
Method Article
光ピンセットを用いた閉じ込めにおける細胞下力学の直接力測定
要約
ここでは、光学トラップによる直接力測定を用いて、3次元閉じ込めで単離した胚ゼブラフィッシュ細胞の細胞内の機械的特性を調べるプロトコルを提示する。
要約
多細胞生物の開発中に、単一の受精細胞が分裂し、多様な機能を持つ複数の組織を生み出します。組織形態形成は、細胞内の機械的特性の変動をもたらす単一細胞レベルでの分子および構造変化と手をつないで行く。結果として、同じ細胞内であっても、異なるオルガネラとコンパートメントは機械的応力に対して異なる抵抗をします。メカノトランスダクション経路は、その機械的特性を積極的に調節することができます。このように組織ニッチの微小環境に適応する細胞の能力は、機械的ストレスを感知し、それに応答する能力の一部である。我々は最近、核の変形と位置決めが細胞が物理的な3D環境を測定し、細胞形状の変化を解読するプロプリオセプションの感覚を細胞に与えることを可能にする新しいメカノセンセーションパラダイムを提案した。この記事では、接着細胞や機械的に閉じ込められた細胞に例示した、生体細胞内の細胞核を形成する力と材料特性を測定する新しい方法について述べた。測定は細胞内の光学トラップで非侵襲的に行われ、力は光の勢いをキャリブレーションフリーで検出することで直接アクセスできます。これにより、細胞表面の変形とは無関係に核の力学を測定し、外発性および知覚的メカノ伝達経路の解剖を可能にする。重要なことに、トラッピング実験は、細胞骨格、カルシウムイオン、または核形態の蛍光イメージングを用いて細胞応答および細胞下ダイナミクスを調べる光学顕微鏡と組み合わせることができる。提示された方法は適用が簡単で、力の測定のための商業的解決と互換性があり、他の細胞下のコンパートメント、例えばミトコンドリア、ストレスファイバー、およびendosomesの力学を調査するために容易に拡張することができる。
概要
組織形態形成は、生化学的シグナルと物理的な力が時間的に調整される複雑なプロセスです。現像胚では、生化学的シグナル伝達因子の勾配が運命の指定を決定し、正しい組織パターニングを確実に行う1,2。同時に、内因性および外因性の力は、胚のアーキテクチャを構築する役割を果たす3,4。この文脈における細胞皮質力学の影響は、広範囲に研究されてきた5,6。形態形成中のメカノ化学プロセス間の緊密な相互接続は、単一細胞の特性に依存して、組織微小環境における機械的な力を感知し、応答する。細胞は、それによって、細胞挙動、細胞運命、および細胞力学を制御する特定のシグナル伝達経路に機械的情報を伝達する力感受性細胞および分子要素の存在を介して機械的信号を解読する。
発達過程の特徴は、細胞がグループとして組織され、多細胞構造を構築することです。したがって、単一の細胞はめったに再配置して単独で動かないが、彼らは上細胞移行7、(un)妨害遷移8,9または胚盤胞圧縮10のような集団行動を示すタイトな社会トープに関連している。細胞内および細胞間で生成された機械的な力は、集合的な細胞ダイナミクス7,11を指示するための重要な手掛かりとして機能します。しかし、組織シートや狭い組織ニッチの間を圧迫する前駆細胞など、細胞が単独で移動しても、3次元環境をナビゲートする際に広範な異方性機械的力が発生します。細胞上のこれらの機械的なストレスは、細胞の挙動に深い影響を及ぼす12,13。高密度3D組織環境15,16内の移動中に受動的または活動的な機械的要素として、主要なメカノ転写素子14,15として核上に収束するいくつかのメカニズムが検討されている。
我々は最近、弾性細胞内メカノゲージ12として核を用いて形状変形を測定するために細胞を装備するメカニズムを提案した。細胞内で最大の小器官である核は、細胞が機械的なストレッチ、閉じ込め、または浸透応力16,17,18,19の下で細胞の形状を偏光、移動、または変化させると大きな変形を起こします。核エンベロープは、核の細胞内位置と共に、細胞の大きさと細胞の変形の種類(細胞圧縮と細胞の腫脹など)に関する情報を細胞に提供することを発見した。核の伸張は、内核膜(INM)の展開に関連しており、INMでのカルシウム依存性cPLA2(細胞質ホスホリパーゼA2)リパーゼ活性を促進し、続いてアラキドン酸(AA)の放出と細胞皮質でのミオシンIIの急速な活性化を促進する。これは、皮質収縮性6の閾値を超える細胞収縮性およびアメーブイド細胞の移動を増加させる。細胞の変形に対するメカノイア応答は1分未満で起こり、閉じ込め解放時に可逆的であり、核が機械的ストレス条件下での適応細胞挙動を調節する細胞のプロプリオセプションの歪みゲージとして機能することを示唆している。このメカノイアセンブル経路は、ゼブラフィッシュ胚由来の前駆幹細胞において活性であることが示されており、多能性および系統性細胞12の両方において、異なる種および細胞株20において保存されている。
核特性は、細胞メカノセンサとして加え、核アーキテクチャと力学は、開発中および細胞運命仕様21に応答して本質的に調節され、細胞メカノ感受性22,23を調整する。その結果、形態学的な変化と移行前から回遊状態への移行、およびその逆8を可能にする核適合の変化が起こるかもしれません。
原子間力顕微鏡24,25、マイクロピペット吸引26,27、マイクロ流体技術28、マイクロニードル29など、細胞核力学を測定するためのいくつかの技術が応用されている。しかし、これらの技術の多くは、細胞全体が変形しなければならないという意味で侵襲的であり、核自体の機械的特性および力依存応答の測定を制限する。細胞表面とそのメカノセンシティブ細胞の同時変形を回避するため、分離核は様々な文脈31,32で研究された。しかし、核の単離は、機械的核特性の変化とその調節(reference24および独自の未発表の観測)に関連していることは否定できない。
光ピンセット(T)は、細胞メカノバイオロジーにおける多くの実験を可能にし、分子機械が化学薬品を機械エネルギーに変換する方法についての理解に役立つ多目的技術です33,34。光ピンセットは、しっかりと集結したレーザービームを使用して、周囲の媒体33よりも屈折率が高い誘電体粒子に光学力を加えます。このような力は、ピコニュートンの数百の順序であり、レーザートラップフォーカス内の粒子の効果的な閉じ込めをもたらし、3次元で閉じ込められた粒子の操作を可能にする。光の使用は、測定が生細胞の内部で非侵襲的に行うことができるという点で重要な利点を有する。光学操作は、さらにレーザー光のトラップフォーカスに限定される。したがって、この操作は周囲の細胞膜を刺激することなく行われ、イオンチャネルの力依存的活性化などの形質膜におけるアクチン皮質またはメカノイ感受性プロセスを摂動させない。
光トゥイザーアプローチの難しさは、等分割定理に基づく間接力較正に依存する古典的なアプローチを用いて、マイクロスフィアに適用される力を正確に決定すること、またはレーザーパワー依存的な脱出力を測定するために定義されたストークス・ドラッグ力の使用である35。これらの方法はインビトロ実験で簡単に実装できますが、通常は細胞環境に翻訳することはできません。運動量の保全の第一原理から導出された直接力の口径測定に依存する分野にはいくつかの戦略が導入された36,37。他の力分光法とは異なり、力の測定は任意の形の閉じ込められた粒子38,39と光運動量の局所的な交換から推測される。我々の実験セットアップでは、光力から生じる光運動量の変化は、インシチュトラップキャリブレーション40、41、42、43で必要なく直接測定されます。これにより、細胞内部や組織内などの粘性環境下での測定が可能となり、かつ、pNレベルまで容易に力を定量することができる。
本プロトコルでは、細胞内小器官や構造を機械的に操作するアッセイを記述し、光ツイーザーのセットアップによってそれらの機械的特性を定量的に評価する。このセットアップは、細胞行動または細胞内ダイナミクスの並列イメージングを可能にする回転ディスク蛍光顕微鏡に統合されています。このアッセイは、核などの特定の細胞区画の機械的特性の特性を特徴付け、同時に変形自体の結果として可能なメカノ応答と分子シグナル伝達経路の活性化を研究することを可能にする。さらに、細胞内に注入されたマイクロビーズの光学トラップは、核(n~1.35)対細胞質(n~1.38)の固有屈折コントラスト44 と比較して、ポリスチレンビーズの屈折率がかなり高いおかげで、インデント力の増加を可能にする。提示された戦略は、他の細胞内構造やオルガネラの研究、およびアクティブなマイクロ理ロジー、同じ/異なる細胞下構造を同時に探査するための複数の光学トラップの使用、および生きた胚における細胞メカノバイオロジーを標的とする測定に容易に適応することができる。
プロトコル
使用されるすべてのプロトコルは、制度的動物管理・使用倫理委員会(PRBB-IACUEC)によって承認され、国内および欧州の規制に従って実施されています。すべての実験は、3Rの原理に従って行った。ゼブラフィッシュ(ダヨ・レリオ)は、前述のように維持した。
1. 単離胚性ゼブラフィッシュ前駆細胞の作製
- マイクロピペットおよびアガロース製剤
注: 完全なゼブラフィッシュ胚マイクロインジェクションプロトコルについては、reference45 を参照してください。- マイクロピペットプーラーで、1.0mmのガラスキャピラリーを引っ張って2本の針45を得る。未使用の針をプレイドウクッションに取り付けられた150mmのペトリ皿に保管するか、または裏返しのラボテープリングに保管して、輸送中の薄い先端を損傷から保護します。
- E3(5 mM NaCl、0.17 mM KCl、0.33 mM CaCl2、0.33 mM MgSO4)の1%超純粋アガロースを標準キッチン/ラボマイクロ波で10 sで溶融します。アガロースが溶けるまで、短時間(数秒)の混合物を繰り返し加熱します。
- アガロースが完全に溶けたら、少し冷ましてから10cmのペトリ皿に注ぎます。気泡の出現を避けるためにアガロースの上部に三角形のマイクロインジェクション金型( 材料表を参照)をゆっくりと追加します。金型を押し込まないで、アガロース表面に残ります。
- アガロースが完全に固まったら、アガロースの破断を避けるために穏やかな力を加えることによって、三角形のカビを非常にゆっくりと取り除く。プレートは、2〜4週間4°Cで逆さまに保存することができます。
- マイクロインジェクションの30分前に、冷蔵庫からプレートを取り出し、28°Cに予温したE3を加えて室温で安定させます。
- 注射ミックス製剤
- 注入混合物を調製するために、RNase遊離水で1:5の比率で1μmマイクロビーズ(ポリスチレン、非蛍光)を希釈する。
- 蛍光マーカーの一過発現または組換え遺伝子構築物の発現および/またはモルフォリーノの目的の濃度での共噴射のためのmRNAを調製する。
注:マイクロビーズの共注射のための典型的な注射混合物と1胚当たり100 pgのmRNAを標識する、 例えば、H2A-mCherryを有する核は、1μLのビーズ+1μLのmRNA(ストック濃度は1μg/μL)+2.5μLのRNAフリーウォーター+0.5μLのフェノールレッド(ストック溶液0.5%、フェノールレッドは必須ではない;それは注入された滴のより良い視覚化のために使用されるが、非標識注入の視覚化のために使用される。落下は経験豊富な実験者のためにも表示されます)。RNA注射はまた、注入された胚を選択するのに有用である。蛍光マイクロビーズは、非蛍光の代わりにそれらを可視化するために注入することができる。
- マイクロインジェクション針のローディングとキャリブレーション
- タイムゲートオプションを使用してマイクロインジェクタの電源を入れます。この設定は、射出量を正しく調整するために非常に重要です。約 500 ミリ秒で、ゲージ時間を設定します。
- マイクロローダーピペットを使用して注射混合物の3 μLを針にロードします。
- マイクロマニピュレーターに針を挿入し、しっかりと密封します。マイクロマニピュレータが良好な位置にあり、射出プレート上のx-y方向に移動するのに十分な自由度があるかどうかを確認します。
- top45に鉱物油を一滴落とし、注入ミックスの滴を鉱物油に直接排出するマイクロメートルスライド(5 mm/100分割)を使用して、ドロップサイズを測定します。
- 急な角度で鋭い鉗子で針を切り抜き、鋭い尖った先端を生成します。0.5 nLの注入された材料に対応する0.1 mmに滴りのサイズを調節する。
注:針を切断してこのボリュームを超えた場合は、新しい針でキャリブレーション手順をやり直すことをお勧めします。マイクロインジェクターの八方修正時間は、ドロップボリュームに合わせて若干調整できます。しかし、短い座数は大きな針径に相当し、胚に損傷を与える可能性がある。
- 1細胞段階におけるゼブラフィッシュ胚の微小注入
- 最初の細胞分裂が起こる前に、ビーズ混合物を直接1細胞(zygote)段階胚に直接注入するために、ゼブラフィッシュ胚を収集する。
注:これは、マイクロスフェアの適切な分布と実験が行われる後の発達段階(ブラストラガストラステージ)で、細胞あたり少なくとも1つの微小球を有する単離されたブラストミアの十分な高い収率を保証します。インデントの実験は、セル内に 2 つの球がある場合でも実行できますが、ビーズのないセルは除外する必要があります (球のないインデントは可能な場合でも)。AB野生型株はこのプロトコルで使用されたが、他の株、例えばTLを使用することができる。 - プラスチック製のパスツールピペットを使用して、 図1Aに示すように、1細胞段階の胚(zygote)を、前温めの三角形の1%アガロース型に入れます。
- 胚が浮かび上がらないように、同じピペットで余分な培地を取り除きます。ブラシを 介して 三角形の型にゆっくりと胚を押し込みます。正しい向きを容易にするために、胚間にいくらかのスペースを置いてください(図1B)。
- 図1Bに示すように、胚が横方向に向き合うように胚をブラシにそっと合わせ、ジゴテの1つの細胞がはっきりと見えるようにします。マイクロインジェクションの理想的な向きは、図1Cに示すように、胚の1つの細胞が針方向(胚の動物極を介した注射)または黄身細胞に対する反対の方向(胚の植物極を介した注射)に向いているときに到達する。
- 片手で皿を持ち、もう一方の手を使ってマイクロマニピュレーターを使用して針先を配置します。針先を胚に向かって下げます。
- 絨毛を突き刺し、体型顕微鏡を通して手順を監視しながら針で1細胞胚に入ります。 図1Cに示すように、針の正しい配置と、注入されたドロップの正しい位置を確認します。
- すべての胚について繰り返します:針を上に移動し、次の胚が中央に配置されるまで胚で皿をスライドさせ、針を下げ、注射します。
- 胚のセット全体を注入したら、いくつかのE3を洗い流すことによってアガロースカビ/ペトリ皿から胚を取り除き、プラスチック製のパスツールピペットを使用して新しいペトリ皿に入れます。マイクロインジェクションの手順で胚の乾燥を避けるために、注入プレートに十分な媒体を置くことをお勧めします。
- 所望の胚数が注入されるまで手順を繰り返す。胚は、ビーズの最大かつ均質な広がることを確実にするために、1つの細胞段階にある必要があります。
注:この手順は初期の胚胚に最適化されており、異なる発達段階を調査する場合は最適化する必要があります。 - 注入した胚をインキュベーター内に28-31°Cで約4時間、または所望の段階(図1D)まで入れてから、一次細胞培養のプロトコルを進める。
注:必要に応じて、胚が生存を確実にし、毒性アーティファクトを排除するために、ブラストラ段階(または所望の測定時点)を超えて発達させます。幼虫の段階では、0.75%アガロースにトリケーヌを用いて麻酔幼虫を取り付け、様々な組織における微小球の分布を画像化する。ストック液を作るためには、97.9mLの蒸留水にトリケーヌ粉末400mg、約2.1mLの1M TRISベース(pH9)を混合し、pH7に調整します。この溶液は4°Cで貯えることができる。 トリカインを麻酔薬として使用するには、卵の培地(または所望の培地)の100 mLに4.2 mLのストック溶液を希釈する。この場合、E3 が使用されました。詳細については、参照46 を参照してください。
- 最初の細胞分裂が起こる前に、ビーズ混合物を直接1細胞(zygote)段階胚に直接注入するために、ゼブラフィッシュ胚を収集する。
2. 単細胞の調製および染色
- プラスチック製のパスツールピペットを使用してガラス皿に球ステージ胚(4 hpf、受精後時間)を入れます。注入されたビーズのシグナルに陽性であり、mRNA注入の場合に蛍光タンパク質を発現する胚を選択する。一部の胚は、高いビーズクラスタリングを示す可能性があり、除外することができます。
- 鉗子を使用して胚を手動でデコールする。ガラスパスツールピペットを使用して、約10〜15個の胚を1.5mL反応容器に移します。
注:胚がデコールリオネートされるとき、それらはプラスチックに付着し、ガラス製品の使用が必要です。ガラス板の代わりに、1%アガロースの薄層を有するプラスチックペトリ皿を使用することができる。手動デコールリオネーションは、細胞表面タンパク質へのタンパク質分解損傷および機械的細胞および組織特性の潜在的な変化を防ぐために酵素プロナーゼ処理よりも好まれ、回復倍数の延長を防ぐべきである。
- 鉗子を使用して胚を手動でデコールする。ガラスパスツールピペットを使用して、約10〜15個の胚を1.5mL反応容器に移します。
- E3培地を取り出し、500μLのあらかじめ温められたCO2非依存性組織培養培地(DMEM-F12;L-グルタミンと15mM HEPES、重炭酸ナトリウムおよびフェノールレッドを10単位ペニシリンと10mg/Lストレプトマイシンで補うことなく)を加える。
注:顕微鏡インキュベーターを使用しない限り、CO2依存性の培地は使用しないでください。炭酸塩緩衝条件でのRPMIの使用は、培地のpHの変化を引き起こし、細胞生存に影響を与える可能性がある。もう一つの重要な側面は、血清を含む培養培地を避けることです。血清は、リソホスファチジン酸(LPA)、Rho/ROCK経路の強力な活性化剤を含み得る、前駆幹細胞6における細胞収縮性および運動性を制御することができる。核形態や力学12を妨げる可能性のある浸透性の課題を避けるために、媒体の浸透性を300mOsmに維持する必要があります。 - チューブを軽く振って細胞の解化を手動で行います。目に大きな塊が見えないとチューブの内容物が濁っていることを確認してください。細胞の損傷と損失を最小限に抑えるために気泡の形成を避けてください。
- 200 x g で 3 分間遠心分離機。ペレットははっきりと見える必要があります。
- 上清を取り除き、以下の手順の1つを実行します。
- 染色が不要な場合は、500 μLのDMEMを追加します。200 μL ピペットで、液体ジェットをペレットにターゲットにして、徐に再懸濁します。細胞に過度のせん断力を加えない。発泡は、細胞の損傷を示します。
- HoechstなどのDNA染料で核を標識するには、DMEMの1,000 μLに0.5 μLのDNA-Hoechst(ストック2 mg/mL)を混ぜ、最終濃度の1 μg/mL を得ます。この染色液を500μL加えて細胞に加え、穏やかに再懸濁します。暗闇の中で7分間インキュベートします。
- 蛍光化学カルシウムインジケーター Calbryte-520 で細胞を染色するには、カルブライト-520を DMEM で 5 μM 濃度に加えます。暗闇の中で20分間インキュベートします。
注: 手順 2.5.2 および 2.5.3 で示されているプロトコルは、これらの特定の製品に対して最適化されています。その他の染色は、製造業者が示すプロトコルを使用して行うことができる。
- ステップ 2.4 と同じ設定を使用して再び遠心分離機;上清を取り除き、細胞を50μLのDMEMで静かに再懸濁し、懸濁液中のサンプルに対して、または閉じ込め中の細胞の場合は20μLのDMEMを使用します。
3. ポリジメチルシロキサン(PDMS)間隔を用いた光学トラップチャンバの調製
注:光の運動量検出に基づく光力測定では、光トラップ40から出てくるすべての光を捕捉する必要があります。不変量キャリブレーション係数 α (pN/V)のロバスト性については、光力センサの背面焦点面(BFP)での光の分布は、光子の運動量に正確に対応する必要があります。これは、収集レンズの表面から約2mmまでの距離を決定します。
- #1.5ガラス底皿のPDMSスピンコーティング。
注:約40の料理には、以下のレシピが用意されています。結果として得られるマイクロチャンバは、実験が浮遊細胞または閉じ込められた細胞で行われるかどうかによって異なる高さを有するであろう(図1D)。- 50 mL円錐管に9mLのベースポリマーPDMSと1mLのPDMS硬化剤を混合します。2つの製品を積極的に混合して、硬化剤の適切な分布を確保します。
- 真空ポンプを使用して気泡を避けるために混合物を脱気。真空ボトルに円錐チューブを導入し、チャンバーを避難させる。混合物中に泡が存在しないまで待ちます。
注:真空をゆっくりと開けて、PDMSがハヤブサのチューブからこぼれ落ちるのを防ぎます。 - スピンコーターチャックにガラス底皿を置きます(図2A)。傷つけたり、指紋を付けたり、皿を汚したりしないように優しくしてください。スピンコーターボックスをアルミニウムホイルでPDMSリークから保護します。
- 懸濁液中の細胞の実験用のOTチャンバーでは、底皿の中央に約250 μLのPDMS混合物を加え、750 rpmで1分間回転させます。PDMS層の高さは約50μmになります。
- 狭い細胞の実験用OTチャンバの場合は、PDMS(約50μL)を小さな滴で加え、4,000 rpmで5分間回転させます。PDMS層の高さは約10μmとなります。異なる PDMS の厚さを取得する方法の詳細なプロトコルについては、reference48 を参照してください。
- PDMSコーティングされたガラス底の食器を70°Cで1時間硬化させます。
- 1 x 1 cmの正方形をPDMS層にメスで切り、ピンセットで剥がします(図2C)。閉じ込められた細胞の場合、PDMSの破片をイソプロパノールで洗浄する。
- サスペンション中の軽く結合した細胞を用いた実験のためのチャンバーコーティング
- 0.5 mg/mLでコンカナバリンA(ConA)100 μLを加えて、正方形の空洞の全面を覆い、30分間インキュベートします。
注:ConAは細胞表面糖に結合し、カバーガラス表面に個々の細胞を結合するレクチンです。 - ConAの落下を取り除き、ConA処理面を傷つけずにDMEM媒体で表面を慎重にすすきます。
- ウェルに、以前に用意したサンプル(ステップ2.6)の30 μLを加え、穏やかに再中断して細胞クラスターを取り除きます。
- PDMSリムの上に22 x 22 mm #1.5カバーガラスをそっと置いて空洞を閉じます(突然落ちないようにし、可能であれば鉗子を使用してください図2B,C)。
注:カバースリップの厚さは、上部のガラスカバーに対して機能します(収集レンズの動作距離は2mm)。
- 0.5 mg/mLでコンカナバリンA(ConA)100 μLを加えて、正方形の空洞の全面を覆い、30分間インキュベートします。
- 閉じ込められた細胞の実験のためのチャンバーの準備
- 細胞を含む溶液の10μLの低下(ステップ2.6)を正方形の空洞に入れます(図2B)。
- 非常に穏やかに、ドロップが全体の領域に広がり、気泡が観察されないような22 x 22ミリメートルカバーガラスでサンプルを挟みます。ここでも、 図2Cに示すように、カバーガラスが突然落下するのを防ぐために、鉗子を使用するのが便利です。
OTチャンバー間隔のための4.代替オプション
注: マイクロファブリケーション ワークショップやスピン コーターがない場合は、これらの手順に従うことができます。
- 懸濁液中の細胞を用いた実験のためのチャンバー調製
注:スピンコーターが使用できない場合、スペーサーは通常の両面スコッチテープ(高さ約100μm)を使用して作ることができます。- 中央に約10cm x 10cmの正方形の穴をあけた両面スコッチテープを切ります(PDMSと同じ寸法、 図2B)。
- テープの保護層の1つを剥がして取り外し、テープの覆い隠された側面を#1.5 Hガラス底皿の中央に置きます。気泡を避けながら、ガラスに貼り付けたすべての表面を取得し、それを剥がすことによって、テープの残りの保護層を削除するために穏やかに押します。
- 手順 3.2 の指示に従います。
- 閉じ込められた細胞の実験のためのチャンバーの準備
注意:細胞を正確に閉じ込めるために、既知の直径を持つ単分散微粒子を2つのカバーグラスの間のスペーサーとして使用することができます。- 104個のビーズ/μLの濃度で浮遊細胞に10μmのポリスチレンビーズを加えます。
- 22 x 60 mm のカバーガラスに、細胞とビーズを含む溶液を 10 μL 落とします。
- 非常に穏やかに、ドロップが全体の領域に広がり、気泡が観察されないような別の22 x 60ミリメートルカバーガラスでサンプルを挟みます。上部カバーガラスを優しく配置するには(突然落ちないように)、鉗子を使用すると便利です。
- サンプルは乾燥できるため、迅速に調製を行うことを推奨します。
5. 細胞内測定用の光トラップの設定
注:以下のステップは、アクト光学偏向(AOD)に基づく光学マイクロマニピュレーションモジュールと光運動量変化の直接検出に基づく光学力センサーを備えた商用光ピンセットプラットフォームに最適化されています(図2、参照12,40,49)。セットアップの詳細と光学部品は図2Fにあります。光用トゥイザー操作中の力による変形を観察するために、ニプコウ紡糸盤共焦点顕微鏡を反転顕微鏡の左ポートに結合して、二重色蛍光イメージングを行います。一般性の欠如なし、このプロトコルは、光の運動量検出に基づいて直接力の測定を備えた任意の動的なOTsシステムと適用することができる。インビボアプリケーション50用の自家製光学勾配トラップを構築するための詳細なステップバイステップの手順が利用可能です。AOD変調に基づくものは、複数のトラップと高速測定で最終的な実験のために際立っています51,52。光運動量ベースの器具を構築するためのいくつかのプロトコルが文献36、39、40、53に存在し、他の任意のイメージングモダリティ(差動干渉コントラスト、広視野蛍光など)を採用することができる。
- 光ピンセットのスタートアップ
- 出力電力の安定性を最適化するために、実験の30分前に、かなり高い電力(例えば3W)でレーザーをオンにします。
- 光学マイクロマニピュレーションと力測定ユニットの電子モジュールをオンにします。
注:すべてのレーザー安全対策を適用し、機関理事会によって承認された機器のみを使用してください。レーザーがオンのときには、光学顕微鏡の接眼レンズを使用しないでください。承認されたIR保護ゴーグル(950-1080 nm範囲のOD7)を常に使用し、発蛍光ポート2のシャッターでIRレーザー光を遮断し、ステップ5.3の後に光学力センサのアライメントを終了するまで光学トラップソフトウェアを実行しない。一般に、反射性の高いサンプルは、反射が強く、レーザーに損傷を与える可能性があります。 - 光マイクロマニピュレーションモジュールの入口で、回転HWP(図2F)でトラップパワーを制御します。
注: このプロトコルで使用されている商用光マイクロマニピュレーションモジュールには、この機能が既に組み込まれています。自家製の光学トラップシステムでは、より高く、より安定したレーザーパワーを使用できるように、電力制御のためにこのツールを統合します。
- 空のマイクロチャンバーをキャリブレーションに使用する
- 1 x 1 cmの正方形を両面スコッチテープに切り、厚さ1mmの顕微鏡スライドに取り付けます。
- 正方形に水を追加し、#1.5カバーガラス(22 x 22ミリメートル)で上からそれを閉じます。水のわずかに高いボリュームを追加, 例えば, 30-40 μLは、覆われたチャンバー内の泡を避けるためにお勧めします.水がこぼれ落ちる場合は、校正室を静かに拭いてください。
- 光学力センサのアライメント
- 60x/1.2の水浸漬目的に水滴を入れます。目的に向けて#1.5カバーガラスをステージ上に置きます。細胞サンプルが最終的になる下面に焦点を当てます。
- サンプルを覆う上部ガラススライドの上に浸漬油の液滴を加えます(図2D)。オイルの液滴に接触するまで、力センサーユニットの収集レンズを慎重に下げてください。
注:液滴は、トラップから出てくるレーザー光を集めるレンズ全体を覆うほどの大きさでなければなりません。通常、200 μL は表面全体を覆い、安定した浸漬接点を提供するのに十分です。サンプルに漏れる可能性があるため、控えめにして過剰充填を避けてください。 - 光学力センサーの位置合わせに関する製造元のプロトコルに従って、AT の位置を決める補助カメラのサンプル プレーンイメージを確認します (AUX、図 2F)。非常に穏やかに、フィールド停止(FS、図2F-G)がサンプル平面に結合して現れるまで光学力センサーを下げる。これにより、光運動量変化のサンプル不変検出による適切な直接力測定が確実に行われます。
注: FS を十分に閉じて、画像が視野(FOV)よりも小さくなるようにし、表示されます。余分な注意とサンプルに対して光学力センサーの収集レンズを押し込まないでください。光力センサの垂直位置は、BFPでの光の分布の解析から、定義された数値開口(NA)を有する光錐体の分析から決定することができる。 - オイルの液滴に気泡がないことを確認します。これらは、力の測定に直接影響を与えることができます。気泡を確認するには、ベルトランレンズを所定の位置に置き(BL、 図2G)、接眼レンズを通るイメージングパスを観察します。汚れや気泡が目に見える場合や、より多くの油が必要な場合(図S1A)、レンズとチャンバーをダストフリーレンズ組織で洗浄し、手順5.3.2と5.3.3で手順を繰り返します。図 S1B に遮るもののない光路が示されています。
- 光学力センサーのホルダーに置かれた横ねじを使用して、FSをFOVに中央に配置します。精度を高めるには、FS を開いて、補助カメラに表示される FOV をほぼ満たします(AUX、 図 2F)。
6. 光学式トゥイザー最適化
注: 直接力の測定は、閉じ込められたパーティクルに加えられる力から生じる光の運動量の変化のみに依存するため、間接法とは対照的に、トラップの剛性は各実験の前に較正する必要はありません。機器固有の偏向/力係数(α;pN/V,reference41)は、製造業者によって較正されるため、実験不変である。しかし、レーザースポットは70 μm x 70 μmの領域で操作されるため、最適なトラッピングと電力安定性を確保するためには、ステップ6.2~6.5が重要です。以下の手順は、製造ソフトウェアに用意されており、作業領域上での作業領域での作業を半自動で最適化します。
- カメラAUX用の、オッツソフトウェアと取得ソフトウェアを起動します。
- 光ピンセット駆動ソフトウェアのシステムキャリブレーションサブメニューの 「ステップ1:エレクトロニクスオフセット 」ステップをクリックして、初期電圧ベースラインを引きます。
- OT作業領域全体でトラップ電力を平坦化するには、HWP をそれに応じて回転させることで、トラップパワーを最大の半分に設定します。レーザー出力を変更してトラップ電力を変更するのではなく、HWPを回転させます(図2F)。 [ステップ 2: 電源 ] をクリックして、トラップ電力のフラット化の自動化ルーチンを開始します。
注: これは、OT の作業領域(図 S1D)全体にわたるトラップパワーの変動を補正する重要なステップです。正常なルーチンは、トラップの電力変動を、作業領域の2%に下げ、2分後に収束します。 - トラップ位置のキャリブレーションを実行するには、カメラにレーザーからの光が見えるように IR フィルタを取り外します。画像面をマイクロチャンバーの下面に焦点を合わせ、IRスポットを見つけます。カメラAUX取得ソフトウェアで画像プレーン(目標位置)とヒストグラムコントラストを調整することで、可能な限り最小のIRスポットを取得します。必要に応じて、HWPを回転させて光トラップの電力を減らします(図2F)。 [ステップ 3: 自動 ルーチンまたはトラップ位置調整を開始する位置] をクリックします。
注: このルーチンは、カメラ AUX の OT の位置座標と AOD ステアリング角度の正確な対応を可能にします。正常に実行されたルーチンは、数秒で角度から位置へのマッピングを生成します。 - 初期運動量補償
注: サンプル全体で光トラップが動くと、BFP での光運動量分布の変動が生じます(図 S1E、F)。これは、トラップパワーがステップ6.3のように平坦化されているにもかかわらず、作業領域上のレーザー位置に関連する力に依存しない信号の変化につながります。その結果、各実験の前に修正する必要がある位置(光学的に閉じ込められたビーズに作用する実際の力とは無関係)による力ベースラインの変動が起きます。- HWPを回転して実験に使用するトラップパワーを設定します(図2F)。
- [ツール]サブメニューの [グローバル オフセット ]オプションを クリック します。これにより、最初の運動量ベースラインを修正する光ピンセットソフトウェアの オフセットキャンセル アシスタントが開きます。
- オフセット|をクリックします 。補正 して、ポジションバリアント初期運動量を補正します。
注: 進行中の週の間に光学路に影響を与える変更がない場合、トラップパワーの平坦化(ステップ6.3)と位置(ステップ6.4)マップは不変のままです。したがって、レーザートラップ経路に影響を与える可能性のある光学要素(二色性ミラー、フィルタなど)の組み合わせを常に使用するか、新しいトラップパワーフラット化ルーチンを実行することをお勧めします。初期運動量補正(ステップ6.5)に関しては、ATプラットフォームのメーカーは、新しいトラッピングパワーと実験セッションごとに変更する必要があるオンザフライキャリブレーションを提供します。ステップ 6.3 および 6.4 は、ステップ 5.2 で説明した空のキャリブレーション・スライドで実行する必要があります。細胞または他のオブジェクトを含むサンプルでは、ステップ6.5は、作業領域の光散乱を変更する可能性のあるオブジェクトを含まないで行われるべきです。
- 必要に応じて、マイクロスフィアをトラップし、フォース信号を記録しながら、既知の速度でトラップを移動します。たとえば、三角形振動を実行するようにトラップを設定します:記録された力信号は正方形の信号になります。
注: ビードに作用するドラッグフォースに従って、力の値は速度に比例して増加するはずです。このテストは、力の測定が正しく行われているという肯定的な制御として役立つ38。あるいは、光学力センサを使用して、光学トラップ剛性、κ[pN/μm]、および位置キャリブレーション係数β[μm/V]をパワースペクトル解析35から得ることができます。正しいアライメントでは、製造元によって提供される不変量キャリブレーション係数はα = κ·β [pN/V]です。- 製造元ソフトウェアの[メジャー]サブメニューの[プロット1]をクリックして、リアルタイムの力の読み取りを開始します。これにより、現在の光トラップ力とパワーの読み取りが行われます。
- [ツール]サブメニューから[振動パラメータ]ダイアログを開きます。シェイプとタイプのセレクターリングに、それぞれ三角形空間波形を設定します。例として、振幅を10 μm、周波数を3Hzに設定します。これにより、直径1 μm38のマイクロビーズに約1pNの粘性力が生じてしまう。
- カメラの AUX ウィンドウで、マイクロビーズを右クリックし、[ 振動の開始] を選択します。力の読み取りは、±1 pNで高原を有する正方形の力信号となる。
- マイクロビードを右クリックし、[振動を 停止]を選択します。
7. 回転ディスク共焦点顕微鏡
- 回転ディスクの共焦点顕微鏡と付属装置、統合レーザーエンジン、および取得カメラをオンにします。
- イメージングソフトウェアを起動します。
- 細胞原形質膜の核およびGFPのHoechst染色のためのイメージングチャネルを設定する。
- 405 nm および 488 nm の励起レーザーラインをアクティブにします。
- マルチバンド二色性を追加して、サンプルに励起を反映し、放出された光をカメラに渡すことができます。
- 500 nmのロングパスエッジ二色性ミラーで蛍光発光を分割します。
- 2 台の取得カメラの前に、DAPI/BFP (~445 nm) と GFP (約 521 nm) のエミッション フィルタをそれぞれ使用します。図2F,Gを参照してください。
- チャンネルごとに露出時間を 100 ミリ秒に設定します。
- レーザー発光を設定して、サンプル平面で5mWのパワーを得ます。電力を測定するには、商用電源メーターを使用します。
- イメージング プロトコルを設定します。Hoechst チャンネルから GFP チャネルへのスペクトル ブリードスルーを回避するには、2 つの色素を順次にイメージ化する必要があります。
メモ:光トラップのAODとカメラの取得の間にハードウェア同期が存在する場合は、トリガ極性が正しく設定されていることを確認してください。疑わしい場合は、施設管理者または顕微鏡のメーカーに相談してください。
8. 核のインデント実験を行う
メモ:力センサーモジュールを持ち上げてサンプルを交換する場合は、ソフトウェアを使用して光トラップをオフにし、起蛍光ポート2のシャッターを閉じます。そうでなければ、光学素子と実験者に深刻な損傷が発生する可能性があります。レンズがステージ/培養皿にぶつからないように、細胞を探すときは、レンズホルダーと底皿の端の間の横方向の距離に注意してください(図2)。
- サンプルを顕微鏡に入れ、このプロトコルのステップ 5.3 に従います。
- 回転HWP(図2F)を用いて、調査した核または細胞内構造の剛性が不明な場合は、トラップパワーを開始値として200mWに設定する。ステップ6.5を通して最初の運動量ベースラインを補うために(顕微鏡段階を使用して)、細胞のない場所に、OTsの作業領域を翻訳します。
注: 細胞内構造の剛性に応じて、トラップの電力値を低い値または高い値に調整して、同様のインデント深度を得る必要があります。 - 顕微鏡ステージソフトウェアコントローラを使用して、透過した明視野顕微鏡を介して1つまたは2つのビーズを持つセルを探します(図3A)。
- トラップ軌道を定義します。
- [ツール]サブメニューで[軌道]ダイアログを開き、[軌道タイプ]セレクタリングで[変位]を選択します。
- 数値シートに、後続の各軌道ステップの変位と時間を書き込みます。2つの例を次に示します。
- ストレス緩和実験の場合、 図3Bに示すように、プログラム台形荷重。 表S1では、2つの台形のインデントが5μmの移動距離で適用されました。5 μm/sの速度;引き込み前の待ち時間:10 s。
- 一定速度で反復的なインデント実験を行い、核に時間をかけずに三角ルーチンを取得するには、軌道振幅(例えば、5 μm)とステップの時間(例えば、2.5 μm/秒の速度の場合)を設定します。 表S2では、これは同じ速度で8回適用される。
注: これらの値は、各細胞の種類と実験に対して決定する必要がありますが、台形ルーチンの次のパラメータは、ここで示す実験で最も重要なダイナミクスをキャプチャします。待ち時間は、核がインデント後にその完全なストレス緩和を示すために十分でなければなりません
- 微小球のトラップ
- 顕微鏡ステージソフトウェアコントローラでビーズの少し上にイメージプレーンを設定します。
- 「オッツ」ソフトウェアを使用してトラップをアクティブにし、カメラのAUXイメージングウィンドウでビードをクリックします(ステップ6.4に従ってキャリブレーション)。オプティカルトラップによるビードの閉じ込めに成功すると、ビーズの動きが強く低下します。
- ビーズを細胞質に向けてクリックアンドドラッグし、核エンベロープから約2μmの距離に置きます(図3A)。ビードのインデントが核膜に対して垂直になるように、軌跡が設定されていることを確認します。
- 必要に応じて、トラップに対するビードの位置測定に必要な場合は、トラップをスキャンしてトラップ剛性を決定します(k [pN/μm]54、それによりΔxbead = -F/kを参照)。このプロトコルで使用される光マイクロマニピュレーションモジュールには、この目的のための組み込みルーチンがあります。
- [ツール]サブメニューで[パーティクル スキャン]ダイアログを開きます。
- スキャンするトラップを選択し、スキャン方法として高周波数を選択します。ビードスキャン測定のインデント軌道の方向(xまたはy)を選択します。
- トラッピング剛性の測定と共にウィンドウが表示されます。グラフで、2 つのカーソルをドラッグして 、F = -kx に対応する線形トラッピング領域を選択します。選択したデータ部分に線形フィットが自動的に更新されます。
注:セル膜から離れたビーズの初期位置(〜5μm)を設定し、中細胞界面での光運動量の偏向が力の測定の適切性に影響を与えます。核が細胞膜に近すぎる場合は、反対側の部位から核をインデントしてみてください。可能でない場合は、セルを破棄します。
- イメージングソフトウェアの取得ボタンをクリックして、画像取得を開始します。
- [データ|をクリックして、トラップ位置を開始し、測定データを強制的に保存リアルタイムの強制読み取りウィンドウ(ステップ6.6.1のように開いた)に保存します。
メモ:光トラップには、カメラのタイミング出力に接続できるトリガ入力が装備されています。したがって、画像データと力データはハードウェア同期化され、電子は取得中の画像のフレーム数とトラップサイクルをマッピングすることができる。 - 以前にロードした軌道を開始するには、ビードを右クリックし、[ 軌道を開始]を選択します。
- 軌道が終了し、システムが安定するまで待ちます。
- トラップ力測定データ保存を停止します。データ保存ダイアログがポップアップ表示されます。
注: データストレージを最適化するには、このダイアログボックスでデシメーションパラメータ(10、100、または1000)を選択して、データをデシメーションすることができます。 - 画像の取得を停止し、ユーザーの選択の後処理ソフトウェアで結果をプロットします。
- ルーチン中に微小球が失われ、核をインデントできない場合(図S2)、測定を破棄してパワーを上げます。手順 6.5 を繰り返す必要があることに注意してください。私たちの手では、ルーチンの少なくとも95%がトラップからビーズを失うことなく正常に完了しています。
結果
トラップビーズのマイクロインジェクション:
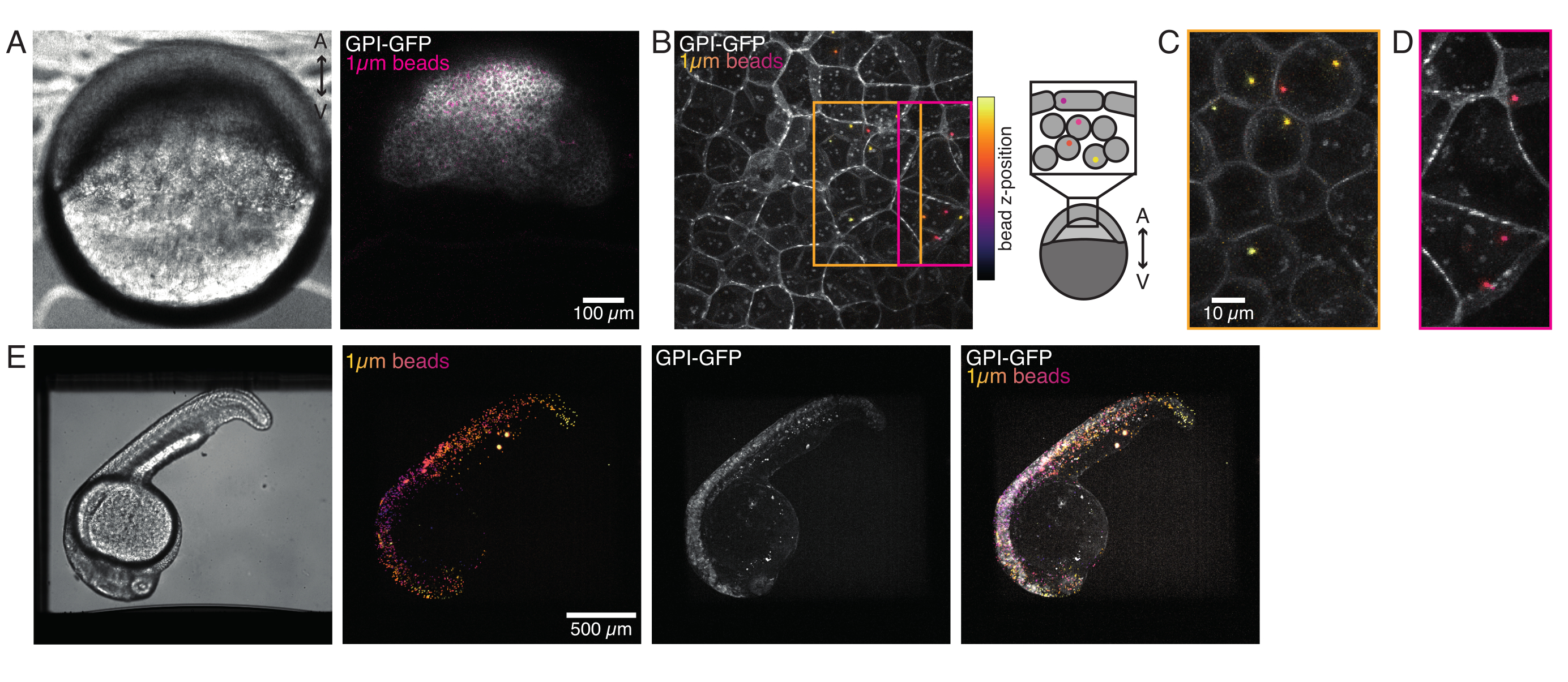
1細胞ゼブラフィッシュ胚に注入された微小球は、形態形成中に動物の帽子全体に広がる。より明確な可視化のために、我々は赤い蛍光マイクロビーズで注入プロトコルを繰り返し、異なる発達段階で我々の共焦点顕微鏡で容積画像を取った。図4A-Dにおいて、注入されたビーズは、5hfpで生体内の前駆細胞幹細胞の細胞質で視覚化される。その後、微小球が24hpfで胚全体に広がって現れた(図4E)。正常に発達した両方の段階の胚および生存率は、対照非注射または模擬注入胚と同等であった(図S3を参照)。これは、受精後5日までビーズ注入ゼブラフィッシュの摂動のない生存を報告する他の研究と一致しています55。
紡績ディスク共焦点顕微鏡は、マルチチャンネル蛍光顕微鏡に対応しています。図5Aでは、細胞質に1個または2個のビーズを含む単離された幹細胞を示す。複数の蛍光標識を使用して、細胞のさまざまな側面を調べることができます(図5B)。核形態は、Hoechst色素またはH2A:mCherry mRNA発現を使用して追跡することができ、内部核膜はLap2b-eGFP12で分析することができます。アキュトミオシン皮質のダイナミクスは、細胞内カルシウムレベルと同様に、それぞれMy12.1:::eGFPトランスジェニックline56およびCalbryte-520インキュベーションで観察することができる。ここで説明したプロトコルは、固定化された野生型細胞の細胞核力学を接着基材(後述サスペンションと呼ぶ)と機械的閉じ込めで比較することを目的としている。高さ10μmの微小室に閉じ込められた単離された幹細胞は、内核膜(INM)の部分的な展開とそれに続くアトミオシン収縮率の増加を示した12。図5Cでは、細胞質内に1個または2個のビーズを有する閉じ込められた細胞が示されている。成功した閉じ込めは、核のより広い断面を有する平坦化された拡張細胞を介して見える。核膜は、閉じ込められた細胞でさらに展開され、懸濁液中の細胞と比較して滑らかに見えるはずです(図5C)。
力時間と力変形解析
得られた結果の分析は、調査対象の検体と関心の問題に強く依存し、したがって、それらはここで一般化することはできません。例えば、インデント測定を分析する一般的な方法は、修正された Hertz モデルを力差し込み data57 に合わせてヤング率を抽出することです。しかし、このような治療の仮定は慎重に評価する必要があり、常に適切に正当化されない可能性があります(調査された構造は等方性で均質で、線形弾力性とインデントはビーズ半径よりも小さい)。したがって、ここでは、調査された構造の機械的挙動を異なる実験シナリオ間で比較できるモデル独立した測定値のみを考慮する。
出発点として、ある種のインデント深度で力変位曲線の傾きを測定することで、核のモデル独立した構造剛性58 の尺度を提供する。この値は複数のサンプルから収集し、さまざまな実験設定とサンプル摂動の間で比較できます。
インデントの測定
以下の行では、閉じ込めにおける細胞変形時の細胞核の機械的応答に注目します。このプロトコルのステップ8での実験は、通常、約2〜3μmのインデント深さのために最大200 pNのピークを強制します。しかし、これらの値は、細胞の種類や実験条件によって大きく異なる可能性があり、より柔らかい核は所定のインデントに対して低い力をもたらす。それにより、核の変形を力と共に正確に測定し、細胞核の正確な機械的特性を測定する必要がある。このセクションでは、代表的な力の字下げ測定から細胞核剛性を得る。
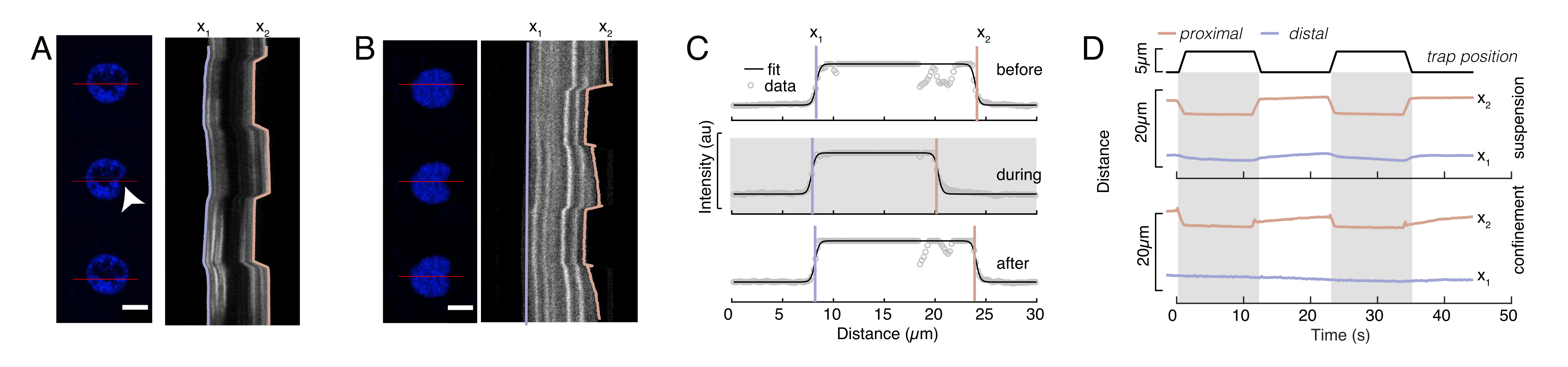
図6では、浮遊および閉じ込められた細胞における核の遠位側および近位側の変形を示す。豊富な機械的挙動を観察することができます。典型的な粘着基質上の懸濁細胞では、核はビーズによって強くインデントされたが、繰り返し押し出す事象の際にもわずかに変位した。我々は、Hoechst染色細胞核の蛍光イメージングから得られたキモグラフを解析することにより、核上のビーズのくぼみを測定した。キモグラフは、インデント方向(図6A、B)に沿ってフィジーのマルチキモグラフプラグインを使用して簡単に計算され、さらに処理するためにMatlab(バージョン2021、Mathworks)にインポートされました。ステップ関数は、インデントルーチンの軌道に沿って核の区切り縁を追跡することを目的として、生の強度プロファイルに取り付けられました。ご確認のとおり、核の形状変化に関する正確な情報を持っています(図6および図S2)。次の二重シグモイド曲線をステップ関数の解析バージョンとして使用しました。
 (方程式1)
(方程式1)
ここで、x1およびx2は核の遠位および近位エッジを示し、AおよびBは画像の青色チャネル(Hoechst色素)の最大および背景の灰色値である(図6B)。エッジ幅が考慮されました(e0 = 0.25 mm)。インデントされた近位核エッジ(x2)が微小球核接触後のオプティカルトラップルーチンによって適用される軌道に従っている間、反対の遠位エッジ(x1)は細胞質などの粘弾性材料に期待される緩和ダイナミクスを表示する(図6D)。対照的に、高いマイクロチャンバ10μmに閉じ込められた細胞内の核は、細胞内のインデント時に核の転位挙動を示さない(図6B,D)。図6Dに示されている、核の後縁は、近位側から押し出すビーズによって変わらず、細胞収縮性とインデント力に対する摩擦に起因するより強い力が原因である可能性が最も高い。正しい変形深さを得るために、変位x1をインデントメジャーx2から差し引いた:Δx = x2-x1(図6Dも参照)。
データ分析を強制する
核変形を引き起こす力は、光学的に閉じ込められたマイクロビードに由来する光運動量の変化から測定した(図7A)。台形軌道を適用したときの力(ステップ8.4.3、図7B)は、トラップが動かなくなるまで直線的に増加したが、その後安定した状態値に緩和された。この挙動は、損失および貯蔵弾性を示す粘弾性材料を示した。インデント イベントの直後に、力はピーク値 Fp に達し、続いて応力緩和(図 7C)に達しました。
 (方程式2)
(方程式2)
ここで、F0 は弾性成分に対して保存された力であり、f(t) は寸法なしの緩和関数です。この動作は、次の 3 つの方法で分析しました。
指数応力緩和を伴う標準線形固形(すなわち、f(t)=e-t/τ、図7Cインセットで概略的に表される。
2. 一般的な二重指数減衰を使用する場合:
F(t) = A + B1e-t/τ1 + B2e-t/τ2.
3. 指数関数的な減衰が続く電力法則の使用59:
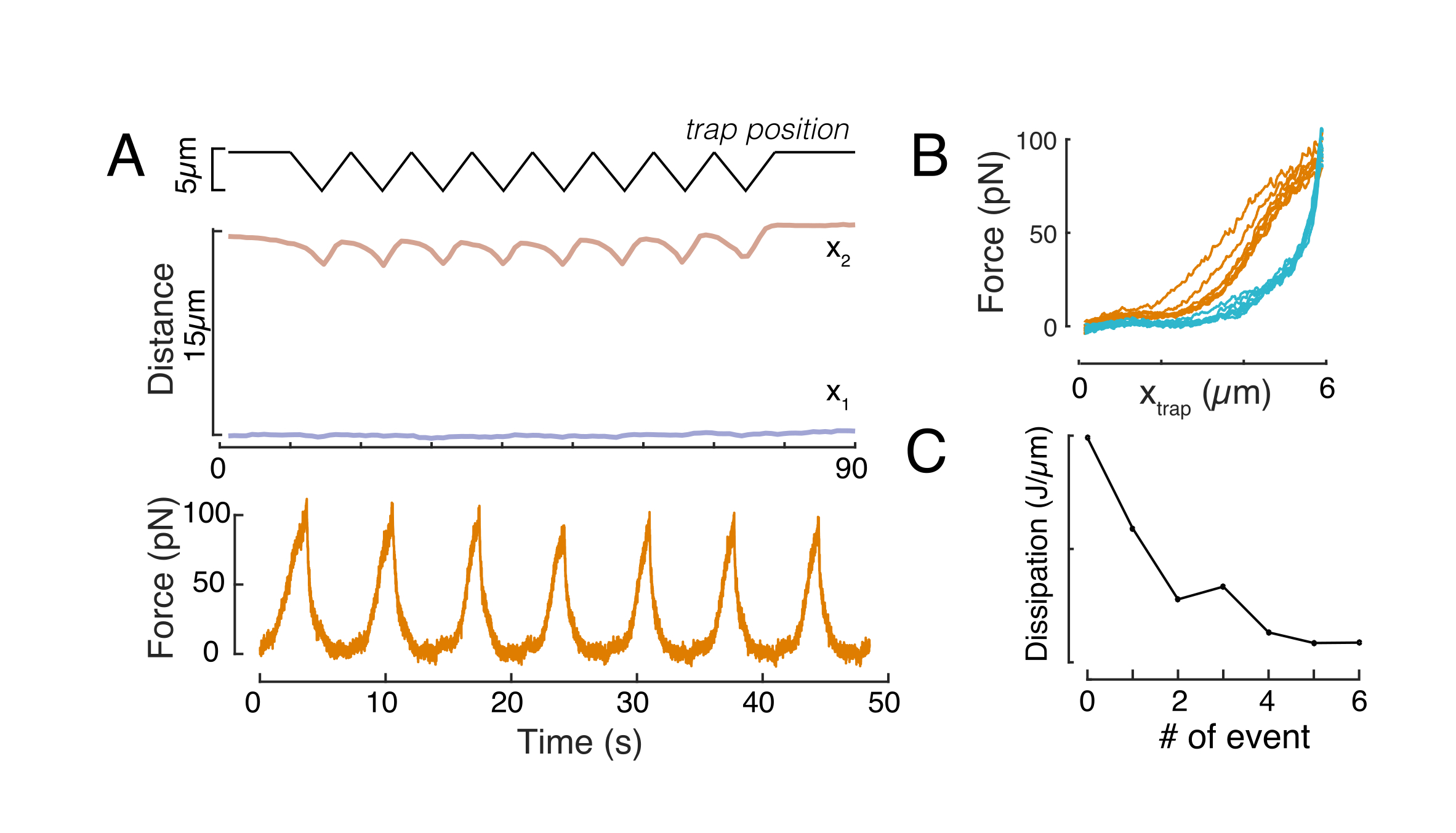
f(t) = t-pe-t/τ(図7Cに適合)
モデル 1 の適合は簡単に行うことができますが、モデル 2 と 3 の初期推定値 (τ1, τ2) と (p, τ) をそれぞれ推定することをお勧めします。この処理は、対数対線形 (図 7D、左) および対数スケールのデータに線を合わせると、それぞれ実行できます。表S3は、図7で分析した例の結果を要約する。次のセクションでは、細胞核力学の特性評価のためのパワー則と指数則の組み合わせを考察する。
強制変位関係
同様に、説明した実験的なセットアップは、複数のインデントイベントの力と変位関係を得るために使用することができる。三角ルーチン(ステップ8.4.4、 図8A)を行うことにより、その力を変形に関連付け、力差し曲線をプロットすることができる。例示的な結果は、ビードが核に接触すると平坦なベースラインが滑らかに斜面を変化させる 図8Bに示されている。騒がしいデータの真の接触点を特定することは難しく、接触領域が弾性モデル60に適合するかどうかを確認するために注意する必要があります。この特定の実験では、その後のインデントにより、より深い接触点を持つ曲線が生じ、ビーズの引き込み後の核形状回復が遅くなり、核粘弾性材料特性によって定義されるヒステリティックサイクルの変化を示す(図8C)。したがって、研究者は、これが起こった場合に注意し、分析パイプラインに組み込むか、この効果が測定を変更しないように、後続の測定の数を制限する必要があります。
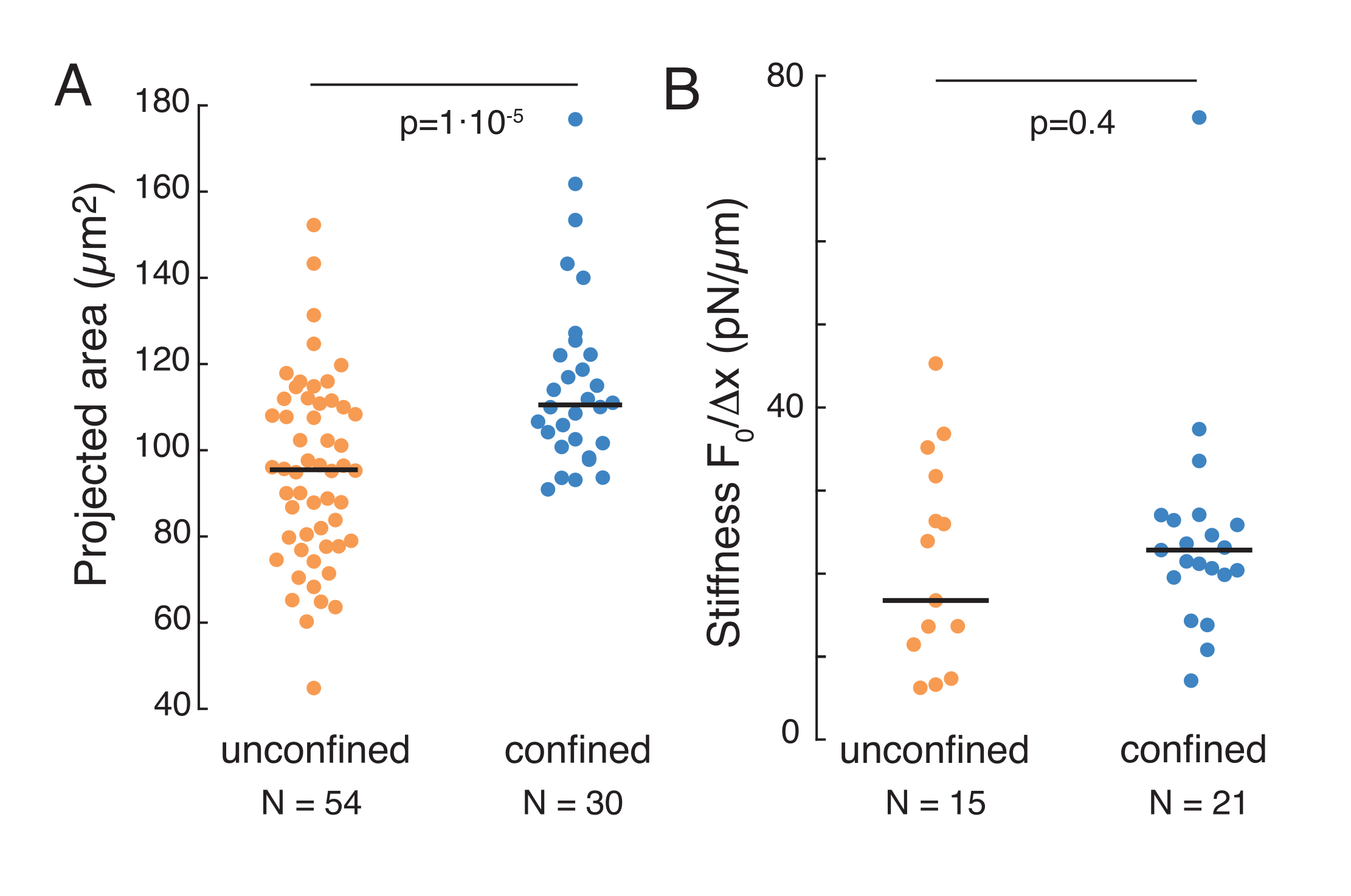
懸濁液および10μm以下の閉じ込めの細胞における核力学
前述のアプローチは、粘着基質および閉じ込められた細胞上の浮遊細胞における核ストレス緩和のダイナミクスを分析するために使用された。我々の結果は、閉じ込めが予想領域の拡大をもたらすことを示している(図9A)が、核剛性の有意な変化(図9B)。我々は、τ= 6.08 ±1.1s(閉じ込められていない)とτ= 4.00±0.6s(閉じ込め)と同様の緩和を測定し、その後核の弾性率に対応する蓄積された力値を示す。インデンテーションルーチンの異なる初期条件によって生成される可能性のある実験的な変動を考慮するために、測定された貯蔵力は、  インデント深度に正規化された。このパラメータは、核剛性を表し、特定のインデントに必要な力、つまり応力を表します。閉じ込め下でも、閉じ込められていない細胞でも同様の剛性を得た: = 20.1 ±12.6 pN/μm、 =24.6±13.6 pN/μm(標準偏差 ±の平均値)をそれぞれ得た。
インデント深度に正規化された。このパラメータは、核剛性を表し、特定のインデントに必要な力、つまり応力を表します。閉じ込め下でも、閉じ込められていない細胞でも同様の剛性を得た: = 20.1 ±12.6 pN/μm、 =24.6±13.6 pN/μm(標準偏差 ±の平均値)をそれぞれ得た。

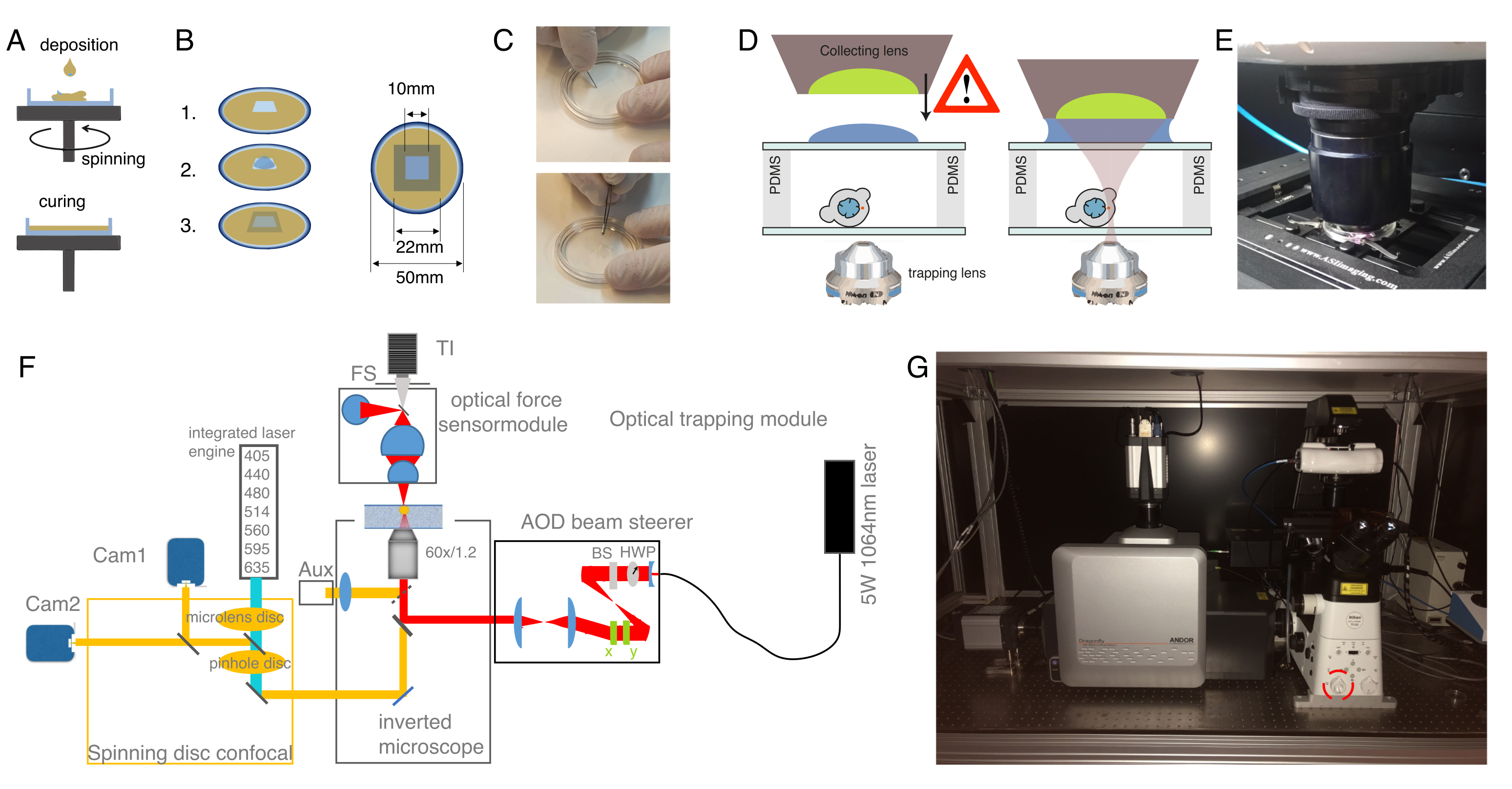
図1:1細胞(ジゴテ)期におけるゼブラフィッシュ胚の微小注入(A)インジェクションプレート:注射用三角形の射出板が用いられる。プレートはE3(卵の培地)で1%超純粋なアガロースで作られています。上と横のビューは右側に表示されます。(B)胚の位置:ブラシを使用して胚を穏やかに向け、1つの細胞が針ではっきりと見え、容易にアクセスできるように向けます。我々は、スケッチに示すように、針の反対側に位置する細胞で胚を向けることを提案する。(C)1細胞段階胚への注入手順:胚を取り巻く絨毛を、針で単一細胞に突き刺す。針の先端が細胞の内側であることを確認し、注入する圧力を解放します。(D)胚をブラスラ(球)段階(4 hpf)まで発達するまで28〜31°Cでインキュベートする。細胞分離プロトコルと細胞染色(ステップ2)を行い、対応する基板表面コーティングと組み合わせた懸濁液および/または閉じ込めで単離された細胞を用いて光学トラップチャンバーを準備する(ステップ3)。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図2:光学用トゥイザー装置の調製. (A)ガラス底皿に定義された高さを有するPDMSのスピンコーティング層。PDMSの低下は遠心力のために均等に広がる。(B)PDMS層からサンプルチャンバの調製。1:メスで正方形をカットし、2:コンカナバリンA(ConA)で内側の井戸をコーティングし、洗浄し、細胞をシード。3:ガラススライドまたはカバースリップで覆い、井戸を密封します。(C)メスで正方形の切断の画像と鉗子でよくPDMSを除去。(D)捕捉チャンバ上に光学力センサの集めるレンズを装着する。浸漬オイルの滴は、集水レンズと上部ガラスカバーの間の浸漬媒体として機能します。スケールしない回路図。サンプル皿のガラスカバーに触れないように収集レンズを下げながら注意してください。(E) サンプルに接触した力検出ユニットの画像。(F)実験セットアップの概略図。光学マイクロマニピュレーションモジュールは、半波板(HWP)と偏光ビームスプリッター(BS)を介した電力制御を備えた連続波レーザービーム(5W、λ = 1064 nm)を使用します。一対のAODで変調された後、逆顕微鏡の上側の起蛍光ポートに結合される。レーザービームは、950 nmのショートパス二色性鏡(IR-DM)によって反射され、蛍光励起および発光の透過を可能にする。捕獲レーザーは顕微鏡の後部、蛍光ポート(上部の砲塔)に導かれる。ATは、水浸し対物レンズ(60x、NA = 1.2)の焦点面で作成されます。光学力センサーは顕微鏡のタレットによって受け取られ、高NA、油浸性レンズとのNATから出てくるレーザー光を捕獲する。同時に、力センサーは明視野の照明を可能にする。回転ディスクの共焦点装置は左ポートに結合される。7つの蛍光励起レーザーと2つのバック照明sCMOSカメラを制御する2つの統合レーザーエンジン(ILE)が搭載されており、並行Abb:TI、トランイルミネーターでの二重蛍光色素イメージングを可能にします。FS、フィールド停止。AOD、アカストオプティカルディフレクター;HWP、ハーフウェーブプレート。CAM、カメラ(G)光学トラップ装置の写真。赤い円はベルトランレンズを示し、手動で光路に切り替えることができる。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図3:適切なサンプルとパラメータを選択する. (A)単一の微小球を有する単一のミクロ球を有する単一のゼブラフィッシュ前駆細胞幹細胞の代表的な画像は、インデント実験を行う。スケールバー= 10 μm. (B) 模範的なトラップ軌道;インデント深さ5 μm;インデント速度 = 5 μm/s;緩和時間 10 s. この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図4:開発中のゼブラフィッシュ胚内のマイクロビーズの局在化。 1 μmの赤色蛍光ビーズの0.5 nLを、WT胚にGPI-GFP mRNA(100pg/胚、形質膜)と一緒に注入し、ビーズの局在を可視化します。(A-D)0.75%アガロースに搭載された胚内部のマイクロスフィア5時間の注入後の分布。(A) ブライトフィールドと蛍光画像。ビーズは、共焦点顕微鏡写真に見られるように胚組織全体に均質に分散している。(B) 共焦点蛍光z-スタックの最大投影ビーズは、イメージ スタック内の Z 位置に応じて、紫色から黄色に色分けされます。紫/マゼンタは、最も外側のビーズ/細胞(EVL;上皮包層、またはEVL表面に近い前駆幹細胞)に対応し、黄色は右側のスケッチに示すように、内側のビーズ(前駆細胞深部細胞)に対応しています。(C)オレンジボックス内の領域に対応する(B)のサブスタックのカットと最大投影:深い細胞の大部分は1〜2ビーズを含む。(D)マゼンタボックスに対応する(B)サブスタックのカットと最大投影:一部のEVL細胞には1〜2個のビーズが含まれる。(E)ブライトフィールド画像と0.75%アガロースに取り付けられ、トリケーヌで麻酔された24 hpf胚のZスタックの最大投影。胚をトリカインで15分間前培養した。左から右へ:微小球(直径1μm)、GPI-GFPおよび画像のオーバーラップ。胚の全身に分布するビーズ。各パネルに示されているスケールバーの寸法。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図5:異なる標識を有する分離されたゼブラフィッシュ前駆幹細胞。 (A)1(上)または2(下)ビーズを注入した懸濁液細胞の透過光顕微鏡画像。シアンの矢印はビーズを指しています。(B)異なる染色を有する懸濁細胞の蛍光共焦点像。左上:Lap2b-eGFP(内核膜、80 pg/胚)およびH2A-mCherry。右上:GPI-GFP(形質膜、100pg/胚)およびDNA-Hoechst(セクション2に記載されているように染色)。左下:MyI12.1-eGFP(トランスジェニックライン)およびDNA-Hoechst。右下:カルブリテ488およびDNA-Hoechst(セクション2に記載されているように染色)。(C)1(上)または2(下)注入ビーズを有する閉じ込められた細胞の透過光顕微鏡画像。シアンの矢印はビーズを指しています。スケールバー= 10 μm. この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図6:回転するディスクムービーからの核変形を推定する(A,B)(A)浮遊細胞および(B)閉じ込められた細胞における核のインデント実験のタイムラプス。スケールバー 10 μm。Hoechst標識核の代表的なスナップショットは、光学的に閉じ込められた微小球(白い矢印)でのインデントの後、5 s前、中、および5 sを示す。インデントセグメント(赤線、右パネル)に沿った動態。x1 と x2 は、強度プロファイルの適合から式 1 に抽出されたインデント実験中の核の遠位境界と近位 (ビーズに近い) 境界です。(C)3つの異なるフレーム(インデントの前、中、後)のインデントセグメントに沿った強度プロファイルを、核エッジの遠位、x1、および近位x2の位置を評価するために式1に適合した。(D) 浮遊細胞と閉じ込められた細胞のインデント実験の間に青でx1(t)およびx2(t)の代表的な軌道(10 μm)。シェーディングされた領域はインデントを示し、x1 と x2 の間の距離は核直径を示します。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図7:力信号処理. (A)細胞核をインデント時に変形させる光学的に閉じ込められた微小球の概略。核膜と光学力は黒矢印で示されます。ビーム運動量の変化は緑色の矢印パウトによって示されます。(B)繰り返し核のインデント実験の間に光学的に閉じ込められた微小球によって経験されたトラップ軌道(上)および力(下)。(C)最大インデント深度で力ピーク後に強制緩和減衰。インセットは、そのダイナミクスがここでのフェノメノ学的観測値に近似する標準線形固体の概略を示しています。(D) 左: 正規化力対時間の対数。影付きの領域は、二重指数減衰(赤線)に適合するために使用されるデータ部分を示します。右: 正規化力と時間の対数の対数。影付き領域は、電力法則に適合するために使用されるデータ部分を示します。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図 8: 三角形トラップ変位を持つ強制インデントルーチン. (A) x1(t) の代表的な軌道 (青) と x2(t) の 3角形のインデント実験中に、セルに対して 10 μm の閉じ込み高さの高さで行われた。上: トラップ位置。中央: 核形状解析。x1 と x2 の間の距離は、核の直径を示します。下: 信号を強制します。(B) 8 つの連続インデントに対して、強制対トラップ位置を指定します。(C)散逸の進化は、f-d曲線のアプローチと離脱部分の間のヒステリシスに由来し、その後の各インデントイベントに対する核の。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図 9.サスペンション(接着面)および台形ルーチンからの閉じ込めにおける細胞の核特性。 (A) 細胞から10μm以下の閉じ込めの細胞から核の突出領域。黒いバーは中央値を表します。(B) 懸濁液および閉じ込め下にある細胞の核剛性。黒いバーは中央値を表します。MatLabを使用したクルスカル-ウォリス試験から得られたP値。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
補足表1:光ピンセットソフトウェアによって定義される台形軌道。 1 行目 (2 行目) は、トラップが直線的に変位する x (y) 距離です。3 行目では、指定したステップの継続時間が秒単位で設定されます。この軌道は7点から構成され、 図7Bの核に対して2回積載された台形に対応している。 このテーブルをダウンロードするには、ここをクリックしてください。
補足表2:光ピンセットソフトウェアによって定義される三角形の軌道。 表2と同様に、この軌道は、深さ5μmの8つのインデントイベントと2.5μm/sの速度に対応する16点で構成されています 。
補足表 3: 図 7 のデータのフィッティング・パラメーターIG:最初の推測。このテーブルをダウンロードするには、ここをクリックしてください。
補足図S1:光学力センサアライメントと運動量ベースライン補正 (A) ベルトランレンズを通して補助カメラ(AUX、 図2)で撮影したフィールドストップ。エアバブルは、接眼を通して見えない浸漬油に見えます。(B) クリーン光路。正確な位置合わせのために、フィールドストップを開き、NA = 1.2ライトコーンと一致させます。(C) サンプル平面の画像。赤い四角はOT作業領域を示しています。スケールバー:20 μm(D)Cで示された白い二重矢印に沿って、FOV全体で測定されたトラップ電力。赤で、補正が適用されていない場合のトラップ電力の変動。青色では、トラップの力が視野全体で補正されます。(E) 同一範囲に沿った運動量ベースラインのX成分。赤で、補正されていないトレース。青色で、トレースはトラップ電源を補正しました。緑色で、製造元のソフトウェアのグローバル オフセット補正を使用して、運動量ベースラインのトレースを修正しました。(F) Y成分の場合はEと同様。通常の操作では、シェーディングされたコンポーネントは、y 軸に沿った移動中に x 座標と力の測定(たとえば、x の力成分)と y 力成分に沿って移動するときに使用されることに注意してください。すべての補正が実施された後、<0.5 pNのRMSDノイズが得られます。 このファイルをダウンロードするには、ここをクリックしてください。
補足図 S2: 弱いトラップによるルーチンの失敗。 (A) 失敗したルーチンからの核のインデントを示すキモグラフ。トラップからのビードのエスケープにより、短い一過性の変形のみが表示されます。重要なことに、トラップレーザーは、事前定義された軌道(緑色の点線)を完了するためにビーズなしで動いています。スケールバー = 10 μm. (B) トップ: トラップ位置対時間。中央: インデントされた近位および遠位の核エッジのエッジ追跡結果。遠位エッジは、接着基板上の単離された細胞上の完了したルーチンに対して一般的に観察されるように、インデントなしで移動しないことに注意してください。下:熱ノイズの減少とゼロフォースへの急激な低下によって示される微小球の損失を示す力対時間。 このファイルをダウンロードするには、ここをクリックしてください。
補足図S3:注入胚の生存率。 プロトコルで概説された濃度で1μmビーズおよび100pg/胚のmRNAを注入した胚は、未注入胚と比較され、受精後24時間で有意差を示さなかった。各実験の条件ごとにN>21胚を用いた3つの独立した実験の平均および標準偏差。 このファイルをダウンロードするには、ここをクリックしてください。
ディスカッション
このプロトコルでは、生細胞内の細胞核の機械的特性を問い合う独自の方法を説明する。他の力分光法とは異なり、非侵襲的な光学トラップは、細胞膜および細胞骨格の寄与を細胞核硬直から切り離すことを可能にした。重要なことに、光学顕微鏡操作はマルチモーダル顕微鏡と互換性があり、実験者は細胞核メカノバイオロジーに関与する異なるプロセスを研究することができます。代表的な結果として、数百個のピコニュートンの程度の力によって行われたインデント時の核変形を測定するためにDNA-Hoechst染色を使用した。
このプロトコルで概説されている例を超えた、当社のメソッドの潜在的な用途
外部の摂動なしに生体細胞内の測定から定量的な機械的情報を抽出する可能性は、探求され始めたばかりの前例のない機会の多くを可能にします。したがって、当社の光学マイクロマニピュレーションプラットフォームの提示プロトコルは、非常に多用途性を備えたより複雑な実験に拡張することができます。アクロスオプティカルディフレクター(AOD)は、異なるセルの位置をまたがって同期力を測定するための複数の光トラップを生成でき、また広い周波数範囲51,61の活動的なマイクロロジーにも使用できます。前述のように、インデント時の力の応答は最大の捕捉力を克服し、光トラップからのビードの脱出につながる。この場合、光力をクランプするために、フォースフィードバックをAODで構成することができる。全体として、このプロトコルで説明されている応力緩和のような複数のミクロレオロジーアプローチだけでなく、アクティブなマイクロレオロジーまたはクリープコンプライアンスも、このプラットフォームで実験的に取得し、新しいソフトウェアパッケージ61,62,63,64,65で徹底的に分析することができます。.さらに、力の適用は核に限定されるものではなく、原発性血管内で流れる赤血球を捕捉したり、葉芽細胞やミトコンドリア68を捕捉して変形させたりするために示されているように、多様な細胞内構造や複雑な組織を測定するために原則的に行うことができる。.光運動量キャリブレーションは、閉じ込められた物体の形状とサイズに依存しないため、任意の形状38,39を有する任意の力プローブで直接力測定を可能にします。注入された微小球を用いることが、細胞構造の直接操作と比較して比較的低いレーザーパワーを有する核に高い力を加えることを可能にした69,70,71。しかし、十分に高い屈折率差を考えると、外部に適用される力プローブは必要なく、細胞内小器官はビーズを注入することなく直接操作することができます(未発表の観察と参照70)。
アプリケーションを拡張するためのメソッドの潜在的な変更
マイクロビーズの異なるサイズは、実験に応じて注入することができますが、相対的な制御を行う必要があります。例えば、後の段階で細胞を研究するには、より小さいビーズを注入することができる。これにより、光トラップ(参考55に示すなど)によって発揮できる最大の力が減少します。より大きなビーズは、より高い力を発揮するために注入することができますが、これらは、そのサイズや関心の段階に応じて胚の発達に影響を与える可能性があります。マイクロビーズ注入が選択肢ではない実験では、屈折率の違いを示す様々なオルガネラは、細胞質と比較して依然として光学的に操作することができ、光運動量変化から測定可能な光学力を生じさせる42。上述したように、これらの方法は、ショウジョウバエ胚70における細胞細胞接合部を変形させるためにバンバルデカールらによって採用されている。同様に、細胞の核は周囲の媒体44よりも屈折率が低く、捕捉強度が低いにもかかわらずビーズを含まないインデント(未発表の観測と参照72)を可能にする。したがって、核は簡単に閉じ込めることができず、トラップをエスケープします。
スピンコーティングされたPDMSスペーサは便利で速い方法 で 製造されますが、マイクロ/ナノファシリテーション施設やエンジニアリングラボにアクセスできないラボでは手の届かないところにある可能性があります。これにより、スペーサは、ラボテープまたはパラフィルムから容易に組み立てることができる(ステップ4)。このプロトコルは、事前定義された測定井戸への単一細胞の送達を自動化するマイクロ流体チャネルを製造したり、同じ標本内の閉じ込め効果を推定するために定義された高さのチャンバーに入ることによっても適応することができる。しかし、このようなマイクロ流体デバイスは、顕微鏡目的と光学力センサの集水レンズとの間の空間に合わせて、2mm程度(ステップ3参照)を設計しなければならない。光力センサは、焦点がずれる光収差がフォトン運動量測定に影響を与えないように、適切な高さに配置する必要があります。
その他の変更には、生物学的記者の変更が含まれる可能性があります。Hoechst蛍光スペクトルがGFPチャンネルにスペクトル的に出血することを発見し、2つの蛍光チャネルで同時測定のための核マーカーとしてmCherryタグ付きヒストンとの組み合わせを好むことがわかりました。あるいは、核変形は、Lap2b-GFPのような内部核膜を対象とする標識で容易に追跡することができる(図2)。
細胞核へのインデントは2~3ミクロンのオーダーで、回折限定回転円盤共焦点顕微鏡の画像解析により正確に測定することができました。硬い核またはより小さな力の場合、インデントは、このアプローチを使用してほとんど測定可能です。しかし、絶対力較正された光ピンセットは、ナノメートル精度のBFP干渉法を用いて 、その場で 閉じ込められたビーズの位置測定のために較正することもできる51。このアプローチを使用すると、電圧信号と光力センサはパラメータ β[ nm/V]を介してトラップされたプローブの位置に変換することができ、インバリアントパラメータα [pN/V]は前述の光運動量キャリブレーション41 (詳細については下記参照)を通じて力の値を得ることができます。
トラブルシューティング
実験中に次の課題が発生することがわかりました。
安定したトラップが形成されず、微小球が容易に脱出する
顕微鏡の目的または不整列の修正首輪の汚れは安定したトラップの失敗につながる可能性がある。直ちに解決策が見つからない場合は、対物レンズの点広がり関数を測定します。目的の標本が光学的に密な組織の奥深くにある場合、レーザーフォーカスは不安定なトラッピングにつながる重度の光学収差を経験する可能性があります(この効果は通常、単離された細胞ではごくわずかですが、より厚い組織でより明らかになります)。高剛性の場合、核の復元力はトラップのエスケープフォースを超える可能性があり、微小球が失われ、インデントルーチンが失敗します。当初、光学トラップに近い核膜縁がほとんどインデントされなくなる(図S2A)。この現象が発生すると、トラッピングレーザーは力やブラウン運動の影響を受けなくなり、力がゼロに低下し、信号ノイズが減少します(図S2B)。これが起こる場合には、レーザーパワーはより強いトラップを有するように増加させることができる、ビーズを核に押し込む台形軌道の振幅を減少させることができる、または閉じ込められたマイクロビーズの初期位置をさらに核から離して設定することができる。
細胞は刺激の間に動いている
細胞が十分に結合していない場合、光学勾配トラップは細胞内の細胞を移動させ、核の力および基礎的な力学が実際的であるような細胞内のインデントルーチンを行う。細胞全体の変位を防ぐために、表面上の細胞接着分子の濃度を高める、例えば、ConA。
初期運動量補償
最初の運動量補正ルーチンがATプラットフォーム(ステップ6.5)で利用できない場合、人工的で力に依存しないベースライン信号を修正する必要があります。これは、ビーズが閉じ込められていなくても力曲線上の傾斜として見える(図S1E)。補正を行うには、まったく同じ位置で細胞の外側にビーズなしで同じ軌道を実行する必要があります。このためには、ステージ コントロールを使用して、トラップからセルを離します。参考として、フォースオフセットは、システム内の200 mWでFOV全体で5pNを変更します。したがって、短い軌道では無視できる。あるいは、ピエゾスキャンステージを使用して、レーザー位置を一定に残して、サンプル上の細胞を移動させることができます。
提示されたプロトコルの重要なステップ
微小球は、胚の上に最大の分布を確保するために、右の1細胞段階で注入する必要があります。ビーズは、イメージングに使用される蛍光チャネルに光が漏れないように蛍光であってはなりません。例えば、典型的な赤蛍光ビーズでさえ、その明るさによるHoechst染色後の細胞核のイメージングに使用される青色チャネルではっきりと見える(励起:405 nm;放出:445nm)。細胞を基板に安定的に取り付け、インデントルーチン中の横変位を防ぐために重要です。ルーチン中にセルが移動すると、力は過小評価されます。このようなことが頻繁に発生する場合は、接続プロトコルを最適化します。組織培養細胞の場合、フィブロネクチン、コラーゲン、ポリL-リジンなどの他の細胞接着タンパク質は、満足のいく付着(未発表の観察)につながります。閉じ込めの間、細胞は突然、激しい機械的ストレスを受ける。これは細胞に損傷を与える可能性があり、手順が慎重に行われない場合、実験者は頻繁に破裂した細胞に遭遇します。また、閉じ込めの高さが小さすぎると、すべての細胞が核の包絡破壊または不可逆的な損傷に苦しむことになります。これらを軽減するには、上部カバースリップをよりゆっくりと下げるか、カバースリップ間の間隔を大きくします。
テクニックの限界とそれらを克服するための提案
技術の明確な制限は、組織の深いセクションにレーザー光の浸透であり、収差および不安定なトラッピングにつながる。したがって、浸透深さの下限は、試料の透明度、採用することができる収差補正73 および適用されたレーザーパワーに依存する。レーザーパワーが高いほど、微小球付近の試料の熱励起につながることを考慮する必要があります。しかし、1064 nm波長レーザースポットによって発生したサンプルの加熱は、当社の生物学的サンプルに対する妥当な熱関連応力を避けるために最小限に抑えられます74。
もう一つの制限は、測定できる最大力です。直接光運動量検出は、光トラップ40,41の線形応答のレジームをはるかに超える力の測定を可能にするが、最大適用力は数百ピコニュートンの順序である。これは、レーザーパワーと軟質生物学的材料の結果的損傷閾値と屈折率の違いによって制限され、通常は0.1または0.344以下です。力検出限界を大きくする方法がいくつか提案されているが、例えば、構造化されたlight75、反射防止被覆微小球76、高屈折率particle77または高度にドープされた量子ドット78を用いる。
OTは、トラップ内のビードの位置がΔx = β Sx、Sxがセンサの電圧信号である、およびβ[μm/V]が異なるプロトコルに従ってオンザフライで較正されるように、BFP干渉測定を通じてナノメートルスケールの位置測定に使用できます35,54。光力センサの場合、電圧対力不変性変換係数α[pN/V]がβとトラップの剛性k[pN/μm]に直接関係していることが証明され、α= kβ 37)を通じて、光イメージングから検出するには小さすぎるビーズ変位の実験では、この戦略を使用して小さな位置検出で測定を補完することができます。例えば、ここで提示される実験ルーチンを非常に硬い核に適用し、合理的なレーザーパワー(200〜500 mW)の力は十分に大きなインデント値を誘導するのに十分ではない。その場合、ビードは核に接触する必要があり、トラッピング剛性は測定前に較正されなければならない(ステップ8.6)。力の関数としての核のインデントdは間接的に次のように決定することができる:
d = xtrap - F/k
ここで、xtrap はトラップ位置です。[pN/V]α不変光運動率係数とは異なり、各実験の前に因子β[μm/V]は、粒子サイズ、光トラップサイズ、相対屈折率指数などのトラップダイナミクスを決定する多くの局所変数に依存するため、各実験の前に較正する必要があります。
開示事項
著者らは開示するものは何もない。
謝辞
MKは、計画ナシオナル(PGC2018-097882-A-I00)、FEDER(EQC2018-005048-P)、セベロ・オチョア・オブ・エクセレンス・オブ・R&D(CEX2019-000910-S)を通じて、スペイン経済競争力省からの財政支援を認めます。RYC-2016-21062)、フンダシオ・プリヴァダ・セレックス、フンダシオ・ミル・プイグ、ジェネラリタット・デ・カタルーニャからCERCAと研究プログラム(2017 SGR 1012)、ERC(メカノシステムズ)とHFSP(CDA0023/2018)を通じた資金調達に加えて。V.R.は、スペイン科学イノベーション省からEMBLパートナーシップ、セントロ・デ・エクセチェンシア・セベロ・オチョア、ミネコのプラン・ナシオナル(BFU2017-86296-P、PID2020-117011GB-I00)、ジェネラリタット・デ・カタルーニャ(CERCA)への支援を認めています。V.V.は、665884マリー・スクウォドフスカ・キュリー交付契約に基づく欧州連合(EU)のHorizon 2020研究・イノベーションプログラムが資金を提供するICFOstepstone PhDプログラムからの支援を認めています。原稿を批判的に読んでくださったアルナウ・ファレに感謝します。マリア・マルサルは、24 hpf胚のイメージングと取り付けの助けと;ゼンダ・ヒメネス=デルガド、ゼブラフィッシュ・ミクロンジェクションのサポート
資料
| Name | Company | Catalog Number | Comments |
| #1.5 22 mm cover glasses | Ted Pella | 260148 | |
| #1.5 22x60 mm Coverglasses | Ted Pella | 260152 | |
| #1.5H glass bottom dishes | Willco | GWST-5040 | |
| 10-um beads | Supelco | 72986 | |
| 1-mm glass capillaries | Harvard Apparatus | 30-0020 GC100F-15 | |
| 1-um polystyrene microbeads | Sigma | 89904 | |
| 1-um red-fluorescent beads | ThermoFisher | F8816 | |
| Agar | ThermoFisher | 16500500 | |
| Aqcuisition cameras sCMOS | Andor | Sona-4BV11-UNI | |
| Auxiliary camera (Figure 3, AUX) | Blackfly, FLIR | BFS-U3-200S6M-C | |
| Calbryte 520 | AAT Bioquest | 520 AM | |
| Centrifuge | Eppendorf | 5453000011 | |
| Concanavalin A | Sigma | C5275 | |
| DMEM | Sigma | D2906 | |
| DNA-Hoechst | ThermoFisher | 33342 | |
| Double scotch tape | Biesse Adesivi | ||
| E3 | 5 mM NaCl. 0.17 mM KCl. 0.33 mM CaCl2. 0.33 mM MgSO4 | ||
| Eclipse Ti2 | Nikon | ||
| Forceps | Fine Science Tools | 11252-20 | |
| GPI-GFP | |||
| H2A-mCh | |||
| Image acquisition software | Fusion-Andor | ||
| Immersion Oil | Cargille | Type B: 16484 | |
| IR protection googles | Thorlabs | LG1 | |
| Lap2b-eGFP | |||
| Micro loader pipette | Eppendorf | GELoader | |
| Microinjector | World Precision Instruments | SYS-PV820 | |
| MicroManager 2.0 | |||
| Micromiter slide | ID5243 GXMGRAT-5 5mm/100 divisions | ||
| Mineral oil | Sigma | M3616 | |
| Motorized stage | ASI | ||
| Needle puller | Sutter instrument Co. | Model P-97 | |
| Optical tweezers platform | Impetux Optics | Sensocell | |
| OTs software (LightAce) | Impetux Optics | ||
| PDMS | Sigma | Sylgard 184 | |
| PDMS Curing agent | Sigma | Sylgard 184 | |
| Post processing software (Matlab) | Mathworks | ||
| RNAse free water | Thermofisher | AM9937 | |
| Short-pass dichroic mirror (Figure 3, IR-F) | Semrock | FF01-950/SP-25 | |
| Spin-coater | Specialty Coating Systems | Spincoat G3P-8 | |
| Spinning-disk confocal microscope | Andor | DragonFly 502 | |
| Stereomicroscope | Leica M80 | ||
| Triangular microinjection mold | Adaptive Science Tools | TU1 | |
| Universal oven | Memmert | UNB 200 | |
| Water immersion objective | Nikon | MRD07602 |
参考文献
- Chan, C. J., Heisenberg, C. P., Hiiragi, T. Coordination of Morphogenesis and Cell-Fate Specification in Development. Current Biology. 27 (18), 1024-1035 (2017).
- Heller, E., Fuchs, E. Tissue patterning and cellular mechanics. Journal of Cell Biology. 211 (2), 219-231 (2015).
- Heisenberg, C. P., Bellaïche, Y. Forces in tissue morphogenesis and patterning. Cell. 153 (5), 948-962 (2013).
- Petridou, N. I., Spiró, Z., Heisenberg, C. P. Multiscale force sensing in development. Nature Cell Biology. 19 (6), 581-588 (2017).
- Krieg, M., et al. Tensile forces govern germ-layer organization in zebrafish. Nature Cell Biology. 10 (4), 429-436 (2008).
- Ruprecht, V., et al. Cortical contractility triggers a stochastic switch to fast amoeboid cell motility. Cell. 160 (4), 673-685 (2015).
- Shellard, A., Mayor, R. Supracellular migration - Beyond collective cell migration. Journal of Cell Science. 132 (8), (2019).
- Mongera, A., et al. A fluid-to-solid jamming transition underlies vertebrate body axis elongation. Nature. 561 (7723), 401-405 (2018).
- Atia, L., et al. Geometric constraints during epithelial jamming. Nature Physics. 14 (6), 613-620 (2018).
- Turlier, H., Maître, J. -. L. Mechanics of tissue compaction. Seminars in Cell & Developmental Biology. 47-48, 110-117 (2015).
- Ladoux, B., Mège, R. M. Mechanobiology of collective cell behaviours. Nature Reviews Molecular Cell Biology. 18 (12), 743-757 (2017).
- Venturini, V., et al. The nucleus measures shape changes for cellular proprioception to control dynamic cell behavior. Science. 370 (6514), (2020).
- Charras, G., Sahai, E. Physical influences of the extracellular environment on cell migration. Nature Reviews Molecular Cell Biology. 15 (12), 813-824 (2014).
- Kirby, T. J., Lammerding, J. Emerging views of the nucleus as a cellular mechanosensor. Nature Cell Biology. 20 (4), 373-381 (2018).
- Lee, H. P., et al. The nuclear piston activates mechanosensitive ion channels to generate cell migration paths in confining microenvironments. Science Advances. 7 (2), (2021).
- Friedl, P., Wolf, K., Lammerding, J. Nuclear mechanics during cell migration. Current Opinion in Cell Biology. 23 (1), 55-64 (2011).
- Versaevel, M., Riaz, M., Grevesse, T., Gabriele, S. Cell confinement: Putting the squeeze on the nucleus. Soft Matter. 9 (29), 6665-6676 (2013).
- Zuela-Sopilniak, N., et al. Measuring nucleus mechanics within a living multicellular organism: Physical decoupling and attenuated recovery rate are physiological protective mechanisms of the cell nucleus under high mechanical load. Molecular Biology of the Cell. 31 (17), 1943-1950 (2020).
- Kim, D. H., Wirtz, D. Cytoskeletal tension induces the polarized architecture of the nucleus. Biomaterials. 48, 161-172 (2015).
- Lomakin, A. J., et al. The nucleus acts as a ruler tailoring cell responses to spatial constraints. Science. 370 (6514), (2020).
- Hampoelz, B., et al. Microtubule-induced nuclear envelope fluctuations control chromatin dynamics in Drosophila embryos. Development. 138 (16), 3377-3386 (2011).
- Heo, S. J., et al. Differentiation alters stem cell nuclear architecture, mechanics, and mechano-sensitivity. eLife. 5, 1-21 (2016).
- Cosgrove, B. D., et al. Nuclear envelope wrinkling predicts mesenchymal progenitor cell mechano-response in 2D and 3D microenvironments. Biomaterials. 270, 120662 (2021).
- Liu, H., et al. In situ mechanical characterization of the cell nucleus by atomic force microscopy. ACS Nano. 8 (4), 3821-3828 (2014).
- Hobson, C. M., et al. Correlating nuclear morphology and external force with combined atomic force microscopy and light sheet imaging separates roles of chromatin and lamin A/C in nuclear mechanics. Molecular Biology of the Cell. 31 (16), 1788-1801 (2020).
- Pajerowski, J. D., Dahl, K. N., Zhong, F. L., Sammak, P. J., Discher, D. E. Physical plasticity of the nucleus in stem cell differentiation. Proceedings of the National Academy of Sciences of the United States of America. 104 (40), 15619-15624 (2007).
- Rowat, A. C., Lammerding, J., Ipsen, J. H. Mechanical properties of the cell nucleus and the effect of emerin deficiency. Biophysical Journal. 91 (12), 4649-4664 (2006).
- Davidson, P. M., et al. High-throughput microfluidic micropipette aspiration device to probe time-scale dependent nuclear mechanics in intact cells. Lab on a Chip. 19 (21), 3652-3663 (2019).
- Lombardi, M., Zwerger, M., Lammerding, J. Biophysical assays to probe the mechanical properties of the interphase cell nucleus: Substrate strain application and microneedle manipulation. Journal of Visualized Experiments: JoVE. (55), (2011).
- Luo, T., Mohan, K., Iglesias, P. A., Robinson, D. N. Molecular mechanisms of cellular mechanosensing. Nature Materials. 12 (11), 1064-1071 (2013).
- Dahl, K. N., Engler, A. J., Pajerowski, J. D., Discher, D. E. Power-law rheology of isolated nuclei with deformation mapping of nuclear substructures. Biophysical Journal. 89 (4), 2855-2864 (2005).
- Guilluy, C., et al. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nature Cell Biology. 16 (4), 376-381 (2014).
- Bustamante, C. J., Wang, M. D. Optical tweezers in single-molecule biophysics. Nature Reviews Methods Primers. , 1-29 (2021).
- Svoboda, K., Block, S. M. Force and velocity measured for single kinesin molecules. Cell. 77 (5), 773-784 (1994).
- Berg-Sørensen, K., Flyvbjerg, H. Power spectrum analysis for optical tweezers. Review of Scientific Instruments. 75 (3), 594-612 (2004).
- Smith, S. B., Cui, Y., Bustamante, C. Optical-trap force transducer that operates by direct measurement of light momentum. Methods in Enzymology. 361 (1994), 134-162 (2003).
- Farré, A., Montes-Usategui, M. A force detection technique for single-beam optical traps based on direct measurement of light momentum changes. Optics Express. 18 (11), 11955 (2010).
- Català, F., Marsà, F., Montes-Usategui, M., Farré, A., Martín-Badosa, E. Extending calibration-free force measurements to optically-trapped rod-shaped samples. Scientific Reports. 7, 1-10 (2017).
- Bui, A. A. M., et al. Calibration of force detection for arbitrarily shaped particles in optical tweezers. Scientific Reports. 8 (1), 1-12 (2018).
- Farré, A., Marsà, F., Montes-Usategui, M. Beyond the hookean spring model: Direct measurement of optical forces through light momentum changes. Methods in Molecular Biology. 1486, (2017).
- Farré, A., Marsà, F., Montes-Usategui, M. Optimized back-focal-plane interferometry directly measures forces of optically trapped particles. Optics Express. 20 (11), 12270 (2012).
- Jun, Y., Tripathy, S. K., Narayanareddy, B. R. J., Mattson-Hoss, M. K., Gross, S. P. Calibration of optical tweezers for in vivo force measurements: How do different approaches compare. Biophysical Journal. 107 (6), 1474-1484 (2014).
- Mas, J., Farré, A., Sancho-Parramon, J., Martín-Badosa, E., Montes-Usategui, M. Force measurements with optical tweezers inside living cells. Optical Trapping and Optical Micromanipulation XI. 9164, (2014).
- Schürmann, M., Scholze, J., Müller, P., Guck, J., Chan, C. J. Cell nuclei have lower refractive index and mass density than cytoplasm. Journal of Biophotonics. 9 (10), 1068-1076 (2016).
- Rosen, J. N., Sweeney, M. F., Mably, J. D. Microinjection of zebrafish embryos to analyze gene function. Journal of Visualized Experiments: JoVE. (25), (2009).
- Westerfield, M. . The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish. (Danio rerio), 5th Edition. , (2007).
- Schubert, R., et al. Assay for characterizing the recovery of vertebrate cells for adhesion measurements by single-cell force spectroscopy. FEBS Letters. 588 (19), 3639-3648 (2014).
- Koschwanez, J. H., Carlson, R. H., Meldrum, D. R. Thin PDMS films using long spin times or tert-butyl alcohol as a solvent. PLoS One. 4 (2), 2-6 (2009).
- Das, R., et al. Mechanical stretch inhibition sensitizes proprioceptors to compressive stresses. bioRxiv. , (2021).
- Chardès, C., Clement, R., Blanc, O., Lenne, P. F. Probing cell mechanics with bead-free optical tweezers in the drosophila embryo. Journal of Visualized Experiments: JoVE. (141), (2018).
- Staunton, J. R., Blehm, B., Devine, A., Tanner, K. In situ calibration of position detection in an optical trap for active microrheology in viscous materials. Optics Express. 25 (3), 1746 (2017).
- Bola, R., Treptow, D., Marzoa, A., Montes-Usategui, M., Martin-Badosa, E. Acousto-holographic optical tweezers. Optics Letters. 45 (10), 2938-2941 (2020).
- Thalhammer, G., Obmascher, L., Ritsch-Marte, M. Direct measurement of axial optical forces. Optics Express. 23 (5), 6112 (2015).
- Vermeulen, K. C., et al. Calibrating bead displacements in optical tweezers using acousto-optic deflectors. Review of Scientific Instruments. 77 (1), 1-6 (2006).
- Dzementsei, A., Barooji, Y. F., Ober, E. A., Oddershede, L. B. Foregut organ progenitors and their niche display distinct viscoelastic properties in vivo during early morphogenesis stages. bioRxiv. , 1-35 (2021).
- Behrndt, M., et al. Forces driving epithelial spreading in zebrafish gastrulation. Science. 338 (6104), 257-260 (2012).
- Krieg, M., et al. Atomic force microscopy-based mechanobiology. Nature Reviews Physics. , (2018).
- A-Hassan, E., et al. Relative microelastic mapping of living cells by atomic force microscopy. Biophysical Journal. 74 (3), 1564-1578 (1998).
- Khalilgharibi, N., et al. Stress relaxation in epithelial monolayers is controlled by the actomyosin cortex. Nature Physics. 15, (2019).
- Crick, S. L., Yin, F. C. Assessing micromechanical properties of cells with atomic force microscopy: importance of the contact point. Biomechanics and Modeling in Mechanobiology. 6 (3), 199-210 (2007).
- Hurst, S., Vos, B. E., Betz, T. Intracellular softening and fluidification reveals a mechanical switch of cytoskeletal material contributions during division. bioRxiv. , 425761 (2021).
- Kaplan, J. L., Bonfanti, A., Kabla, A. RHEOS.jl - A Julia package for rheology data analysis. arXiv. 4, 1-5 (2020).
- Bonfanti, A., Kaplan, J. L., Charras, G., Kabla, A. Fractional viscoelastic models for power-law materials. Soft Matter. 16 (26), 6002-6020 (2020).
- Rivas-Barbosa, R., Escobedo-Sánchez, M. A., Tassieri, M., Laurati, M. i-Rheo: determining the linear viscoelastic moduli of colloidal dispersions from step-stress measurements. Physical Chemistry Chemical Physics: PCCP. 22 (7), 3839-3848 (2020).
- Tassieri, M., et al. i-Rheo: Measuring the materials' linear viscoelastic properties "in a step". Journal of Rheology. 60 (4), 649-660 (2016).
- Zhong, M. C., Wei, X. B., Zhou, J. H., Wang, Z. Q., Li, Y. M. Trapping red blood cells in living animals using optical tweezers. Nature Communications. 4, 1767-1768 (2013).
- Harlepp, S., Thalmann, F., Follain, G., Goetz, J. G. Hemodynamic forces can be accurately measured in vivo with optical tweezers. Molecular Biology of the Cell. 28 (23), 3252-3260 (2017).
- Bayoudh, S., Mehta, M., Rubinsztein-Dunlop, H., Heckenberg, N. R., Critchley, C. Micromanipulation of chloroplasts using optical tweezers. Journal of Microscopy. 203 (2), 214-222 (2001).
- Favre-Bulle, I. A., Stilgoe, A. B., Rubinsztein-Dunlop, H., Scott, E. K. Optical trapping of otoliths drives vestibular behaviours in larval zebrafish. Nature Communications. 8 (1), 630 (2017).
- Bambardekar, K., Clément, R., Blanc, O., Chardès, C., Lenne, P. F. Direct laser manipulation reveals the mechanics of cell contacts in vivo. Proceedings of the National Academy of Sciences of the United States of America. 112 (5), 1416-1421 (2015).
- Ferro, V., Chuai, M., McGloin, D., Weijer, C. J. Measurement of junctional tension in epithelial cells at the onset of primitive streak formation in the chick embryo via non-destructive optical manipulation. Development (Cambridge). 147 (3), (2020).
- Hörner, F., et al. Holographic optical tweezers-based in vivo manipulations in zebrafish embryos. Journal of Biophotonics. 10 (11), 1492-1501 (2017).
- Zhong, M. -. C., Wang, Z. -. Q., Li, Y. -. M. Aberration compensation for optical trapping of cells within living mice. Applied Optics. 56 (7), 1972 (2017).
- Català, F., Marsà, F., Montes-Usategui, M., Farré, A., Martín-Badosa, E. Influence of experimental parameters on the laser heating of an optical trap. Scientific Reports. 7 (1), 1-9 (2017).
- Taylor, M. A., Waleed, M., Stilgoe, A. B., Rubinsztein-Dunlop, H., Bowen, W. P. Enhanced optical trapping via structured scattering. Nature Photonics. 9 (10), 669-673 (2015).
- Bormuth, V., et al. Optical trapping of coated microspheres. Optics Express. 16 (18), 13831-13844 (2008).
- Sudhakar, S., et al. Germanium nanospheres for ultraresolution picotensiometry of kinesin motors. Science. 371 (6530), (2021).
- Shan, X., et al. Optical tweezers beyond refractive index mismatch using highly doped upconversion nanoparticles. Nature Nanotechnology. 16 (5), 531-537 (2021).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved