.2.7に進みます。
- ステップ3.2.2.1から3.2.2.5を繰り返し、実験の総量を徐々に10μLまで増やします。
- 容量が10 μL以上になったら、ステップ3.2.3の遠沈管に進みます。
- 遠沈管でのスケーリング
注:エンドチアペプシンバッチ条件の改良は、主に200μL容量の時点で行われました(結果、スケーリングを参照)。このプロセスは、0.1 M Tris-HCl pH 7.0、0.15 M MgCl2、および30%(w / v)PEG 6,000の結晶化条件で開始されました。しかし、PEG濃度は最終的に40%(w / v)に変化しました。種子はまた、Xnを制御する必要があり、結晶が大きくなりすぎるのを防ぐために、結晶成長を急冷する必要がありました。ステップ 3.2.3.1 から 3.2.3.7 では、条件最適化のプロセスについて詳しく説明します。ステップ 3.2.4.最終的なバッチプロトコルについて説明する。- 1 mL の結晶化バッファーを調製します: 0.1 M トリス塩酸 pH 7.0、0.15 M MgCl2、および 30% (w/v) PEG 6,000。
- 解凍したばかりの100 mg/mLエンドチアペプシンを使用して、25 μLのタンパク質を1.5 mLの遠沈管に添加します。
- 結晶化バッファーとタンパク質溶液をピペットチップで1:1の比率で完全に混合します。チューブを20°Cで高撹拌のリボルバー/ローテーターに入れます。
- 定期的に(5、10、30、60分、2、5、10、24時間)2.5 μLのアリコートを取り、血球計算盤で表示します。Xn とXs の範囲を記録します。
- 変更が発生した場合は、手順 3.2.3.1 を繰り返します。を 3.2.3.4 に置き換えます。ただし、タンパク質、沈殿剤、および/または種子の濃度を増減して、XsおよびXnの範囲への変化を復元します
- XsとXnの範囲が許容できる場合は、手順3.2.3.7に進みます。
- ステップ3.2.2.1から3.2.2.5を繰り返し、必要に応じて実験の総量を徐々に200μL以上に増やします。
- 最終シードバッチプロトコル
- シードストックを準備します。
- 2 mLの結晶化バッファーを準備します:0.1 M トリス塩酸pH 7.0、0.15 M MgCl2、および40%(w / v)PEG 6,000。
- 解凍したばかりの100 mg/mLエンドチアペプシンを使用して、1.5 mLの遠沈管に100 μLのタンパク質を加えます。
- 結晶化バッファーとタンパク質溶液をピペットチップで1:1の比率で完全に混合します。チューブを20°Cで24時間高撹拌しながらリボルバー/ローテーターに入れ、50 μmの結晶を成長させます。
- 50 μmの結晶スラリーに10〜15個の1 mmガラスビーズを追加します。
- スラリーとビーズを入れた遠沈管を1000rpmで30秒間ボルテックスします。
- 遠沈管を氷に30秒間戻します。
- 手順 3.2.4.1.5 と 3.2.4.1.6 をさらに 10 回繰り返します。
- これは現在、1xシードストックの200μLです。1.8 mLの結晶化バッファーを添加してシードストックを10倍に希釈します。10xシードストックを50 μLバッチで分注し、-20°Cで保存します。
- シードバッチプロトコル。
- 結晶化バッファーを調製します:0.1 M トリス塩酸pH 7.0、0.15 M MgCl2、および40%(w / v)PEG 6,000。
- 遠沈管内で、100 μLの結晶化バッファーを50 μLの解凍したばかりの10xシードストックと混合します。
- 解凍したばかりの100 mg/mLエンドチアペプシンを使用して、1.5 mLの遠沈管に150 μLのタンパク質を加えます。
- 結晶化バッファー/シード混合物をピペットチップでエンドチアペプシン溶液と完全に混合し、チューブを20°Cで高撹拌のリボルバー/ローテーターに入れます。
- 定期的に2.5 μLのアリコートを採取して結晶化をモニタリングし、血球計算盤で結晶を観察します。Xn とXs の範囲を記録します。
- 約80分後、結晶が15 μmのXs に達したら、150 μLの0.05 M Na Acetate pH 4.6、0.05 M Tris-HCl pH 7.0、0.075 M MgCl2、および20%(w / v)PEG 6,000(エンドチアペプシンバッファーと結晶化バッファーからなる溶液、1:1混合)を添加して反応を停止します。
- 結晶は20°Cで保存します。
- プロトコルは、意図した実験に許容できる結晶サイズの範囲と数を生成しましたか?はい - 完了 - いいえ - ステップ 3.1 に戻ります。をクリックし、別のスケーリング オプションを試します。たとえば、異なるタンパク質:沈殿剤の比率が可能であったり、以前に行われていなかった場合は種子を追加したりできます。これらがすべて使い果たされたら、ステップ1で新しい条件を見つける必要がある場合があります。
- シードストックを準備します。
- 96ウェルプレートでのスケーリング。エンドチアペプシンモルフォグラムから、結晶化条件0.1 M Tris-HCl pH 7.0、0.15 M MgCl2、および30%(w / v)PEG 6,000を使用したストレートバッチ法をスケーリングに最初に選択しました。100 mg/mL エンドチアペプシンを結晶化バッファーと 1:1 の比率で混合しました。
結果
結晶形態の最適化
結晶形態を最適化するステップ1は、読者にその重要性を思い出させるために含まれています。回折が不十分な針球から完全な微結晶を作成することは可能かもしれません。ただし、著者は、2つを別々に最適化する方が良いと示唆しています。まず、蒸気拡散 によって よく回折する単結晶を生じる条件を見つけ、次にこれらの条件をバッチに変換し、2つのステップを組み合わせることを試みるのではなく、バッチに変換します。この段階では、高度に核形成する条件を発見する必要はありません。形態と回折品質が主な目標です。
エンドチアペプシンの微小結晶化を開始する前に、PDBから析出構造の結晶化条件の解析を行った。結晶化条件とおおよそのプロトコルは、エントチアペプシンの48の沈着のうち47について得ることができました。これらはすべて、Moews and Bunn (1970)46によって行われたエンドチアペプシンの最初の結晶化に大まかに基づいていました。これらの条件の類似性とそれらの「古典的な」起源を考慮して、96ウェルの蒸気拡散スパースマトリックススクリーンを実行して、より多様な結晶化条件を調査しました。エンドチアペプシンを70 mg/mLまで濃縮し、PACTスパースマトリックススクリーン47 を96ウェルシッティングドロッププレート中で20°Cで実施し、100 nLのタンパク質と100 nLのウェル溶液を混合しました。36時間後のこの実験からのすべての条件は結晶を生じさせた。しかし、結晶形態の分析は、いくつかの条件が微小結晶化の最適化に適していることを証明するかもしれないことを示しました。
図4A は、プレートの大部分で観察されたものを広く代表するPACTスクリーンからのドロップを示しています。一見すると、これらの結晶は微小結晶化のためにさらに最適化する価値があるかもしれないと考えたくなるかもしれません。結晶は大きく、かなりの核生成があるようです。しかしながら、全体的な結晶形態は理想的ではない。第一に、複数の結晶が単一の核形成点から成長しているように見えるため、結晶は観察可能なシングルトンではありません。第二に、結晶サイズは非常に非対称であり、成長は主に単一軸を下って発生します。このような結晶は、理論的には、X線ビームに送達されたときに優先的に整列する可能性が高い。どちらの特性も、シリアル結晶学的データの収集および処理中に問題を提示します。
しかしながら、図4Bは、MgCl2の存在下で成長したエンドチアペプシン結晶を示す。この形態は、MgCl2を含むすべての条件で一貫していたため、それらの形態はMgCl2によるものであることが示唆されました。MgCl2条件は、究極の連続実験のためのより良い標的を表す単一の、より箱のような結晶を生成した。
PACTスクリーン内には、MgCl2を含む4つの条件がありました。エンドチアペプシン結晶化に対するこれらの条件のすべての異なる成分の影響をよりよく理解するために、ランダム最適化を実行しました。緩衝液と沈殿剤をさまざまな濃度およびpHでランダムに組み合わせたスクリーンを作成しました。MgCl2 濃度も変化させ、得られた液滴を、視覚的な結晶品質と沈殿レベルの観点から、0〜5(0は結晶または沈殿なし)から任意に等級付けしました。
図5A は、降水レベルと結晶品質、およびスクリーン変数との間のピアソンの相関分析の結果のヒートマップを示しています(この実験からの低下の例は 図5B、 C 、 およびDに示されています)。その結果,溶液のpHは沈殿量と高い相関があり,アルカリ性緩衝液は沈殿量を増加させることが示された。MgCl2 濃度は、結晶品質に対するpHおよび沈殿剤濃度と同様に、沈殿レベルとわずかに相関していた。
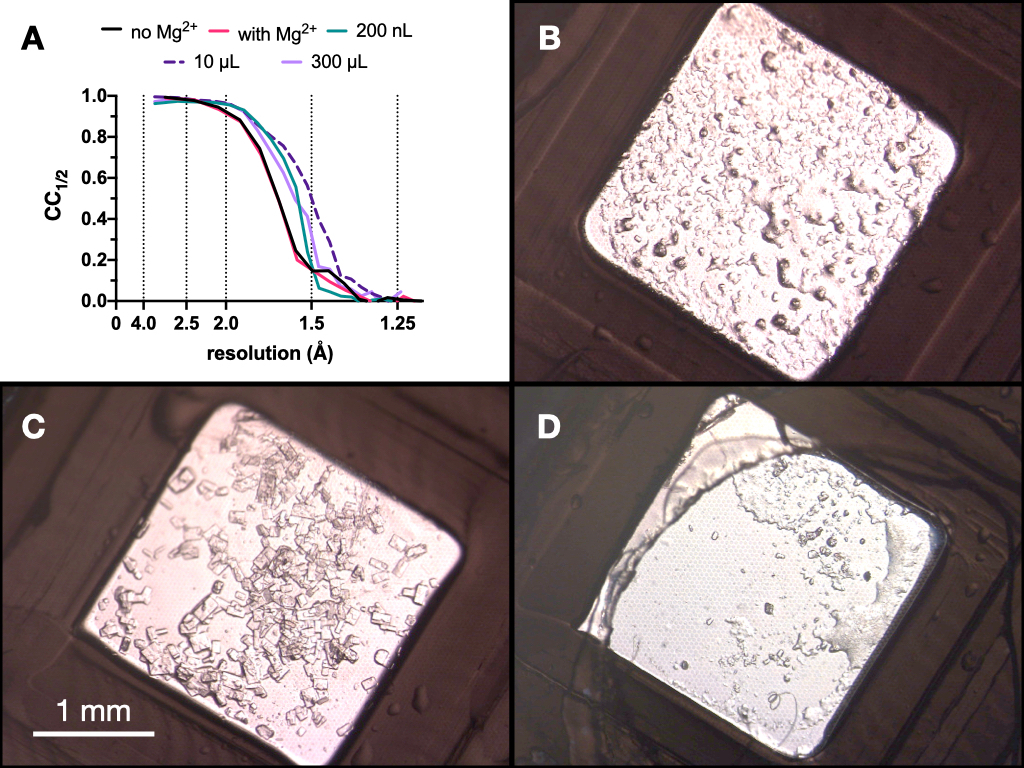
これらの結果に基づいて、0.1 M Tris-HCl pH 7.0、0.15 M MgCl2、20%(w / v)PEG 6,000で成長させた結晶をプロトコルの次のステップであるバッチに移行することを決定しました。結晶の形態は許容範囲であり、これらの結晶からのX線回折およびデータ品質メトリックの分析は、Mg2+ の存在下で成長した結晶の間に有意差がないことを示唆した(図9)。
バッチへの移行
多くのシリアル結晶構造解析のマイクロ結晶化の最適化では、ステップ2が出発点になります。目的のタンパク質はすでにクライオ結晶構造解析のために結晶化されており、結晶化プロトコルは微結晶スラリーを作成するために変換する必要があります。このプロトコルでは、蒸気拡散がPDBエントリ26の95%で使用されている結晶化方法であるため、バッチへの変換を実行するために96ウェル蒸気拡散プレートのみを使用しています。プロトコルは、この移行によって同様の最適化が発生する可能性があるため、マイクロバッチ34、35、37への移行を回避しています。これは、このプロトコルが蒸気拡散プレートでのみ実行できるということではありません。提示されたすべてのステップは、これが元の結晶化方法である場合、マイクロバッチでも機能します。
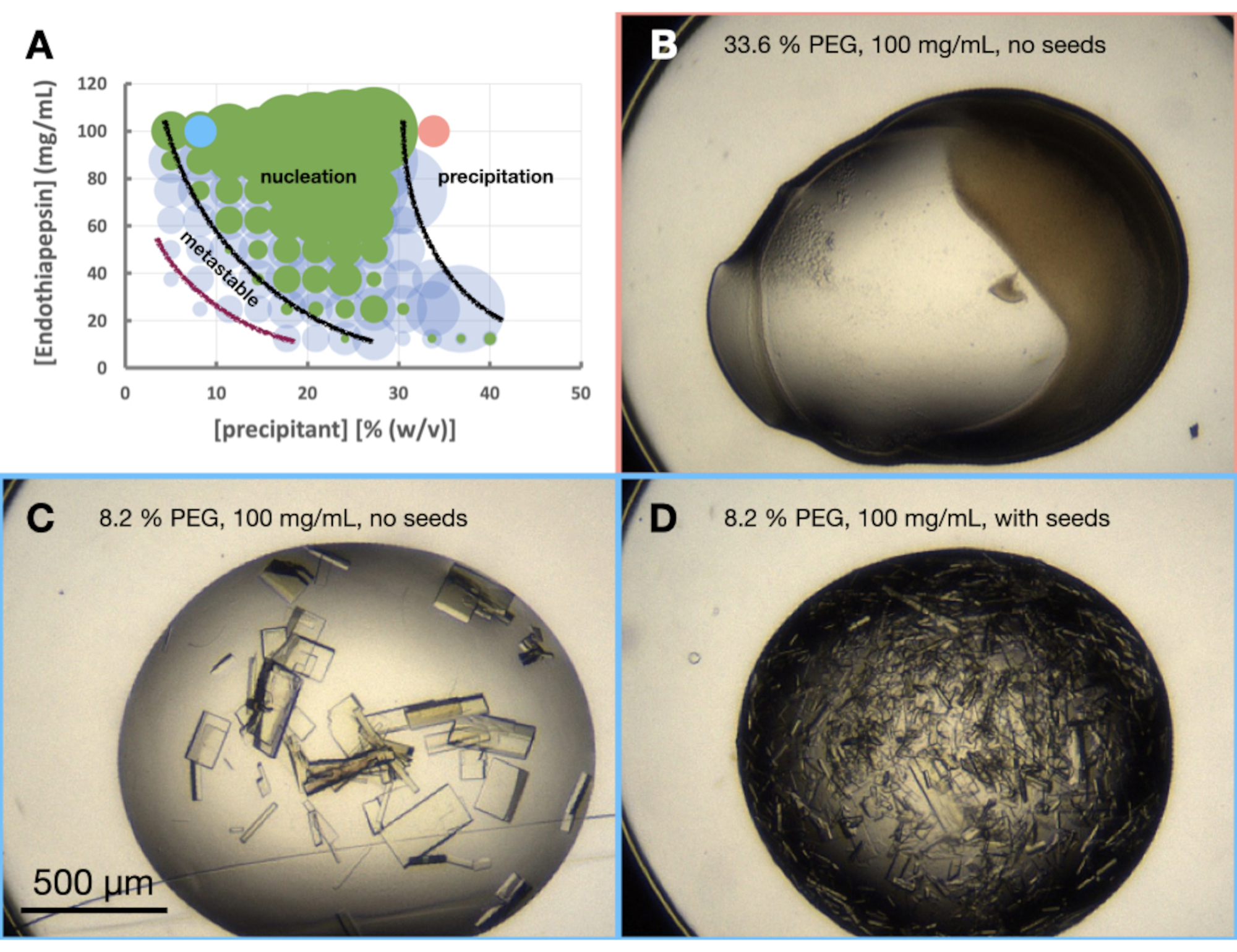
選択された条件におけるエンドチアペプシンの結晶化を評価するために、モルフォグラム - または大まかな状態図 - が作成された。モルフォグラム実験の目的は3つあります。まず、モルフォグラムの分析は、ステップ3 - スケーリングでスケーリングルートを評価するときに非常に役立ちます。第二に、モルフォグラムは最適化ツールとして機能し、バッチ(すなわち、急速に現れる結晶(<24時間))を介して結晶を生じる蒸気拡散条件を発見するのに役立ちます。第三に、結晶が急速に現れていない場合、播種された液滴の分析により、結晶学者は相図上の現在の状態のおおよその位置を知ることができます。たとえば、播種条件が結晶を与えるが、播種されていない条件が結晶を与えない場合、それらの条件は準安定領域にある可能性があります。
エンドチアペプシンのモルフォグラム実験は、0.1 M Tris-HCl pH 7.0、0.15 M MgCl2、20%(w / v)PEG 6,000条件に基づいて実施されました。タンパク質およびPEG濃度は、それぞれ100〜12.5 mg / mLおよび5〜40%(w / v)の範囲で変化しました。滴を分析し、提供されたワークシートを使用して結果をプロットした(図6A)。
また、結晶 形態の最適化 段階から、この条件で、これらのタンパク質濃度でのエンドチアペプシンの結晶成長は、24時間以内に結晶成長をもたらすこともすでに明らかでした。これは、結晶化が蒸気拡散駆動プロセスではなくバッチ を介して 発生していることを示していました。したがって、これらの条件で成長した結晶は、より大きな体積へのスケーリングに適していました。
24時間後に非播種液滴に結晶が見えなかった場合、結晶化は依然として遷移に依存しており(図1B)、したがってバッチに依存していない可能性があります。この場合、モルフォグラム実験の結果は依然として興味深いものです。それらは、相図上の結晶化の推定開始点を示し、したがって、その後の最適化がどのように進むべきかを示します。種をまいた滴を見てください。シードは、核生成に関係なく、準安定領域での結晶成長を可能にします。たとえば、播種された液滴では24時間以内に結晶が現れ、播種されていない液滴では現れない場合、これは準安定領域の一部が観察されることを示しています。シード液滴または非シード液滴のいずれにも結晶が観察されない場合、すべてのウェルは低飽和のままである。
スケーリング
モルフォグラム(図6A)を見ると、いくつかの観察を行うことができました。核生成の量は、タンパク質濃度と沈殿物濃度の両方に影響されるようでした。また、タンパク質の沈殿につながる液滴の非常に明確な境界があり、液滴には何も含まれていないか、結晶または沈殿物が含まれていました(図6B)。種子の添加(図6D)はまた、種子なしの滴(図6C)と比較してXnを大幅に増加させた。これらすべての結果をまとめて、バッチプロトコルとシードバッチプロトコルの両方を30%(w / v)PEG 6,000および100 mg / mLエンドチアペプシンでスケーリングすることを試みることが決定されました。
最初のテストスケーリングは、24ウェルハンギングドロッププレートで行われました。結晶化挙動の変化を観察できるように、液滴量を徐々に増加させました(図7)。図からわかるように、非播種液滴と播種液滴の両方で結晶成長が起こっていた。播種されていないすべての液滴は、さまざまな結晶サイズを成長させましたが、主に大きな結晶(100〜200μm-最長寸法)でした。しかし、播種された液滴は、より小さな結晶(5〜50μm-最長寸法)を生成しました。これらの初期テストは、種子がXsを減らすために必要であることを示唆しましたが、この条件はより大きなボリュームに適しているはずであることも示唆しました。
体積を200μLに増加させると、結晶成長中に結晶化体積が連続的に攪拌された。この攪拌の主な理由は、結晶化溶液が均質なままであり、成長する結晶がチューブの底部または側面に沈殿しないようにするためでした。結晶の沈降は、非常に大きな結晶と小さな結晶の両方を持つ不均一な結晶集団につながる可能性があります。結晶化溶液を攪拌することは、核生成を促進することもできる44、45。
残念ながら、播種されていない30%(w / v)のPEG 6,000では結晶が生成されなかったため、PEG濃度は35%(w / v)に増加しました。この増加により結晶化が著しく改善され、最終的なXnとXsの範囲はそれぞれ3.6 ± 1.2 x 106結晶・mL-1と42 ± 4.1 μmになりました(図8AとB-黒)。大幅な改善と許容可能な結晶濃度でしたが、最終的な結晶は計画された実験には大きすぎたため、さらなる最適化が行われました。最終結晶のサイズを小さくするために、タンパク質濃度を下げて最終的な結晶成長を制限すること(図8AおよびB-ホットピンク)、およびPEG濃度を上げて核生成を増加させること(図8AおよびB-緑)の2つの方法が検討されました。
残念ながら、タンパク質濃度の低下はXnも劇的に減少させ、最終的にはさらに大きな結晶を生成しました。PEG濃度を40%に上げると、最終的なX、n、Xsの範囲はそれぞれ3.1 ± 0.7 x 106結晶・mL-1および39 ± 2.3 μmになりました。これらは35%と有意差はありませんでしたが、最終的な結晶サイズが小さくなったため、この条件をさらに最適化して継続しました。
Xnを増加させるために、種子を添加した。これにより、Xn(1.1 ± 1.8 x 108結晶・mL-1)が劇的に増加し、Xsが小さくなりました(4.2 ± 4.0 μm)(図8AおよびB-紫色の破線)。これらの結晶は、いくつかの連続結晶構造解析実験に非常に適していますが、小さすぎると見なされたため、添加されたシードの濃度が変更されました。
しかし、追加された種子ストックのこの調整は、確実に繰り返すことが困難であることが判明しました。そのため、急冷を試みた。シードストックの添加後、結晶サイズをモニターし、適切な結晶サイズ(約10〜20μm)が達成されたら、バッチ結晶化を急冷した(図8C および D)。急冷は、微小結晶化に関して、Kupitz et al. (2014)25で提案されている。おそらく理想的な方法ではないが、タンパク質溶液は最終的に無駄になるので26、結晶成長を制御することが困難であったため、この技術はこの状況で非常に有用であった。急冷の背後にある考え方は、結晶化混合物を溶解度線のすぐ上の点に迅速に戻すことです(図1F)。溶液が溶解度ラインに戻ると、溶液は安定した飽和溶液に戻り、それ以上の結晶成長は起こりません。
結晶化反応をクエンチしようとすることにはリスクがないわけではありません。添加するクエンチング溶液が多すぎると、溶液中のタンパク質が溶解度線を通過するほど希釈される可能性があります。この場合、溶液は過飽和になり、結晶は溶解し始めます。これを防止するために、モルフォグラムの結果に基づいて必要な急冷溶液の量を推定することができる。急冷の時点で、タンパク質溶液の濃度を取ります。溶解度線でのタンパク質濃度と溶液中のタンパク質濃度を比較することにより、必要な希釈率を推定できます。
40%(w / v)PEG 6,000、10 x希釈シード実験の急冷バージョンでは、最終的な結晶濃度とサイズ範囲は、それぞれ2.6 ± 3.1 x 106 結晶・mL-1 および15 ± 3.9 μmでした。
プロセス全体を通して、エンドチアペプシン結晶の試験X線データを収集し、スイス光源PXIIビームラインで、10 x 30 μmの焦点、80%減衰した12.4 keVのエネルギー、および低温条件下で収集しました。データはダイヤルを使用して処理され、 図9 はCC1/2の比較を示しています。最適化の過程でCC1/2 の劇的な変化は観察されませんでした。

図1:転移結晶化とバッチ結晶化の概要、および相図にマッピングされたスケーリング方法。 A.いいえ。原型タンパク質結晶化のゾーンと限界 状態図。沈殿剤とタンパク質の濃度は、純水を原点として、それぞれx軸とy軸にプロットされます。紫色の線はタンパク質の過飽和境界を示し、準安定、核生成、沈殿帯はそれぞれ青、緑、ピンクで示されています。イ.蒸気拡散などの'遷移相'結晶化法の核生成帯浸透限界の一例。この理論的実験では、滴下沈殿剤とタンパク質濃度は溶解度線のすぐ下で始まります-まだ過飽和ではありません。液滴が平衡化している間、液滴成分の濃度は上昇し、液滴が過飽和になり、核生成ゾーンに移動し続けます。結晶核形成により、溶液中のタンパク質濃度は低下し始める。濃度は、結晶が成長するにつれて低下し続け、最終的に溶解度線で停止します。青い点線は、核生成帯への移行の理論的限界を示しています。核形成が始まるとすぐに、タンパク質濃度は低下し、それ以上の浸透を防ぎます。ウ. バッチおよびシードバッチ結晶化軌道の例。バッチでは、タンパク質と沈殿剤の混合は、結晶成長が起こることができるように、核生成ゾーン内に過飽和溶液を生成する必要があります。シードバッチでは、マイクロシードの添加により核生成ゾーンにいる必要は厳密にはないため、準安定領域の場所も探索できます。D.Bに示す結晶化実験の蒸気拡散からバッチへの仮想的な最適化。元の蒸気拡散開始点は、結果の最適化ベクトルを介して新しい開始位置に遷移しました。核生成帯の内側。結果として得られるベクターは、タンパク質濃度と沈殿剤濃度の両方の増加という2つの最適化の産物です。E.バッチ条件をスケーリングして最終的な Xn と Xs を調整する場合の最適化の例。F.結晶化バッファーの添加による結晶化実験の急冷。クエンチは、タンパク質濃度を準安定領域から取り出さず、したがってタンパク質過飽和点を下回らないことが不可欠です。そうしないと、結晶が溶液に溶解し始めます。イ. およびC.は、著者の許可を得てBeale et al. (2019)26から改作されています。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図2:Xnの増加とXsの減少。 結晶化実験から生成された結晶の数とその平均最長寸法との間の理想的な関係。このグラフを作成するために、仮想の10 kDaモデルタンパク質の結晶化を使用しました。タンパク質は10 mg/mLの濃度で結晶化し、49x50x51 Åの寸法のP21 2 1 21結晶を生成しました。すべての核生成イベントは結晶を生成すると仮定されました。結晶成長はすべての面から均質であると仮定した。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図3:小容量(<500 nL)の蒸気拡散実験で成長させた結晶を大容量(> 100 μL)のバッチ実験に最適化する手順を示すフローチャート。 結晶の最適化は、(1)結晶形態の最適化の3つの段階に分けられます。(2)バッチへの移行。(3)スケーリング。ステージ1では、微小結晶化に適した結晶を特定することが重要です。一部のタンパク質は、結晶化条件に関係なく単結晶形態でのみ存在します。ただし、単一の立方体のような結晶、または人間が可能な限りこれらに近い条件を探す価値があります。単一の立方体のような結晶は、仮説的および逸話的に、一般に、連続結晶学実験からより良い結果をもたらします。結晶形態を選択し、回折を確認したら、結晶化実験を蒸気拡散からバッチ(ステージ2)に移行する必要があります。ここで、結晶は核生成時間によって最適化されるべきである。目標は、急速に現れる結晶(> 24時間)を生成する条件を見つけることです これらの条件はすぐに核生成ゾーンに当たる可能性があり、したがってバッチです。核生成帯の条件が見つかると、モルフォグラムを作成できます。モルフォグラムにより、核生成ゾーンの大部分が、ステージ3で特定されたマッピングされた潜在的なスケーリングルートが可能になります。同定されたバッチ条件の容量は、徐々にまたは急速にサイズをスケーリングして、>100 μLの最終容量を得ることができます。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図4:PACTスパースマトリックススクリーンからのエンドチアペプシン結晶化条件の分析。 A .および B. は、それぞれPACTスクリーンからのウェルA4およびC10の24時間後の写真である。結晶化バッファー成分は図上で強調表示されています。SPGバッファーは、コハク酸、リン酸二水素ナトリウム、およびグリシンを2:7:7のモル比で混合したものです。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図5:PACTMgCl2条件からのエンドチアペプシン結晶化最適化の解析。 A.いいえ。バッファーのpH、MgCl2濃度、沈殿剤濃度と沈殿レベルおよび結晶品質との間のピアソンの相関分析の結果のヒートマップ。沈殿レベルと結晶品質は、24時間後に0〜5のスケールで任意に評価されました(0は結晶または沈殿なし)。D.は、3つの異なる液滴における結晶化および沈殿の例を示す。結晶化条件と沈殿レベルと結晶品質の評価も示されています。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図6:0.1 M Tris-HCl pH 7.0、0.15 M MgCl2およびPEG 6,000で結晶化したときのエンドチアペプシンモルフォグラム。 A.いいえ。提供された「相図ジェネレータ」スプレッドシートから作成されたモルフォグラム。各液滴中の結晶の相対数は円の大きさで示され、滴1(タンパク質および沈殿剤)および滴2(タンパク質、沈殿剤および種子)の結果はそれぞれ緑および青で強調表示される。x軸とy軸のタンパク質濃度と沈殿剤濃度の値は、それぞれ最終体積ではなく、それぞれの事前混合値を示します。この結果に基づき、核生成帯と準安定帯の境界をそれぞれ示すために黒線と紫線が引かれました。紀元前 D.は実験の結果の例をいくつか示しています。A.にマークされた赤と青の点は、B.とC.の位置を示しています。 とD.、それぞれ。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図7:24ウェルハンギングドロッププレートでのエンドチアペプシンの初期スケーリング試験。 すべてのトレイルで同じタンパク質および沈殿剤濃度を使用しました:0.1 M Na 酢酸pH 4.6および0.1 M Tris-HCl pH 7.0、0.15 M MgCl2、および30%(w / v)PEG 6,000中の100 mg/mLエンドチアペプシン。表示されたすべての画像は24時間後に撮影されたもので、最終的なドロップボリュームは各画像にラベル付けされています。左のパネル(A、 D、および G)はタンパク質と沈殿剤の1:1の混合物であり、中央のパネル(B、 E、 およびH)は種子、沈殿剤、およびタンパク質の1:2:3の混合物であり、右のパネル(C、 F、 およびI)は中央のパネルの拡大画像です。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図8:200〜300 μLの容量でのエンドチアペプシン微小結晶化の分析。 A.およびC.は、実験時間にわたってXnがどのように変化したかを示す。イ. D.は、Xs(最長次元)が時間とともにどのように変化したかを示しています。実験の結果は、わかりやすくするために分離されています。C.とD.の赤い点線は、急冷が行われたポイントを示しています。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図9:回折品質を評価するための微結晶化プロセスの各段階で得られた結晶のCC1/2結果と画像。 A.いいえ。成長した結晶から収集されたデータからの分解能に対してプロットされたCC1/2:Mgの有無-ステージ1の最適化の一部、200 nL容量、10 μL容量、および最終300 μL容量。B.C.およびD.は、それぞれ200nL、10μLおよび300μLの容量からの結晶を示す。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
| タンパク質情報 | |
| 蛋白質 | エンドチアペプシン |
| 分子量(kDa) | 33.8 |
| スペースグループ | P12 11 |
| a, b, c (Å) | 45.2, 73.3, 52.7 |
| α, β, ɣ (°) | 90.0, 109.2, 90.0 |
| 固定ターゲット パラメーター | |
| チップあたりの負荷量(μL) | 150 |
| チップあたりのアペラチャ | 25,600 |
| 必要結晶濃度(結晶/mL) | 500,000 |
| サンプル情報 | |
| 200 μLのサンプルを作るのに使用したタンパク質量(mg) | 10 |
| 結晶最長寸法(μm) | 15 |
| 結晶濃度(結晶/mL) | 2,500,000 |
| 実験変数 | |
| 必要な時間ポイントの数 | 5 |
| 時点ごとに必要な画像の数 | 50,000 |
| ヒット率(統合されたパターン/画像が収集されます) | 0.3 |
| 時点ごとに必要な固定目標 (切り上げ) | 7 |
| サンプル要件 | |
| 時点ごとに必要なサンプル量(μL) | 1,050 |
| 実験に必要な総サンプル量(mL) | 5.25 |
| 必要なタンパク質の総質量(mg) | 52.5 |
表1:固定ターゲットを使用して実行された架空の光ポンププローブ実験のサンプル要件の例。この理論実験で使用したタンパク質はエンドチアペプシンでした。固定ターゲットパラメータは、Ebrahim et al. (2019)48およびDavy et al. (2019)49で報告された実験に基づいている。サンプル情報は、このビデオ記事で報告されたプロトコルからのものであり、実験変数は、実際の経験に基づく控えめな推定値でした。以下のサンプル要件は、前述の仮定を考慮して後で計算されました。
ディスカッション
提示された方法は、スパースマトリックス96ウェルスクリーンで成長させた大きな結晶(最長寸法≥100μm)から、バッチ を介して 遠沈管(300μL容量)で成長させた微小結晶へのエンドチアペプシンの結晶化を最適化する方法を示しています。プロトコルの背後にある考え方は、エンドチアペプシンを最適化するために取られたステップを他のタンパク質にも使用できるということです。最終的には、XFELやシンクロトロンでの連続結晶学実験のために大量(>100μL)の微結晶(10〜20μm)を作成するという問題に答えます。
このプロトコルは、大量の微小結晶化のタスクを、(1)結晶形態の最適化、(2)バッチへの移行、および(3)スケーリングの3つのステップに分割します。ステップ1では、タンパク質が生成できる結晶形の範囲を蒸気拡散プレートで調査する必要があります。必要な分解能まで回折する単一の箱状の結晶を生じる条件が目標であるべきです。ステップ2では、選択した条件を蒸気拡散からバッチに変換できます。ここで、最適化基準は結晶成長時間と、24時間以内にタンパク質結晶を生じる条件を見つけることです。モルフォグラムをプロットして、実験者に溶解度線と核生成ゾーンの境界の位置を把握させることもできます。このモーフォグラムは、ステップ 3 のスケーリングで非常に役立ちます。モルフォグラムは、核生成だけでXn を増加させ、Xsを低下させることができるかどうかを示します。実験の量が増加するにつれて、Xn およびXs は、スケーリング成功の重要な基準として継続的に評価することができる。
エンドチアペプシンの場合、ステップ1は、これまで知られていなかったエンドチアペプシンの結晶形態である可能性のあるものを発掘しました。この形態は、以前に報告されたものと同じ空間群を持っていましたが、連続結晶学にとって重要なことに、より箱のような形状でした。単結晶も、他の条件で作成されたファンとは異なり、単一の核生成点から成長しているように見えました(図4)。選択した条件については、<24時間で結晶成長が発生したため、ステップ2はすでに部分的に満たされていました。このモーフォグラムは、ステップ3でスケーリングすると、ストレートまたはシードバッチプロトコルの両方が成功する可能性があることを示しました。ストレートバッチでの初期スケーリングにより、X、n、Xsの範囲がそれぞれ3.6 ± 1.2 x 106結晶・mL-1および42 ± 4.1 μmの結晶を生成する条件を作成しました。これらの結晶は、いくつかの連続結晶学実験には許容できるものの、大きすぎると見なされました。そのため、追加の最適化が実行されました。最終的なプロトコルでは、それぞれ3.1 x 106結晶・mL-1および15 ± 3.9 μmの濃度およびサイズ範囲の結晶が生成されました。これは、計画された実験にとって理想的以上のものでした。
この方法は、蒸気拡散プレートで成長させた「可溶性」タンパク質結晶のバッチへの変換に焦点を当てています。この焦点の理由は、可溶性タンパク質結晶の大部分が蒸気拡散 を介して 成長するためです26。しかしながら、提示された概念は、マイクロバッチなどの他の方法を用いて成長させた可溶性タンパク質結晶にも適用することができる。この概念は、LCPで成長させた膜タンパク質結晶にも適用できます。これもバッチ結晶化プロセスです。
プロトコルの重要な側面は、蒸気拡散板で成長させた結晶の条件をバッチで成長できるように変換する方法です。この変換のために、この方法はBealeら(2019)26によって提案された基準を使用する。バッチプロセス で 成長した結晶は、蒸気拡散板でも急速に形成されます(<24時間)。この基準は、蒸気拡散液滴平衡化の速度に基づく近似値であり、PEGベースの沈殿物条件に最も当てはまります。ただし、結晶化条件には、平衡化時間に影響を与えるさまざまな化合物が含まれます。塩ベースの結晶化条件、 例えば 高濃度塩化アンモニウムの平衡化は、1〜2日で起こり得る。したがって、24時間の基準は、塩ベースの条件には当てはまらない可能性があります。塩ベースの条件はまた、このプロトコルで提示された原型に適合しないかもしれないより複雑な状態図26、30 を有することができる。塩ベースの条件の時間基準を12時間または6時間に減らすことは、より大きなボリュームへのスケーリングが不可能であることが判明した場合に必要になる場合があります。
この方法の別の制限は、その明らかな複雑さです。エンドチアペプシンの微小結晶化を最適化するために従ったプロトコルは、実際にはスパースマトリックススクリーンから元の状態を比較的ほとんど変化させませんでした。PACTスクリーンで観察された最初のヒットは、0.1 HEPES pH 7.0、0.2 M MgCl2、および20%(w / v)PEG 6,000でした。最終的なスケーリング結晶化バッファーは、0.1 Tris-HCl pH 7.0、0.15 M MgCl2、および40%(w / v)PEG 6,000でした。HEPESからTris-HClへの緩衝液の変化、およびMgCl2 濃度の変化が、プロセスの成功にほとんど寄与しなかった可能性も非常に高いです。PEG 6,000濃度の増加を唯一の最適化のままにしておくことは、非常に簡単に達成できたはずです。
ただし、この評価も単純すぎます。スケーリング中に遭遇する問題(つまり、シードの使用とクエンチング)を軽視するだけでなく、このタンパク質が簡単であることが証明されたからといって、 次のタンパク質も証明される保証はないという事実も無視します。プロトコルでアドバイスされたステップは、タンパク質結晶化体積のスケーリングを最適化することが非常に高価なタンパク質になり得るので考案された。示されている7つのエンドチアペプシンスケーリング試験で、100 mgのタンパク質が消費されました。確かに、これらの手順のいくつかは、このプロトコルに照らして結果を示すために実行されました。それでも、100 mgのタンパク質に加えて、実験中に消費されるタンパク質にさらに50 mgを追加する可能性があります(表1)は、時間または費用の大幅な投資になる可能性があります。
幸いなことに、この大量の必要なサンプルがすべてのタンパク質に遍在しているかどうかは明らかではありません。エンドチアペプシンは溶解性が高いため、過飽和に達するには高いタンパク質濃度が必要でした。他のもの(現在最適化中)では、過飽和は10または5 mg / mLでさえ到達する可能性があります。このような変数はタンパク質特異的であり、出現時に受け入れる必要があります。
この方法のその他の制限には、スクリーンやプレートの作成のための液体処理ロボットや、必要に応じてプレートを自動的に画像化するイメージャーなどの複雑な機器への依存が含まれます。これらの機器のいくつかの必要性を制限するために代替ルーチンが提供されていますが、プロトコルはそれらなしで従うのに時間がかかります。このプロトコルは、最適化された結晶の回折をテストすることも提案しています。シンクロトロンに定期的にアクセスできない結晶学者にとって、これらのテストは困難になる可能性があります。すべてのステップでコントロールを行う必要はないかもしれませんが、ヒットが特定されたら、スケーリングの前後にこれらのテストを行うことを強くお勧めします。XFELでの非回折結晶は、残念ながら珍しいことではありません。これを考えると、結晶回折に関する仮定に関して注意を怠る方が良いでしょう。
最終的に、ここで提示されたこのプロトコルと結果は、連続結晶学実験用のサンプルの作成に苦労している人々にガイド、アイデア、および例を提供します。うまくいけば、連続結晶構造解析がさらに開発されるにつれて、このようなプロトコルの必要性が減るように、この技術のサンプル需要が減るでしょう。ただし、このイベントでも、ここで紹介する戦略は、タンパク質の結晶化空間を探索したい人にとっては依然として有用です。
開示事項
著者は、開示すべき利益相反はありません。
謝辞
このプロジェクトは、マリー・スクウォドフスカ・キュリー助成契約No 701647の下で、欧州連合のホライズン2020研究およびイノベーションプログラムから資金提供を受けています。スイス光源ビームラインX10SA-PXIIのビームライン研究者の支援と支援に感謝します。
資料
| Name | Company | Catalog Number | Comments |
| Swissci 96-well 2-Drop plates | Molecular Dimensions | MD11-002 | 96-well 2-drop crystallisation plate |
| Swissci 96-well 3-Drop plates | Molecular Dimensions | MD11-003 | 96-well 3-drop crystallisation plate |
| mosquito LCP liquid handling robot | sptlabtech | mosquito LCP | Crystallisation robot |
| ClearVue Sheets | Molecular Dimensions | MD6-015 | 96-well crystallization plate seals |
| Safe-Tube 1.5 mL | Eppendorf | 30120086 | 1.5 mL centrifuge tubes |
| Scaple | Swan and Morton | No. 3 scalple and No. 3 handle | Scalple for cutting open plate seals |
| MS 3 Vortex | IKA | 3319000 | Vortex for mixing solution and making seed stocks |
| 24-well XRL Plate | Molecular Dimensions | MD3-11 | 24-well hanging-drop plates |
| Tube revolver/rotator | Thermo Fischer Scientific | 88881001 | Tube revolver for mixing solution during scaling |
| Eppendorf Research plus pipettes | Eppendorf | Range of manual pipettes, 0.5-10, 1-20, 10-100, 100-1000 µL | |
| Eppendorf pipette tips | Eppendorf | Range of tip sizes for manual pipettes | |
| Suparen 600 | Prochem AG | Suparen 600 | Endothiapepsin solution |
| Sodium Acetate | Sigma-Aldrich | 241245-1KG | Sodium Acetate |
| Tris | Merck | 8382T014 | Tris |
| Magnesium Chloride | Sigma-Aldrich | M2670-1kg | Magnesium Chloride |
| PEG 6,000 | Sigma-Aldrich | 81255-1kg | PEG 6,000 |
| Ethelyene glycol | Sigma-Aldrich | 324558-1L | Ethelyene glycol for cyro-protecting the crystals |
| PACT Premier HT screen | Molecular Dimensions | MD1-36 | PACT Premier 96-well crystal screen |
| DOW CORNING high vacuum grease | Molecular Dimensions | MD6-02 | Grease for sealing 24-well plates |
| Hirschmann 22 x 22 mm glaser cover slides | Hirschmann | 8000104 | Cover slides for sealing 24-well sitting drop plates |
| Crystal pins | PSI | Manufactured inhouse | Thin-film supports for micro-crystals. |
| 1-1.3 mm SiLibeads Type S | Faust | 6239547 | Glass beads for making mico-seed stocks |
| Macbook Pro | Apple | Macbook Pro | Computer for performing data analysis |
| CCP4 software suite | CCP4 | Diffraction pattern data processing software | |
| Excel | Microsoft | Microsoft Office | Plotting tool for phase diagram |
| Hausser Scientific Bright-Line counting chamber | Thermo Fischer Scientific | 02-671-51B | Tool to calculate crystal concentration |
| PACT Premier | Molecular Dimensions | MD1-29-ECO | Sparse-matrix crystallization screen |
| Rock Imager | Formulatrix | Rock Imager | Temperature controlled crystal plate storage and imager |
| Rock MakerWeb | Formulatrix | Rock MakerWeb | Crystal plate creation and image storage stoftware |
| Formulator | Formulatrix | Formulator | 96-well crystal screen creation liquid handling robot |
| Leica MZ16 Microscope | Leica | Leica MZ16 | Light microscope |
| LAS V4.6 | Leica | LAS V4.6 | Software for Leica microscopes |
| Spectra/Por 3.5 kDa dialysis tubing | Spectrumlabs | Spectra/Por 3 Dialysis Membrane | 3.5 kDa dialysis membrane |
| Dialysis tubing closures | Spectrumlabs | Spectra/Por 3 Duniversal Closures | Clips to seal the dialysis tubing ends |
| Amicon 10 kDa centrifugal concentrator | Merck-Millipore | Amicon Ultra-15 10 kDa centrifugal concentrator | 10 kDa centrifugal filter |
| 5810 R swing bucket centrifuge | Eppendorf | 5810 R Centrifuge | Swing bucket centrifuge |
参考文献
- DePonte, D. P., et al. Gas dynamic virtual nozzle for generation of microscopic droplet streams. Journal of Physics D: Applied Physics. 41 (19), 195505 (2008).
- Hunter, M. S., et al. Fixed-target protein serial microcrystallography with an x-ray free electron laser. Scientific Reports. 4 (1), 6026 (2014).
- Weierstall, U., et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nature Communications. 5 (1), 1-6 (2014).
- Roessler, C. G. G., et al. Acoustic Injectors for Drop-On-Demand Serial Femtosecond Crystallography. Structure. 24 (4), 631-640 (2016).
- Sherrell, D. A., et al. A modular and compact portable mini-endstation for high-precision, high-speed fixed target serial crystallography at FEL and synchrotron sources. Journal of Synchrotron Radiation. 22 (6), 1372-1378 (2015).
- Roedig, P., et al. A micro-patterned silicon chip as sample holder for macromolecular crystallography experiments with minimal background scattering. Scientific Reports. 5 (1), 1-11 (2015).
- Botha, S., et al. Room-temperature serial crystallography at synchrotron X-ray sources using slowly flowing free-standing high-viscosity microstreams. Acta Crystallographica Section D Biological Crystallography. 71 (2), 387-397 (2015).
- Weinert, T., et al. Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nature Communications. 8 (1), 542 (2017).
- Tenboer, J., et al. Time-resolved serial crystallography captures high-resolution intermediates of photoactive yellow protein. Science. 346 (6214), 1242-1246 (2014).
- Nango, E., et al. A three-dimensionalmovie of structural changes in bacteriorhodopsin. Science. 354 (6319), 1552-1557 (2016).
- Suga, M., et al. Light-induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature. 543 (7643), 131-135 (2017).
- Mehrabi, P., et al. Liquid application method for time-resolved analyses by serial synchrotron crystallography. Nature Methods. 16 (10), 979-982 (2019).
- Halle, B. Biomolecular cryocrystallography: structural changes during flash-cooling. Proceedings of the National Academy of Sciences of the United States of America. 101 (14), 4793-4798 (2004).
- Fraser, J. S., et al. Hidden alternative structures of proline isomerase essential for catalysis. Nature. 462 (7273), 669-673 (2009).
- Fenwick, R. B., van den Bedem, H., Fraser, J. S., Wright, P. E. Integrated description of protein dynamics from room-temperature X-ray crystallography and NMR. Proceedings of the National Academy of Sciences of the United States of America. 111 (4), 445-454 (2014).
- Keedy, D. A., et al. Mapping the conformational landscape of a dynamic enzyme by multitemperature and XFEL crystallography. Elife. 4, (2015).
- Thomaston, J. L., et al. XFEL structures of the influenza M2 proton channel: Room temperature water networks and insights into proton conduction. Proceedings of the National Academy of Sciences of the United States of America. 114 (51), 13357-13362 (2017).
- Haas, D. J., Rossmann, M. G. Crystallographic studies on lactate dehydrogenase at -75 °C. Acta Crystallographica Section B Structural Crystallography and Crystal Chemistry. 26 (7), 998-1004 (1970).
- Hope, H. Cryocrystallography of biological macromolecules: a generally applicable method. Acta Crystallographica Section B Structural Science. 44 (1), 22-26 (1988).
- Wu, W., et al. Batch crystallization of rhodopsin for structural dynamics using an X-ray free-electron laser. Acta Crystallographica Section:F Structural Biology Communications. 71 (7), 856-860 (2015).
- Ishchenko, A., Cherezov, V., Liu, W. Preparation and delivery of protein microcrystals in lipidic cubic phase for serial femtosecond crystallography. Journal of Visualized Experiments. (115), e54463 (2016).
- Andersson, R., et al. Well-based crystallization of lipidic cubic phase microcrystals for serial X-ray crystallography experiments. Acta Crystallographica Section D: Structural Biology. 75 (10), 937-946 (2019).
- Luft, J. R., et al. The detection and subsequent volume optimization of biological nanocrystals. Structural Dynamics. 2 (4), 041710 (2015).
- Lee, D. B., et al. Supersaturation-controlled microcrystallization and visualization analysis for serial femtosecond crystallography. Scientific Reports. 8 (1), 1-10 (2018).
- Kupitz, C., et al. Microcrystallization techniques for serial femtosecond crystallography using photosystem II from Thermosynechococcus elongatus as a model system. Philosophical transactions of the Royal Society of London. Series B, Biological Sciences. 369 (1647), 20130316 (2014).
- Beale, J. H., et al. Successful sample preparation for serial crystallography experiments. Journal of Applied Crystallography. 52, 1385-1396 (2019).
- Rayment, I. Small-scale batch crystallization of proteins revisited: An underutilized way to grow large protein crystals. Structure. 10 (2), 147-151 (2002).
- García-Ruiz, J. M. Nucleation of protein crystals. Journal of Structural Biology. 142 (1), 22-31 (2003).
- McPherson, A., Kuznetsov, Y. G. Mechanisms, kinetics, impurities and defects: Consequences in macromolecular crystallization. Acta Crystallographica Section F:Structural Biology Communications. 70 (4), 384-403 (2014).
- Rupp, B. Origin and use of crystallization phase diagrams. Acta crystallographica. Section F, Structural biology communications. 71, 247-260 (2015).
- Luft, J. R., DeTitta, G. T. A method to produce microseed stock for use in the crystallization of biological macromolecules. Acta Crystallographica Section D Biological Crystallography. 55 (5), 988-993 (1999).
- Ireton, G. C., Stoddard, B. L. Microseed matrix screening to improve crystals of yeast cytosine deaminase. Acta crystallographica. Section D, Biological crystallography. 60 (3), 601-605 (2004).
- Forsythe, E. L., Maxwell, D. L., Pusey, M. Vapor diffusion, nucleation rates and the reservoir to crystallization volume ratio. Acta Crystallographica Section D Biological Crystallography. 58 (10), 1601-1605 (2002).
- Chayen, N. E., Shaw Stewart, P. D., Maeder, D. L., Blow, D. M. IUCr An automated system for micro-batch protein crystallization and screening. Journal of Applied Crystallography. 23 (4), 297-302 (1990).
- Chayen, N. E., Shaw Stewart, P. D., Blow, D. M. Microbatch crystallization under oil - a new technique allowing many small-volume crystallization trials. Journal of Crystal Growth. 122 (1-4), 176-180 (1992).
- Chayen, N. E. Comparative studies of protein crystallization by vapour-diffusion and microbatch techniques. Acta Crystallographica Section D: Biological Crystallography. 54 (1), 8-15 (1998).
- D'Arcy, A., Mac Sweeney, A., Stihle, M., Haber, A. The advantages of using a modified microbatch method for rapid screening of protein crystallization conditions. Acta Crystallographica - Section D Biological Crystallography. 59 (2), 396-399 (2003).
- Darmanin, C., et al. Protein crystal screening and characterization for serial femtosecond nanocrystallography. Scientific Reports. 6, 25345 (2016).
- Gati, C., et al. Atomic structure of granulin determined from native nanocrystalline granulovirus using an X-ray free-electron laser. Proceedings of the National Academy of Sciences of the United States of America. 114 (9), 2247-2252 (2017).
- Cheng, R. K. Y. Towards an optimal sample delivery method for serial crystallography at XFEL. Crystals. 10 (3), 215 (2020).
- Schmidt, M. Mix and Inject: Reaction Initiation by Diffusion for Time-Resolved Macromolecular Crystallography. Advances in Condensed Matter Physics. 2013, 1-10 (2013).
- Oberthuer, D., et al. Double-flow focused liquid injector for efficient serial femtosecond crystallography. Scientific Reports. 7 (1), 44628 (2017).
- McPherson, A., Gavira, J. A. Introduction to protein crystallization. Acta crystallographica. Section F, Structural biology communications. 70, 2-20 (2014).
- Yaoi, M., et al. Effect of stirring method on protein crystallization. Japanese Journal of Applied Physics, Part 2: Letters. 43 (10), 1318 (2004).
- Castro, F., Ferreira, A., Teixeira, J. J., Rocha, F. Influence of Mixing Intensity on Lysozyme Crystallization in a Meso Oscillatory Flow Reactor. Crystal Growth & Design. 18 (10), 5940-5946 (2018).
- Moews, P. C., Bunn, C. W. An X-ray crystallographic study of the rennin-like enzyme of Endothia parasitica. Journal of Molecular Biology. 54 (2), 395-397 (1970).
- Newman, J., et al. Towards rationalization of crystallization screening for small- to medium-sized academic laboratories: the PACT/JCSG+ strategy. Acta crystallographica. Section D, Biological. 61, 1426-1431 (2005).
- Ebrahim, A., et al. Resolving polymorphs and radiation-driven effects in microcrystals using fixed-target serial synchrotron crystallography. Acta Crystallographica Section D Structural Biology. 75, 151-159 (2019).
- Davy, B., et al. Reducing sample consumption for serial crystallography using acoustic drop ejection. Journal of Synchrotron Radiation. 26 (5), (2019).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved