
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
ゼブラフィッシュガストラの2光子顕微鏡を用いた深部および空間制御体積アブレーション
要約
胚発生には、細胞運動の大規模な調整が必要です。2光子励起媒レーザーアブレーションは、深部細胞の大群の空間制御3次元アブレーションを可能にする。さらに、この技術は、生体内で細胞を機械的環境 で 摂動する一括的に遊動する反応をプローブすることができる。
要約
形態形成は、組織や器官に細胞を整理するために多くの細胞の動きを伴います。適切な開発のためには、これらすべての動きを緊密に調整する必要があり、証拠を蓄積することは、少なくとも部分的には機械的相互作用を通じてこれが達成されることを示唆している。胚でこれをテストするには、直接的な物理的摂動が必要です。レーザーアブレーションは、機械的な制約を緩和したり、2つの細胞集団を互いに物理的に分離することを可能にする、ますます使用されるオプションです。しかし、多くのアブレーションは、限られた軸解像度と組織の浸透を提供する紫外線(UV)レーザーで行われます。2光子顕微鏡を用いて深く、有意で、空間的に明確に定義された容積を取り下げるための方法をここに記載する。アブレーションは、軸性メセンドームの緑色蛍光タンパク質を発現するトランスジェニックゼブラフィッシュラインで実証され、上にある外胚葉または下層の黄身細胞に影響を与えることなく軸性の間皮を切断するために使用される。細胞の挙動は、アブレーションの前後のライブイメージングによって監視されます。アブレーションプロトコルは、数ミクロンから100ミクロン以上のスケールで、任意の細胞タイプまたは組織上の異なる発達段階で使用することができます。
概要
細胞間相互作用は、開発において重要な役割を果たします。細胞は、直接の隣人、または遠く離れた細胞が知覚できる信号を提供し、それによってその運命および/または行動に影響を与えます。これらの信号の多くは、自然の中で化学的です。例えば、よく特徴付けられる誘導事象では、ある細胞群は、別の細胞集団の運命に影響を与える拡散性分子を生成する1。しかし、他の信号は機械的です。細胞は、隣人が知覚し、2に応答する彼らの隣人に力と制約を発揮します。
生体内での細胞間相互作用の重要性を研究する一つの方法は、細胞を除去し、その後の発達を観察することです。残念ながら、細胞を除去または破壊する利用可能な技術は限られています。細胞は、針または小さなワイヤーを使用して外科的に3,4を除去することができるが、そのような治療は侵襲的であり、あまり正確ではなく、通常は実体顕微鏡下で行われ、顕微鏡下での即時のイメージングを防ぐ。さらに、深部細胞を標的とすることは、上層部の組織に穴を開け、望ましくない摂動を生み出すことも意味します。遺伝的にコード化された光化物は、KillerRedのような、光照明を介して細胞死を誘導するために使用されてきた5。光化剤は、光照射時に活性酸素種を生成するクロモフォアです。彼らの主な制限は、細胞が動いている場合に達成するのが難しいかもしれない長い光のイルミネーション(約15分)を必要とし、即座ではないアポトーシスを通じて細胞死を誘発することです。
最後に、レーザーアブレーションは、過去15年間で開発され、広く使用されています 6,7,8,9,10,11,12.レーザービームは標的細胞/組織に焦点を合わせる。それは加熱、光切除、またはプラズマ誘発アブレーションを通してそのアブレーションを誘発する;関係するプロセスは、電力密度と露光時間13に依存します。ほとんどのアブレーションプロトコルは、高エネルギーにUVレーザーを使用します。しかし、UV光は両方とも生体組織によって吸収され、散乱される。したがって、深部細胞を標的化するには高いレーザーパワーが必要であり、その後、より表面的で平面外の組織に損傷を与える。これは、表面構造にUVレーザーの使用を制限し、その比較的低い軸解像度を説明します。非線形光学(いわゆる2光子顕微鏡)は、非線形の光特性を使用して、赤外線領域内の約半エネルギーの2つの光子を有する蛍光色素を励起する。アブレーションに適用する場合、これには3つの主な利点があります。まず、赤外線は生体組織14によるUV光よりも散乱が少なく、吸収が少なく、必要なレーザーパワーを増加させることなく、より深い構造に到達することを可能にする。第二に、フェムト秒パルスレーザーの使用は、プラズマ誘導を介してアブレーションを作成し、加熱に反して、空間的に拡散しない非常に高い電力密度を提供します15。第3に、プラズマ形成を誘導する電力密度は、焦点のみで到達する。これらの特性のおかげで、2光子レーザーアブレーションは、周囲の組織環境に影響を与えることなく、深部細胞を正確に標的化するために使用することができます。
集団移動は、細胞と細胞の相互作用が基本的な発達過程の優れた例である。集合的な移行は、隣接するセルが 1 つの cell16 の動作に影響を与えるセル移動として定義されます。これらの相互作用の性質(化学的または機械的)およびそれらが細胞の移動に与える影響は大きく異なり、多くの場合、完全には理解されていません。細胞を除去し、これが他の人にどのような影響を与えるかを観察する能力は、これらの集合的なプロセスをさらに解明する上で重要です。数年前、ゼブラフィッシュの胃の中でポルスターの移動が集団移住であることを、外科的アプローチを用いて確立しました17。ポルスターは、胚18の後側の第1の内在化細胞を構成する細胞群である。 Tg(gsc:GFP) トランスジェニックラインで緑色に標識されたこれらの細胞は、胚の深部、エピブラスト細胞のいくつかの層の下に位置する。胃の間、このグループは軸性中胚の延長を導き、胚の形成者から動物の極19、20、21、22、23 に移動する(図1A)。私たちは、細胞が動物の極の方向に彼らの移動を向けるために彼らの隣人との接触を必要とすることを確立しました。しかし、この集団移動の細胞および分子塩基をよりよく理解するには、いくつかの細胞を除去して、これが残りの細胞にどのような影響を与えるかを確認する必要があります。そこで、2光子顕微鏡を用いて大体積と深いボリュームのアブレーションを開発しました。ここでは、このプロトコルを使用して、その真ん中にポルスターを切断し、Histone2B-mCherryで標識された核を追跡することによって細胞移動の結果を観察することを実証する。
プロトコル
すべての動物の仕事は、倫理委員会N 59とミニステア・ド・レヒスメント・ナショナル、ド・ランセワニエメント・スペリエール・エ・ド・ラ・レシェルシュのファイル番号APAFIS#15859-2018051710341011v3によって承認されました。以下に説明する手順の一部は、当社の機器とソフトウェアに固有のものですが、さまざまな機器に簡単に適応することができます。
1. 注射準備
- 胚培地(EM)で1%アガロース溶液の75 mLを調製します。
- 90 mm ペトリ皿に注入金型を入れ、約 50 mL のアガロースを注ぎ、金型が浮くのに十分です。アガロースを固め、注入型を取り除きましょう。
- アガロースを30mmペトリ皿に1mL入れ、アガロースコーティングした料理を用意します。
- RNaseフリー水でストック溶液を希釈し、氷の上に保つことによって、30 ng /μLヒストン2B-mCherry mRNA溶液の4 μLを調製します。
注:RNase媒介性の劣化を避けるためにmRNAを操作しながら手袋を着用するように注意してください。 - マイクロピペットプーラーを使用して毛細血管から注射針を引っ張ります。
2. 胚の準備
- 魚が卵を産んだら、EMで90mmのペトリ皿に収穫し、収集し、すすべり、収穫します。胚を28.5°Cインキュベーターに入れる。
- 最初のセルが見えるように 20 分待ちます。
- 30個の胚をEMで満たされた注入プレートに移す。わずかに鈍い鉗子を使用して溝の中の胚を絞り、動物のポールを上に向けます。
- マイクロローダーチップを使用して、注射針に2 μLのmRNA溶液を充填します。ポリテトラフルオロエチレン(PTFE)チューブに接続されたマイクロマニピュレータに入れたキャピラリーホルダーに針をエアインジェクタに挿入します。
- 実体顕微鏡の下で、針の先端を慎重に折ります。
- 細胞に針を挿入して、1細胞段階の胚にmRNA溶液を注入する。
メモ:注入される容積はセル容積の約3分の1である。 - 28.5°Cインキュベーターに戻って注入された胚を入れる。
3. 二光子顕微鏡の作製
注: このプロトコルでは、2 つのレーザーが使用されています。1 つは、GFP (920 nm) をイメージし、アブレーション (820 nm) を実行するために使用されます。グリーン/アブレーションレーザーと呼ばれます。もう一つは、mCherryをイメージするために1160 nmで使用されます。赤レーザーと呼ばれます。
- 緑/アブレーションレーザーを820 nm(アブレーション波長)に、赤色レーザーを1160 nm(mCherry励起)に設定します。
- 光路上の可動ミラーを使用して、スキャンヘッドの入り口と出口の両方で緑色/アブレーションと赤のレーザービームを整列させます。
注: これにより、レーザービームの焦点が増加し、励起とアブレーションの焦点量が最小限に抑えられます。 - 目標の下で820 nmでグリーン/アブレーションレーザーの最大電力を測定します。そのためには、目的の下にパワーメーターを置き、黒い部屋を閉じ、緑/アブレーションレーザーパワーを100%に設定し、シャッターを開きます。300 mW に達するために必要なレーザーパワーの割合を計算します。
- グリーン/アブレーションレーザーを920 nm(GFP励起)に戻し、レーザーパワーを7%に設定します。赤いレーザーパワーを15%に設定します。
- 緑と赤の線のためのエピフォトマルチプライヤチューブ(PMT)検出器をアクティブにします。65 に緑と赤の線 PMT 感度を設定します。
- 視野を400 x 400 μmに、画像解像度を512 x 512ピクセルに設定し、スキャン周波数を800 Hzに設定します。
- [3D タイムラプス イメージング モード]を選択します。次に、フォルダを作成し、取得後にデータの自動保存を有効にします。
- 加熱室を組み立て、28°Cに設定します。 チャンバーと目的が暖かくなるのに少なくとも10分待ちます。
4. 胚の取り付け
- 蛍光体型顕微鏡の下で、GFPを発現する70%のエピボリーで胚を同定する。
注:より良いイメージング品質のための軸中胚および背景蛍光のない明るい信号を有する胚を選択する。 - プラスチック製のパスツールピペットを使用してアガロースコーティング皿(ステップ1.3)で3〜4個の選択した胚を移し、細かい鉗子を使用して慎重にデコリオネートします。
注意:デコリオネート胚は非常に繊細であり、空気またはプラスチックと接触すると破裂します。 - 小さなガラスバイアルに1xペニシリンストレプトマイシンEMで0.2%アガロースの1 mLを注ぎます。バイアルを予熱した42°Cのドライブロックヒーターに入れる。
注:アガロースが設定される前に胚の向きを可能にするために、次の手順を迅速に実行する必要があります。 - 火で磨いたガラスピペットを使用して、0.2%のアガロースガラスバイアルでデコリオネート胚を移す。アガロースにEMを加えすぎないように注意して、希釈しないように注意してください。残りのEMをピペットから捨て、胚がピペットから落ちる前にガラス底皿のスライドを覆うのに十分なアガロースと一緒に胚を吸引する。
- アガロースと胚を皿のガラススライドで吹きます。胚が空気や皿のプラスチック側に触れないように注意してください。次に、ガラススライドの周りのチャンバーをアガロースで満たします。
- まつげを使用して、ターゲット領域が上部になるように胚の向きを指定します(図1B)。
注:胚の配向時には、非常に壊れやすい黄身ではなく、ブラトデベルムだけに触れるのに注意してください。アガロースは室温に応じて約1分で設定されます。 - アガロースが完全に設定されるまで〜5分待ってから、ペニシリンストレプトマイシンEMを数滴加えます。
5. 胚の位置とアブレーション前のイメージング
- 熱い部屋の目的の下にガラス底皿を置きます。ペニシリンストレプトマイシンEMに目的を浸し、加熱されたチャンバーを閉じます。
- スライダを動かして、光のパスをオキュラーに設定します。次に、眼、蛍光灯、およびステージコントロールを使用して、胚を見つけ、胚の表面に焦点を当てる。
- 蛍光灯を消し、PMTへの光路を設定し、黒い部屋を閉じます。
注:PMTが損傷する可能性があるため、黒い部屋のすべての光源をオフにするように注意してください。 - ライブイメージングを開始し、軸中軸を見つけます。グリーン/アブレーションと赤レーザーのパワーを調整して、良好な信号を得ます(GFP表現領域の場合、ピクセルあたり1,000~20,000光子)。赤いチャネルを使用して、ステージを胚の最上部に移動し、この位置を Z = 0 に設定します。
- 1分の時間ステップと2μmのZステップを選択してください。110 μmのZコースは、ポルスター全体を包含するのに十分であり、これらの設定で1分未満で獲得されます。軸中隔の上に15μmの最初のスライスをセットします(より表面的な外視)。
注:ポルスターは曲線に沿って移動するので、Zスタックの底部のスライスは、タイムラプスイメージング中の動きに対応するために、最も深い位置を30μm深く設定する必要があります(図1E)。 - アブレーション前の映画の記録10-15分。

図1:レーザーアブレーションの成功した結果.(A)後視で70%のエピボリーでの胃切れ胚のスキーム。pAM: 後軸中隔;黒い矢印はポルスター移行の方向を示します。黒い四角は、ポルターのアブレーションの典型的な視野を示します。(B)ポルスター断絶のための胚取付のスキーム。横表示。胚は、ポルスターの平面が光軸に対して垂直になるように取り付けられる。(C)生存および(D)受精後24時間での対照およびアブレート胚の形態。スケールバーはヒストン2B-mCherryを発現するTg(gsc:GFP)胚のポルスターにおけるレーザーアブレーションからの時間配列です。緑のチャンネルのみで表示されるビューは、最大投影です。クローズアップは、細胞の破片を含むアブレート領域を表示します。緑と赤(マゼンタとして表示)チャンネルを持つビューは、アブレーションの前後に XY と XZ スライスです(緑色の稲妻はアブレーションを表します)。XZスライスは、上にある組織(GFP発現のないマゼンタ核)が基礎構造のアブレーションの影響を受けないことを示しています。黄色の破線ボックスは、レーザーアブレーション処理のために選択されたROIに対応します。スケールバーは、大きなビューで50μm、クローズアップで25μmです。この図の大きなバージョンを表示するには、ここをクリックしてください。
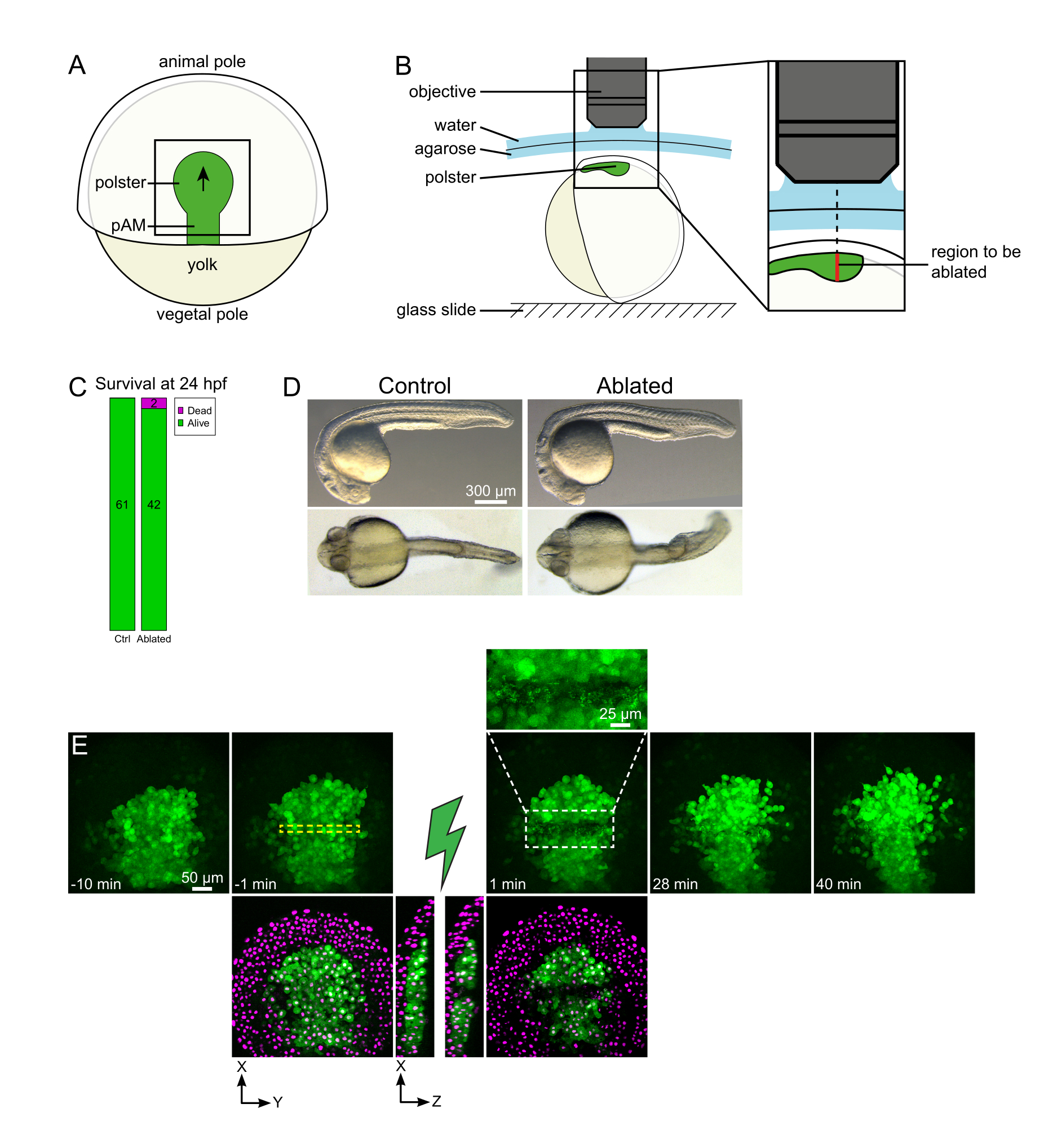{kind=link}
6. ターゲット位置とレーザーアブレーション
- ライブイメージングでポルター輪郭を見つけ、電気光学変調器の関心領域(EOM ROI)ツールを使用して、ポルスターの幅にまたがる20ピクセル(15 μm)の大きな長方形を描きます。この長方形をポルスターの中央に配置します(図1E)。
- ポルテル セルを含む最も高い平面と最も低い平面の軸方向の位置に注意してください。アブレーションは、これら2つの平面の間で10μmごとに行われます。ROI がこれらの平面の卵黄セルに重ならないように注意してください。
- 間隔の最も低い Z 位置にステージを配置します。アブレーションは、破片が光を吸収するので、ボトムアップを実行する必要があります。
- 緑/アブレーションレーザー波長を820 nmに設定し、 出力の割合 を設定して300 mWの出力電力を得ます(ステップ3.3)。
- イメージング周波数を200 Hzに設定します。
- 緑/アブレーションレーザーイメージングEOMを0に設定し、 ROI-Treatモードを 選択します。
- EOMをオンにし、(0フレーム後)すぐに処理を開始するように設定します。
- イメージングモードをタイムラプスに設定し、自動保存を解除します。
- [タイム ステップ]を[高速]モードに設定します。
- [処理フレーム数] と [フレーム数] を、対象の深さに対応する値に設定します(表 1)。
| 深さ(μm) | 治療フレーム |
| -30 | 1 |
| -35 | 1-2 |
| -40 | 1-2 |
| -45 | 2 |
| -50 | 2-3 |
| -55 | 3 |
| -60 | 3-4 |
| -65 | 4 |
| -70 | 4 |
| -75 | 4-5 |
| -80 | 4-5 |
| -85 | 5 |
| -90 | 5 |
| -95 | 5-6 |
| -100 | 6 |
| -105 | 6 |
表1:胚における標的細胞深度の関数としてのレーザー処理フレーム数の提案(0は胚の表面である)。
- イメージングを開始します。EOM治療中にPMTへのシャッターが閉じると、取得は黒になります。
- ステージをリストの次の Z 位置に上げる (ステップ 6.2)。
- ステップ 6.10 ~ 6.12 を繰り返して、ポールスターの上部に到達します。
7. アブレーション後の検証とイメージング
- グリーン/アブレーションレーザーを920 nmおよび5%の電力に設定します。緑/アブレーションレーザーイメージングEOMを100に設定し、 フルフィールド モードを選択します。
- イメージング周波数を800 Hzに設定し、EOMをオフにします。
- すべてのプレーンがアブレートされているかどうかを確認するために、ライブモードでスタック全体を通過します。この場合は、手順 6.2 に戻ります。
注: アブレーションは、Zスタックを再定義しなければならないように、隣接する組織の垂直シフトを誘発することがあります。 - イメージングモードを3D Timelapseに設定し、自動保存を再びアクティブにします。アブレーション後の映画の記録40-60分。
- アブレーション後のムービーで、標的細胞が効果的に失われたかどうかを確認します。蛍光回復、または領域を占有し、フォロワー細胞が通過するのを妨げる標的細胞は、標的細胞が光漂白されただけで、アブレートされないことを示す(図1Eおよび図2A)。

図2:レーザーアブレーションの負の結果(A)レーザーアブレーションにおける潜在的な故障の典型的な例。大きな XY ビューは最大投影であり、XZ ビューは再構築された断面です。レーザー処理領域は、2つの白い矢印の間にあります。3 つの焦点平面が再構築されたセクションでハイライト表示され、右側に表示されます。これらは、3 種類の障害に対応します。平面1は、ポルスターの上の細胞がアブレートされたことを示しています。これは、ポルスターの上のこの焦点面(クローズアップを参照)に自己蛍光破片が存在することによって識別することができます(再構築されたセクションの平面1の位置を参照してください)。これは、省略する領域の定義が正しくないことが原因である可能性があります。平面 2 は、漂白されたが、アブレートされていない細胞を示しています。それらは低い蛍光シグナルとして識別することができるが、それでも無傷の細胞輪郭を明らかにする(クローズアップ参照)。平面3は、レーザー処理によってほとんど漂白されていない無傷の細胞を表示します。これは、アブレートされる領域の誤った定義または貧しい治療から生じる可能性があります。平面2および3に描かれた状況では、アブレーション処理を非アブレート対象細胞に再適用することができる。スケールバーは、大きなビューで50 μm、クローズアップで20 μmです。(B)あまりにも激しいレーザー処理のためにキャビテーションによって形成された気泡(白いアスタリスクでマークされる)の典型的な例。このような気泡はZ面に限らず、時にはポルスターの全高にまたがって隣接組織を変形させることもある。スケールバーは50 μmです。この図の大きなバージョンを表示するには、ここをクリックしてください。
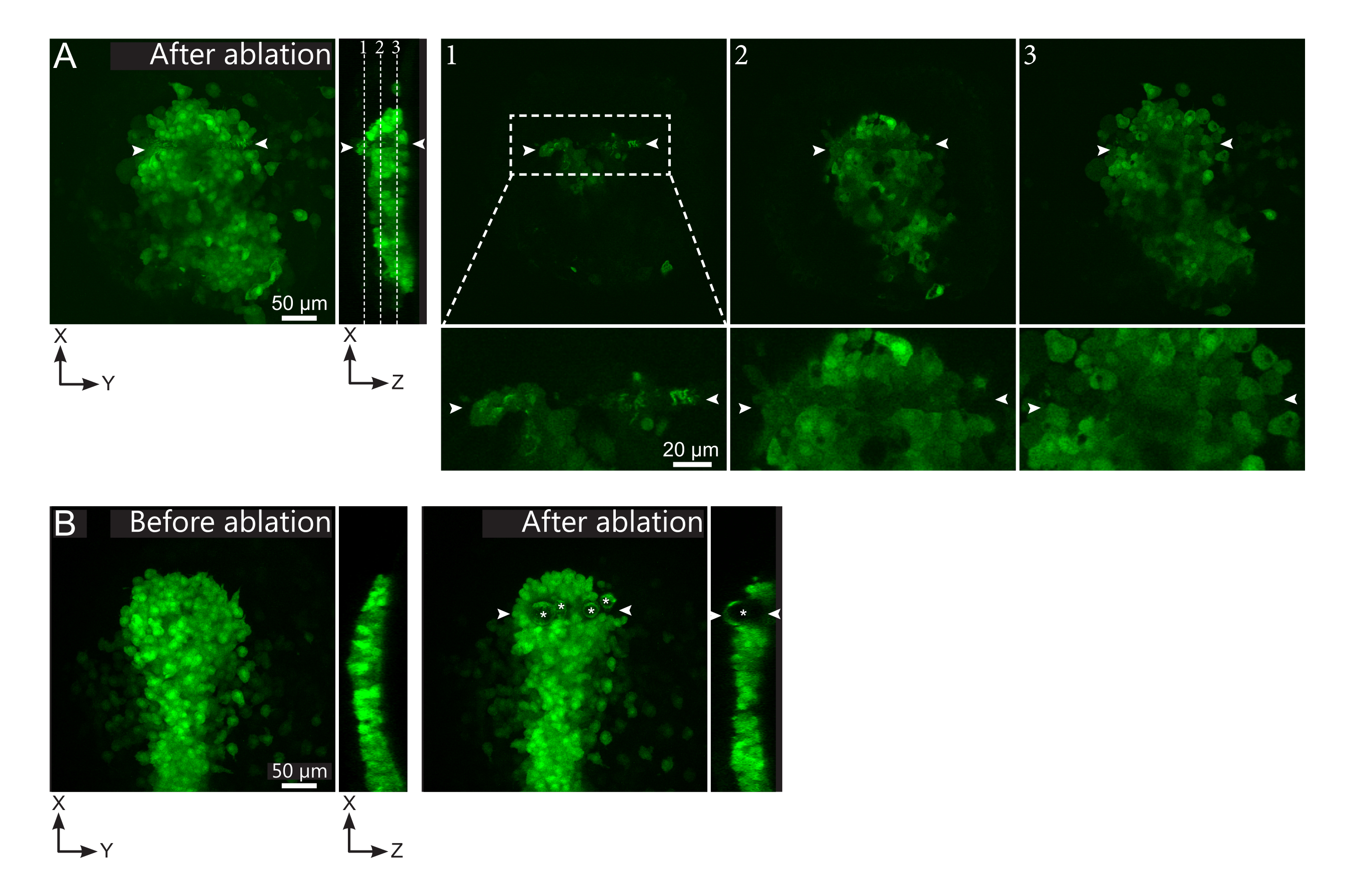{kind=link}
8. データ分析
- 画像解析ソフトウェアでタイムラプスシリーズを開き、正しいピクセルサイズを設定します。
- スポット関数で、オブジェクトサイズを 10 μm に設定します(これは、ガストルレーション中の平均核サイズ)。次に、スポット関数を実行して、核を検出して追跡します。
注: 検出は、Z軸に沿って長さ12μmの楕円体形状を合わせ、下軸分解能を考慮することで若干改善される場合があります。 - フィルターを使用して、誤検出を削除します。 Tg(Gsc:GFP) 線では、胚軸の細胞と一部の内皮細胞は緑色で標識されます。したがって、緑色の強度でフィルタリングすると、これらのセルを素早く選択できます(図3A)。
- 連続する点の最大距離を、セルの速度と互換性のある値に設定します。
注: 2 つのフレーム間の時間間隔を考慮するように注意してください。ポルスター細胞は2.8±0.8 μm/minで移動します。したがって、1分の時間ステップで最大変位の4 μmを許可すると、ほとんどのアーテフ実測のトラックが削除されます。 - 1 つまたは 2 つのタイム ポイントにギャップを許容すると、連続したトラックが長くなりますが、トラッキング エラーが発生する可能性があります。1 回限りのポイントで核が正しく検出されない場合は、異なるパラメータ/フィルタでスポット検出を再実行することを検討してください。
- トラックを視覚的にチェックし、必要に応じて修正します。
- 結果を.xlsxファイルとしてエクスポートします。公開されたスプレッドシートルーチン24 (図3B)とデータ分析ソフトウェアのカスタムルーチン(要求に応じて利用可能)を使用してファイルを処理します。

図3:ポルスターの前半分の分離は細胞の方向性に影響を与える。 (A)3D再構成は、ヒストン2B-mCherry(マゼンタに表示される )を発現するTg(gsc:GFP) 胚を、その真ん中にポルスターを切断する前後に起こる。ポルスターの前半分に属する核はマゼンタドットでマークされ、アブレーションの前後に時間をかけて追跡されます( Movie S1を参照)。スケールバーは50μm(B)移動持続性の尺度として、アブレーション前後のポルスターの前部に属する細胞の方向自動相関である。細胞はアブレーション前の連続運動を示し、アブレーション後に大幅に減少し、集合指向の移動の喪失を示します。方向自己相関はまた、コントロールとして非アブレート胚のポルスターの前半分を形成する細胞についても測定した。グラフエンベロープは標準誤差を示します。 この図の大きなバージョンを表示するには、ここをクリックしてください。
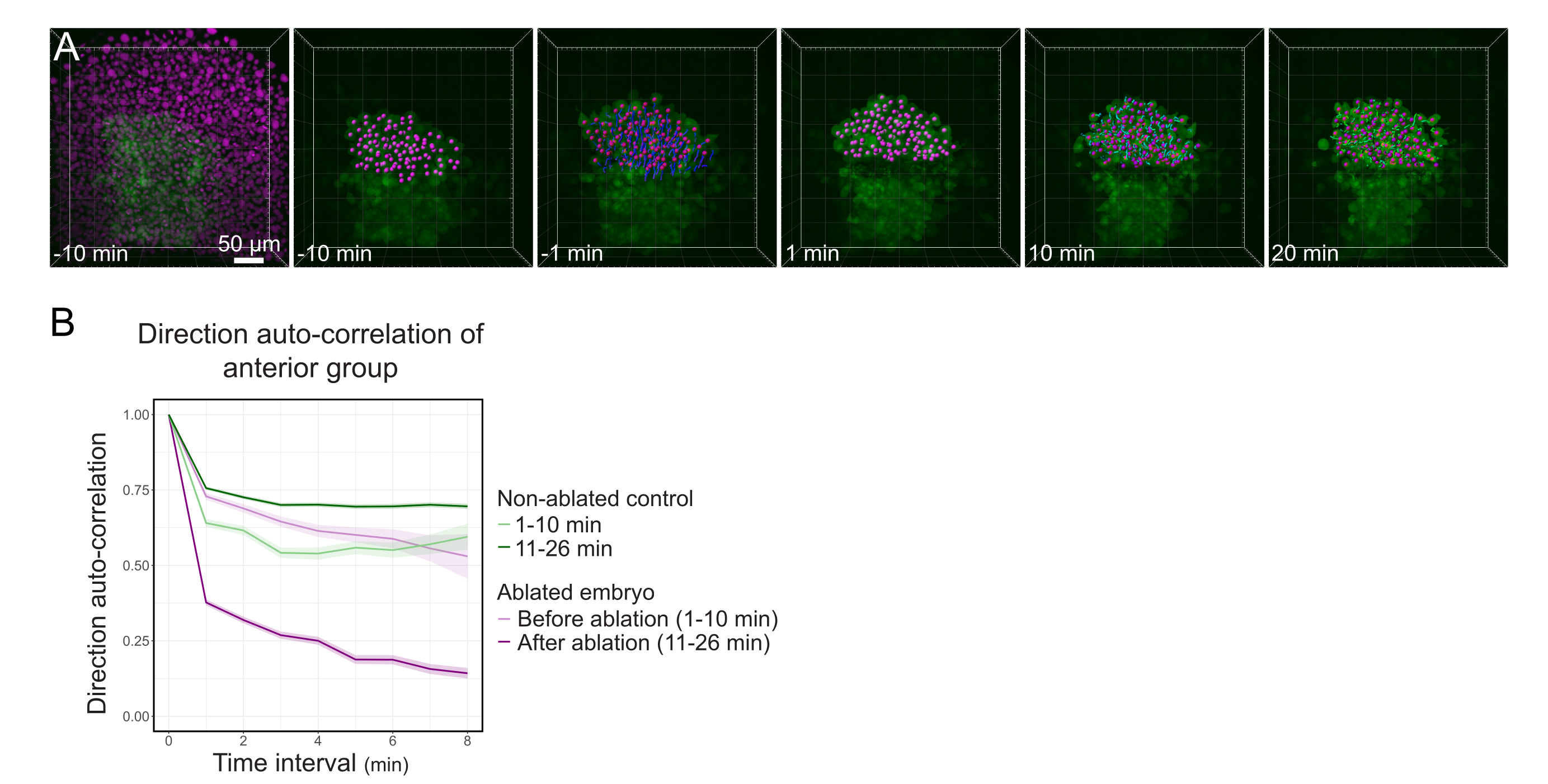{kind=link}
結果
その途中でポルスターを切断するために、ヒストン2B-mCherry mRNAを注入した Tg(gsc:GFP) 胚を、ステップ4で説明したように70%エピボリー段階に取り付けた。このポルスターはGFPの発現により同定され、その胚は、ポルスターの平面が光軸に対して垂直になるように取り付けた(図1B)。胚をこの位置から離すと、手順が複雑になります。光はアブレーション平面に到達す?...
ディスカッション
ここでは、非線形光学を使用して深く、空間的に明確に定義されたボリュームアブレーションを実行するプロトコルについて説明します。プロトコルの最も重要なステップは、過度の破片やキャビテーションを避けるために、アブレーションを可能にするのに十分なエネルギーを提供するが、あまりにも多くのエネルギーを提供する治療条件を見つけることです。標的部位での送達エネルギ?...
開示事項
著者らは競合する利益を宣言しない。
謝辞
私たちは、魚のケアのためのエミリー・メナント、特にピエール・マホウのポリテクニックバイオイメージング施設に、レギオン・イル・ド・フランス(interDIM)とアジェンス・ナショナル・ド・ラ・レシェルシュ(ANR-11-EQPX-0029モルフォスコープ2、ANR-10-INBS-04フランス)によって部分的にサポートされている機器のライブイメージングの支援に感謝します。この作業は、ANR助成金15-CE13-0016-1によってサポートされました。 18-CE13-0024、20-CE13-0016、およびマリー・スクウォトフスカ・キュリー交付金契約なし 840201、ミニステア・ド・ランセニュメント・スペリエール・エ・ド・ラ・レシェルシュとセンター・ナショナル・ド・ラ・レヴェンチャーシェ・シフィケの下での欧州連合のホライゾン2020研究とイノベーションプログラム。
資料
| Name | Company | Catalog Number | Comments |
| 25x water immersion objective | Olympus | XLPLN25XWMP2 | |
| Agarose | PanReac AppliChem | A8963,0500 | |
| Data analysis software : Matlab | Math Works | ||
| Electro-optic modulator (EOM) | ConOptics | 350-80LA | |
| Embryo Medium (EM) solution | Westerfield, M. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio), 5th Edition. University of Oregon Press, Eugene (Book). (2000). | ||
| Environmental chamber chamber | Okolab | H201-T-UNIT-BL | |
| EOM driver | ConOptics | 302RM | |
| Fluorescence source | Lumencor | SOLA | |
| Glass bottom dishes | MatTek | P35G-0-10-C | |
| Glass capillaries | Harvard Apparatus | 300085 | Outside diameter 1.0 mm, inside diameter 0.58 mm |
| Glass pipettes | Volac | D810 | Tip should be fire polished |
| Green/ablation laser | Spectra Physics | Mai Tai HP DeepSee | |
| Histone2B-mCherry mRNA | Synthesized from pCS2-H2B-mCherry plasmid (Dumortier& al. 2012) | ||
| Image analysis software: IMARIS | Bitplane | ||
| ImSpector software | Abberior Instruments Development Team | ||
| Injection mold | Adapative Science Tools | I-34 | |
| Microloader tips | Eppendorf | 5242956003 | |
| Micromanipulator | Narishige | MN-151 | |
| Micropipette puller | Sutter | P-1000 | |
| mMESSAGE mMACHINE SP6 Transcription Kit | Invitrogen | AM1340 | |
| Penicillin-Streptomycin | Thermofisher | 15140-122 | 10 000 units penicillin and 10 mgstreptomycin per ml |
| Photomultiplier tube (PMT) | Hammamatsu | H7422-40 | |
| PicoPump (Air injector) | World Precision Instrument | PV820 | |
| Red laser | Spectra Physics | OPO/Insight DeepSee | |
| RNAse free water for injection | Sigma | W3500 | |
| Spreadsheet software: Excel | Microsoft | ||
| Stereomicroscope | Nikon | SMZ18 | |
| Tg(gsc:GFP) zebrafish line | Doitsidou, M. et al. Guidance of primordial germ cell migration by the chemokine SDF-1. Cell. 111 (5), 647–59, doi: doi.org/10.1016/S0092-8674(02)01135-2 (2002). | ||
| TriM Scope II microscope | La Vision Biotech |
参考文献
- Slack, J. M. W. Embryonic induction. Mechanisms of Development. 41 (2-3), 91-107 (1993).
- Fernandez-Sanchez, M. -. E., Brunet, T., Röper, J. -. C., Farge, E. Mechanotransduction's impact on animal development, evolution, and tumorigenesis. Annual Review of Cell and Developmental Biology. 31, 373-397 (2015).
- Shih, J., Fraser, S. E. Characterizing the zebrafish organizer: microsurgical analysis at the early-shield stage. Development. 122 (4), 1313-1322 (1996).
- Selleck, M. A. J. Culture and microsurgical manipulation of the early avian embryo. Methods in Cell Biology. 51 (51), 1-21 (1996).
- Bulina, M. E., et al. A genetically encoded photosensitizer. Nature Biotechnology. 24 (1), 95-99 (2006).
- Fang-Yen, C., Gabel, C. V., Samuel, A. D. T., Bargmann, C. I., Avery, L. Laser microsurgery in Caenorhabditis elegans. Methods in Cell Biology. 107, 177-206 (2012).
- Colombelli, J., Grill, S. W., Stelzer, E. H. K. Ultraviolet diffraction limited nanosurgery of live biological tissues. Review of Scientific Instruments. 75 (2), 472-478 (2004).
- Smutny, M., Behrndt, M., Campinho, P., Ruprecht, V., Heisenberg, C. -. P. UV laser ablation to measure cell and tissue-generated forces in the zebrafish embryo in vivo and ex vivo. Methods in Molecular Biology. 1189, 219-235 (2015).
- Behrndt, M., et al. Forces driving epithelial spreading in zebrafish gastrulation. Science. 338 (6104), 257-260 (2012).
- Volpe, B. A., Fotino, T. H., Steiner, A. B. Confocal microscope-based laser ablation and regeneration assay in zebrafish interneuromast cells. Journal of Visualized Experiments: JoVE. (159), (2020).
- Bonnet, I., et al. Mechanical state, material properties and continuous description of an epithelial tissue. Journal of the Royal Society, Interface. 9 (75), 2614-2623 (2012).
- Rauzi, M., Lenne, P. F., Lecuit, T. Planar polarized actomyosin contractile flows control epithelial junction remodelling. Nature. 468 (7327), 1110-1115 (2010).
- Niemz, M. H. . Laser-Tissue Interactions. Encyclopedia of Biomaterials and Biomedical Engineering, Second Edition - Four Volume Set. , (2019).
- Smith, A. M., Mancini, M. C., Nie, S. Bioimaging: second window for in vivo imaging. Nature Nanotechnology. 4 (11), 710-711 (2009).
- Rauzi, M., Lenne, P. -. F. Cortical forces in cell shape changes and tissue morphogenesis. Current Topics in Developmental Biology. 95, 93-144 (2011).
- Theveneau, E., David, N. B. Migrations cellulaires collectives. Medecine/Sciences. 30 (8-9), 751-757 (2014).
- Dumortier, J. G., Martin, S., Meyer, D., Rosa, F. M., David, N. B. Collective mesendoderm migration relies on an intrinsic directionality signal transmitted through cell contacts. Proceedings of the National Academy of Sciences of the United States of America. 109 (42), 16945-16950 (2012).
- Solnica-Krezel, L., Stemple, D. L., Driever, W. Transparent things: cell fates and cell movements during early embryogenesis of zebrafish. BioEssays. 17 (11), 931-939 (1995).
- Montero, J. -. A., Kilian, B., Chan, J., Bayliss, P. E., Heisenberg, C. -. P. Phosphoinositide 3-kinase is required for process outgrowth and cell polarization of gastrulating mesendodermal cells. Current Biology. 13 (15), 1279-1289 (2003).
- Ulrich, F., et al. Slb/Wnt11 controls hypoblast cell migration and morphogenesis at the onset of zebrafish gastrulation. Development. 130 (22), 5375-5384 (2003).
- Kai, M., Heisenberg, C. -. P., Tada, M. Sphingosine-1-phosphate receptors regulate individual cell behaviours underlying the directed migration of prechordal plate progenitor cells during zebrafish gastrulation. Development. 135 (18), 3043-3051 (2008).
- Smutny, M., et al. Friction forces position the neural anlage. Nature Cell Biology. 19 (4), 306-317 (2017).
- Johansson, M., Giger, F. A., Fielding, T., Houart, C. Dkk1 controls cell-cell interaction through regulation of non-nuclear β-Catenin pools. Developmental Cell. 51 (6), 775-786 (2019).
- Gorelik, R., Gautreau, A. Quantitative and unbiased analysis of directional persistence in cell migration. Nature Protocols. 9 (8), 1931-1943 (2014).
- Grill, S. W., Howard, J., Schäffer, E., Stelzer, E. H. K., Hyman, A. A. The distribution of active force generators controls mitotic spindle position. Science. 301 (5632), 518-521 (2003).
- Desprat, N., Supatto, W., Pouille, P. -. A. A., Beaurepaire, E., Farge, E. Tissue deformation modulates twist expression to determine anterior midgut differentiation in Drosophila embryos. Developmental Cell. 15 (3), 470-477 (2008).
- Farhadifar, R., Röper, J. -. C., Aigouy, B., Eaton, S., Jülicher, F. The influence of cell mechanics, cell-cell interactions, and proliferation on epithelial packing. Current Biology. 17 (24), 2095-2104 (2007).
- Willier, B. H., Oppenheimer, J. M. . Foundations of Experimental Embryology. , (1964).
- Ashby, W. J., Zijlstra, A. Established and novel methods of interrogating two-dimensional cell migration. Integrative Biology: Quantitative Biosciences from Nano to Macro. 4 (11), 1338-1350 (2012).
- Bosze, B., et al. Pcdh18a regulates endocytosis of E-cadherin during axial mesoderm development in zebrafish. Histochemistry and Cell Biology. 154 (5), 463-480 (2020).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved