
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
細胞外小胞サブセット解析のための多重化表面プラズモン共鳴イメージングチップ上のマルチモーダル解析プラットフォーム
要約
この論文は、細胞外小胞サブセットの特性評価のためのスループットを向上させた新世代のマルチパラメトリック分析プラットフォームを提案します。この方法は、多重化されたバイオセンシング法と、原子間力顕微鏡による計量学的および形態力学的分析とラマン分光法の組み合わせに基づいており、マイクロアレイバイオチップにトラップされた小胞ターゲットを認定します。
要約
細胞外小胞(EV)は、直径50ナノメートルから数百ナノメートルの範囲のすべての細胞によって産生される膜由来の小さな小胞であり、細胞間コミュニケーションの手段として使用されます。それらは、さまざまな疾患の有望な診断および治療ツールとして浮上しています。サイズ、組成、内容が異なるEVを生成するために細胞が使用する2つの主要な生合成プロセスがあります。サイズ、組成、および細胞起源が非常に複雑であるため、それらの特性評価には分析技術の組み合わせが必要です。このプロジェクトには、EVの亜集団の特性評価のためのスループットが向上した新世代のマルチパラメトリック分析プラットフォームの開発が含まれます。この目標を達成するために、研究グループは、マイクロアレイバイオチップに閉じ込められた小胞ターゲットの原子間力顕微鏡(AFM)による計量および形態力学的分析と多重バイオセンシング法の組み合わせに基づいてEVの独自の調査を可能にする、グループが確立したナノバイオ分析プラットフォーム(NBA)から始まります。目的は、ラマン分光法による表現型および分子分析でこのEV調査を完了することでした。これらの開発により、臨床的可能性を秘めた体液中のEVサブセットを識別するためのマルチモーダルで使いやすい分析ソリューションの提案が可能になります。
概要
診断および治療におけるEV研究への関心の高まり1,2,3,4,5は、この分野が直面する課題と相まって、これらの小胞を定量化または特徴付けるための多種多様なアプローチと技術の開発と実装をもたらしました。EVの同定に最も広く使用されている方法は、EVの起源を確認するためのタンパク質特異的イムノブロッティングとプロテオミクス、その構造を確認するための透過型電子顕微鏡(TEM)、およびサンプルボリューム中のそれらの数とサイズ分布を定量化するナノ粒子追跡分析(NTA)です。
ただし、これらの手法だけでは、EVサブセットの特性評価に必要なすべての情報を提供するものではありません。生化学的および物理的特性の多様性によるEVの固有の不均一性は、特に混合物(粗サンプル)に含まれるEVの場合、信頼性が高く再現性のあるグローバルな分析を妨げます。したがって、検出および特性評価の方法は、EVに個別にも一般的にも、より高速であるが選択的ではない他の方法を補完するために必要です6。
TEM(またはクライオTEM)またはAFMによる高解像度イメージングにより、ナノメートル分解能7,8,9,10,11,12でEVの形態と計測を決定できます。ただし、EVなどの生物学的物体に電子顕微鏡を使用することの主な制限は、サンプルの固定と脱水を必要とする研究を実行するための真空の必要性です。このような調製は、観察された構造から溶液中のEV形態に変換することを困難にします。サンプルのこの脱水を避けるために、クライオTEMの技術はEVの特性評価に最も適しています13。EVの微細構造を決定するために広く使用されています。生体機能化金ナノ粒子による小胞の免疫標識は、EVの特定の亜集団を同定し、複雑な生物学的サンプル中に存在する他の粒子と区別することも可能にします。しかし、電子顕微鏡で分析するEVの数が少ないため、複雑で不均一なサンプルを代表する特性評価を行うことはしばしば困難です。
このサイズの不均一性を明らかにするために、国際細胞外小胞学会(ISEV)は、高解像度の個々のEVを明らかにするために、十分な数の広視野画像を分析して、より小さな画像を伴うことを提案しています14。AFMは、EVの研究のための光学的アプローチと電子回折技術の代替手段です。この手法では、柔軟なカンチレバーによって保持される鋭い先端を使用して、1つの支持体に堆積したサンプルを1行ずつスキャンし、先端とフィードバックループを介して存在する要素との間の距離を調整します。これにより、試料のトポグラフィーを特徴付け、形態力学情報を収集することができる15、16、17、18。EVは、原子レベルで平坦な基板上に堆積した後、または抗体、ペプチド、またはアプタマーによって機能化された特定の基板上に捕捉された後のいずれかでAFMによってスキャンされ、様々な亜集団を特徴付けることができる18,19。AFMは、前処理、標識、脱水を必要とせずに、複雑な生物学的サンプル内のEVの構造、生体力学、および膜状の生体分子含有量を定量し、同時にプローブできるため、温度と媒体の生理学的条件下でEVを細かくマルチパラメトリックに特性評価するためにますます使用されています。
本論文では、多重化されたフォーマットで(バイオ)化学的に機能化できるコア金バイオチップを使用する方法論を提案します。この基板は、表面プラズモン共鳴によるEVサブセットのバイオ検出を組み合わせた強力な分析プラットフォームの基礎であり、EVがチップに吸着/グラフトまたは免疫捕捉されると、AFMはEVの計測学的および形態機械的特性評価を可能にします。チップ上に取り込まれたEVサブセットのラマンシグネチャと組み合わせることで、この分析プラットフォームは、分析前のステップを必要とせずに、ラベルフリーの方法で生物学的サンプルに存在するEVの認定を可能にします。この論文は、基板調製とデータ収集における非常に厳密な方法論に支えられた強力な技術の組み合わせにより、EV分析が深く、決定的で、堅牢になることを示しています。
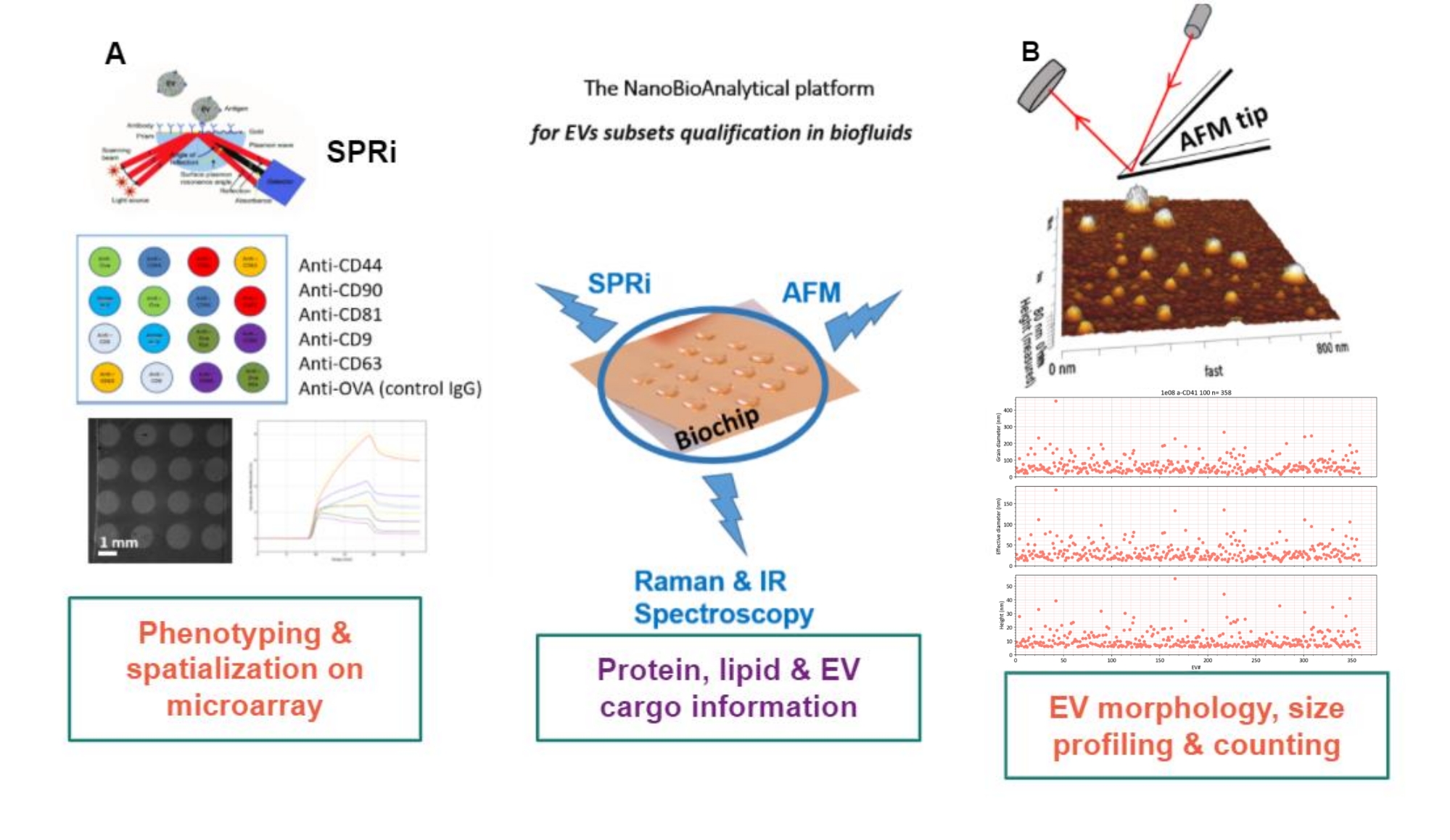
提案されたアプローチの原理は、金基質を準備し、EVサブタイプを吸着/グラフトまたは捕捉し、それらをAFMでスキャンして、各EVサブセットのサイズと形態を推定することです。さらに、吸着されたEVはラマン分光法によって分析されます。実際、この基板は、複雑化する3種類の界面、すなわち裸、化学的に機能化された、またはリガンドマイクロアレイを提示することができる。プロトコルのさまざまなステップを説明する前に、読者は、表面プラズモン共鳴画像法(SPRi)、AFM、および分光法を組み合わせた、 図1のナノバイオ分析プラットフォーム(NBA)アプローチの概略図を参照します。

図1:ナノバイオアナリシスプラットフォーム。このアプローチは、(A)表面プラズモン共鳴イメージング、(B)原子間力顕微鏡、および赤外線/ラマン(ナノ)分光法を組み合わせており、すべて同じ基板(マルチプレックス金チップ)にかみ合っています。略語: NBA = ナノバイオ分析プラットフォーム;SPRi =表面プラズモン共鳴イメージング;AFM = 原子間力顕微鏡;EV =細胞外小胞。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
コアゴールドバイオチップは、すべてのラベルフリーの特性評価技術がこのバイオチップで実施されるため、プラットフォームの心臓部を構成します。EVの特性評価(グローバル/トータルEVまたはEVサブセットのいずれか)のニーズと使用される方法の制限/要求に応じて、3種類の金バイオチップ表面が開発されました:「裸」の化学的に機能化された「C11 / C16」、または「リガンド」金表面と呼ばれるリガンド生体機能化のいずれか。
「ネイキッド」と呼ばれるネイキッドバイオチップは、EVを金に簡単に吸着することを可能にします。使用するバッファーを選択し、受動的な方法(インキュベーションとその後のすすぎステップ)またはフロー下でのモニタリング(SPRi)のいずれかでこの吸着を実現することができます。さらに、この受動吸着は、チップ全体(マクロアレイとして)またはマイクロピペットスポッターを使用してマイクロアレイに局在化のいずれかで実現できます。「アンダーフロー手順」により、研究者はEV吸着の動態とレベルを追跡できます。裸の金基板上でのこのアプローチは、化学層界面が分析方法に干渉する可能性がある場合(例えば、ラマン分光法の場合)に採用される。
「C11/C16」と呼ばれる化学的に機能化されたバイオチップは、EVサンプルのグローバルビューを持つことを目的としている場合、チオレートと第一級アミド結合を形成することにより、金表面に共有結合したEVの高密度で堅牢な「カーペット」を作成するために使用されます。実際、この場合、金はメルカプト-1-ウンデカノール(11-MUOH:「C11」)とメルカプト-1-ヘキサデカン酸(16-MHA:「C16」)のチオレート混合物によって官能化され、チオレートの一部が化学的に活性化されてターゲットとの共有結合が確立されます。繰り返しになりますが、この戦略は、受動的(「マクロアレイ」またはマイクロピペットスポッターを使用した複数のマイクロアレイのいずれかでインキュベーションしてからすすぎのステップ)または流速下(SPRi)のいずれかで実現でき、金表面へのEVグラフトの動態とレベルを追跡します。
「リガンド」と呼ばれるリガンド 生体機能化バイオチップ は、化学的に活性化され、異なるリガンド(抗体、受容体など)を共有結合的に移植し、生物学的サンプル中に共存する異なるEVサブセットを選択的に(親和性で)捕捉します。
Access restricted. Please log in or start a trial to view this content.
プロトコル
1.金基板の準備
注:金チップ上には、1)裸の表面、2)化学的に機能化、3)生体機能化(C11C16層にグラフトされた配位子)の3種類の表面が生成されます。これ以降、それぞれ「ネイキッド」、「C11C16」、「リガンド」と呼ばれます。
- 金基板の準備:
注:このプロトコルでは、金バイオチップはクリーンルームで社内で製造されました。自家製のバイオチップは、クロム(2 nm Cr)と金(48 nm Au)のコーティングを施したスライドガラス(SF11)で構成されていました。バイオチップの長さは28mm、幅は12.5mm、厚さは0.5mm20であった。- DCマグネトロンスパッタリング15 を使用して、物理蒸着(PVD)によってスライドをコーティングします。
- 化学機能化:
- 裸のチップを、メルカプト-1-ウンデカノール(11-MUOH:C11)とメルカプト-1-ヘキサデカン酸(16-MHA:C16)の90%/10%モル混合物中で、無水エタノール中で1 mM、攪拌下、室温で一晩インキュベートすることにより、機能化します。
注:このステップは、リガンドのグラフト化に役立つ安定した自己組織化単分子膜(SAM)を形成します。 - バイオチップを無水エタノールと超純水で洗浄(やさしく洗浄)し、窒素乾燥後、クリーンルーム状態で保管してください。
- 化学的に機能化されたバイオチップの活性化:
注:このステップ以降、実験は分析ラボで実行する必要があります。- バイオチップを超純水でやさしく洗浄し、穏やかな気流下でチップを乾燥させます。C16 カルボキシ基を活性化するには、200 mM エチル (ジメチルアミノプロピル) カルボジイミド/N-ヒドロキシスクシンイミド (EDC) と 50 mmol/L N-ヒドロキシスクシンイミド (Sulfo-NHS) の混合物中で、室温の暗所で少なくとも 30 分間、バイオチップをインキュベートします。その後、接ぎ木実験の前に水ですすいでください。
- 裸のチップを、メルカプト-1-ウンデカノール(11-MUOH:C11)とメルカプト-1-ヘキサデカン酸(16-MHA:C16)の90%/10%モル混合物中で、無水エタノール中で1 mM、攪拌下、室温で一晩インキュベートすることにより、機能化します。
- 機能化されたバイオチップへのリガンドのグラフト化:
注:チップ上のリガンド(または一部の実験ではEV)の固定化は、SPRi装置の外部で受動的に行うか(活性化チップ上の液滴のインキュベーション)、またはSPRi装置への動的にアンダーフローして行うことができます。これは、 EVまたは リガンド修飾チップを構成します。 図2 は、金バイオチップ、マイクロピペットスポッター、およびそれぞれ300 nLの16個のリガンド液滴でスポットした後のバイオチップを示しています。- リガンド移植には、抗体(免疫グロブリン-抗CD41[ N-PEVと呼ばれる天然血小板由来のEVに特異的]、抗CD61、抗CD62P、抗CD9、および抗OVA[オボアルブミンに対する対照抗体])およびアネキシンVなどの分子を使用します。 酸性溶液(リガンドの活性または機能に最適なpHに応じてpH 4.5から6)で200 μg/mLで希釈します。
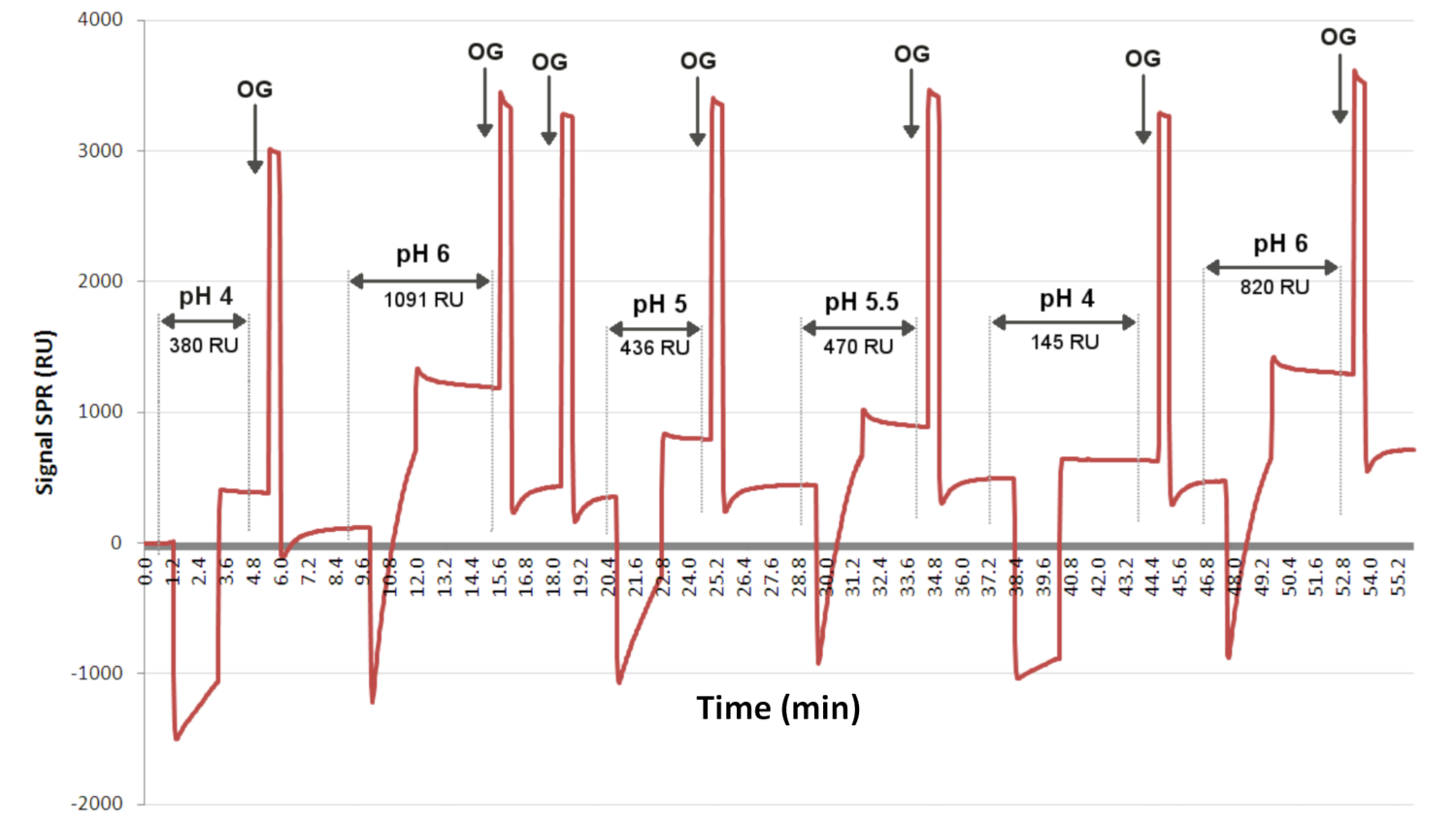
注:抗体のグラフトに最適なpHは、リガンドグラフトに最適なpHを決定するためにSPR装置で実施した予備濃縮実験によって以前に決定されました( 図3を参照)。使用する抗体のクローンによって移植条件が変化するため、SPRi実験に進む前にこれらの条件を決定することをお勧めします。 - グラフト化手順では、スポッターを使用して300 nLのEV/リガンド溶液を追加します。
注意: 水に浸した紙は、液滴の蒸発を防ぐために、左右両方のウェルに保管する必要があります。このステップは、EV/リガンドを安定性と機能性のために最適な状態に維持するために重要です。 - スポッティング後、バイオチップをソニックバス(周波数:37 kHz、電力:30%)の下に置き、30分間インキュベートします。バイオチップを上から超純水で洗浄し、バイオチップと同じ屈折率(RI)のプリズムにそっと置きます。プリズム上部のバイオチップを調整しながら、プリズムと同じRIのオイルを液滴(~2.3μL)加え、バイオチップとプリズムの間に均一で薄い層を作ります。
メモ: この手順により、光路内に同じRIの連続媒体が確保されます。このステップでは、気泡を油層に取り込まないようにすることが重要です, これは経路の光学特性を変化させ、さらなる分析を妨げるため.
- リガンド移植には、抗体(免疫グロブリン-抗CD41[ N-PEVと呼ばれる天然血小板由来のEVに特異的]、抗CD61、抗CD62P、抗CD9、および抗OVA[オボアルブミンに対する対照抗体])およびアネキシンVなどの分子を使用します。 酸性溶液(リガンドの活性または機能に最適なpHに応じてpH 4.5から6)で200 μg/mLで希釈します。

図2:バイオチップと手動スポッター。 金バイオチップ(左)、マイクロピペットスポッター(中央)、およびそれぞれ300nLのリガンド液滴でスポッティングした後のバイオチップ(右)。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図3:リガンドグラフトに最適なpHを決定するための予備濃縮試験。 センサーグラムは、相互作用のレベルを、表面に同じ濃度で2分間にわたってランダムに(異なるpH値で)注入された1つのリガンドの時間の関数として示します。OGは洗剤であり、各注射の間にベースラインを回収することができます。ここで、センサーグラムは、pH 6が最も多くのリガンドグラフトを可能にし、1091RUのSPRiシグナルを有することを示している。略語:OG =オクチルグルコシド;RU = 応答単位。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
2. 表面プラズモン共鳴イメージング
- バイオチップをSPRiシステムにマウントします。PBSバッファー(ランニングバッファー)の 流速 を 50 μL/minに保ちます。
注:気泡がある場合は、流量を 500〜1,000 μL / minまで増やし、40 mMオクチルグルコシド(OG)を頻繁に注入して、できるだけ早く除去してください。 - SPRi装置の金バイオチップのコンディショニング:作動角度の選択
- ソフトウェアの左側にあるドロップダウンメニューをクリックし、 作業ディレクトリ をクリックして、実験データを保存するフォルダーを定義します。その後、プラ ズモン|画像取得。
- さまざまなスポットが表示されている画像(図4Aを参照)を見つけ、クリックしてこの画像を選択します。図4に示すように、関心領域(ROI)を定義するには、スポット内の自動検出または手動定義(図4B)をクリックするか、チップをクリックしてスポットがなくても特定の場所を参照します(図4C)。
- マルチプレックスバイオチップを使用する場合は、リガンドファミリーの名前を書き、対応するスポットをクリックします。
注:リガンドファミリーは、同じリガンドで官能化されたいくつかのスポットに対応します。通常、チップは各リガンドの少なくとも重複、さらにはしばしば三重を示す。 - 終了種定義をクリックし、得られたプラズモン曲線を含むウィンドウに移動するのを待ちます。
- 作業角度を選択します。カーソルで黒い線を最適な作業角度にドラッグし、[ミラーを作業角度に移動]をクリックします。
注:プラズモン曲線は反射率(%)対角度の値で構成され、ソフトウェアは角度に対する傾き(%)の値を含む別の曲線を提供します。適切な作業角度を選択するには、勾配の値が最も高い角度を選択します。- 装置内部で行われる不動態化工程(アルブミンのため)の場合、表面に対して最適な感度を得るために 作動角度 を選択し、表面反応性の品質管理を確立します。
注:このパッシベーションステップは、サンプルとバイオチップ表面間の非特異的相互作用を減らすために、チップがアフィニティ/キャプチャバイオ検出用に準備されている場合に重要です。
- 装置内部で行われる不動態化工程(アルブミンのため)の場合、表面に対して最適な感度を得るために 作動角度 を選択し、表面反応性の品質管理を確立します。
- [キネティクス]をクリックして、リアルタイムの キネ ティックモニタリングを開始します。ソフトウェアがユーザーにネガティブコントロールを定義するように促したら、この時点でネガティブコントロール を選択しません (これにより、ネガティブコントロールスポットの動力学を観察できるため)。
- ラット血清アルブミン(RSA、200 μg/mL、酢酸バッファー、pH 4.5で調製)を 50 μL/minで4分間 注入して、スポットの周囲の表面を不動態化し、場合によってはリガンドスポット内の空きスペースを埋めます。
注:RSAは、リガンドに結合していないバイオチップを覆うように注入されます。 - エタノールアミン(1 M)を20 μL/minで10分間注入し、表面にまだ存在し反応性のあるカルボキシル基を失活させます。
- バイオチップを洗浄するには、 40 mM OG を 50 μL/min で 4 分間注入します。
注意: パッシベーションステップの後、作業角度は(サンプル注入前の新しいベースライン決定として)調整され、関心のあるスポットで最高の感度になります。
- ラット血清アルブミン(RSA、200 μg/mL、酢酸バッファー、pH 4.5で調製)を 50 μL/minで4分間 注入して、スポットの周囲の表面を不動態化し、場合によってはリガンドスポット内の空きスペースを埋めます。
- サンプル注入:
- パッシベーション後のプラズモン曲線を再定義し、今度は配位子に応じて作動角を選択します。
- キネティックモニタリングでは、流量を 20 μL/minに減らし、 ベースラインが安定するのを待ちます。
- 選択した濃度(EVとグラフトリガンド間の親和性に応じて1 x 10 8 EVs/mLから1 x10 10 EVs/mL)でサンプルを注入し、手動注入または自動注入のいずれかをクリックします。手動注入の場合、200 μLのサンプルを注入した後、注入の停止をクリックします。注入の持続時間は一般に10分であるため、相互作用の速度論に従い始め、速度論モニタリング中に観察される注入の開始と終了の間の反射率の差を計算することによって反射率の変化を測定します(図5)。
注:注入されたさまざまなサンプルは、代表的な結果のセクションで説明されています。
- サンプル注入後、次の2つの方法のいずれかに従ってSPRi実験を終了します。
- 非固定/液中アプローチでは、バイオチップをSPRi装置から取り出し、その上に液滴を追加して、液体中の表面のさらなるAFM特性評価に進みます。
- 固定アプローチでは、水で希釈したグルタルアルデヒド(0.5%)を20 μL/minで10分間注入し、バイオチップに捕捉された分子を固定します。水を注入して表面をすすぎ、バイオチップを取り出し、蒸留水で非常に穏やかに洗浄し、風乾してAFM下でさらに分析します。

図4:バイオチップのSPRi CCD画像。 (A,B)アルブミンパッシベーション後の多重バイオチップ。(A)デフォルトのないチップ。(B)チップに現れたいくつかの欠陥:スポットの融合(i)、弱いグラフト(ii)、またはほこりまたは「汚染物質」(iii)。スポット内の色(リガンドファミリーごとに1色)のROIは、これらの「汚染物質」を回避するために選択されました。スポットが合流すると、それらは記録され、無視されるか、「リガンド1と2の混合物」と命名されました。(C)EVの金への吸着を調べる実験のためのマイクロアレイなしの裸の金チップ。青い矢印はフロー方向を示します。このチップはスポットを示さず、サンプル注入中にライン1(L1、赤い円)からライン4(L4、紫色の円)までの反射率信号を登録するためにROIが選択されました。スケールバー = 3 つの画像すべてで 1 mm。略語:SPRi =表面プラズモン共鳴イメージング;CCD = 電荷結合素子;ROI = 関心領域。EV =細胞外小胞。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

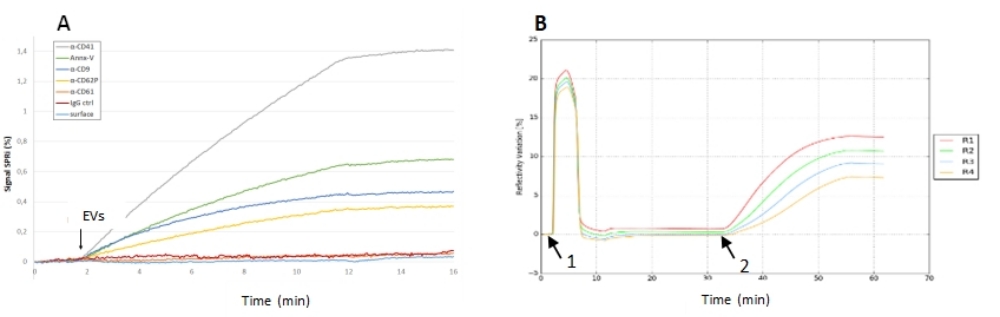
図5:バイオチップへのEV注入のSPRi実験 。 (A)異なるリガンドの反射率シグナルを示す多重バイオチップでのキャプチャ実験。ここでは、陰性対照の応答が無視できたため、異なるリガンドの信号対雑音比は非常に良好でした(特に抗CD41スポットで)。(B)裸のバイオチップへのEVの吸着実験。バッファとOGクリーニングの2回のフラッシュ(1)、EVサンプル注入(2)、およびEV相互作用後の反射率信号(3)によるチップのコンディショニングを示すセンサーグラム。このバイオチップでは、ネガティブコントロールはありませんでしたが、反射率信号(その動力学、注入後の安定性)は高く、これらのEVは金チップ上で吸着して安定を維持することができました。略語:EV =細胞外小胞;OG =オクチルグルコシド。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
3. 原子間力顕微鏡
- 接触モードを使用して空気中のバイオチップをスキャンし、定量イメージングモードを使用して液体状態でバイオチップをスキャンします。
- スライドガラス(ラボで開発)のマスクの上部にバイオチップを合わせ、バイオチップ上のそれぞれのマイクロスポットの位置を特定します(図6A)。
注:マスクには、使用するスポッターに応じたリガンドのスポットの位置に対応するスポットのマーキングが含まれています。このマスクは、2つの垂直ウェッジがチップの配置を可能にするスライドガラスで構成されています。さらに、スライドガラスには、チップ上のスポット局在化に対応する16個のフェルトドットがマークされており、透明化によってスポットの位置を特定し、目的の領域をスキャンすることができます。 - 先端の位置:
- AFMの上部にあるCCDカメラを使用して、スキャンする必要のある正しい場所にカンチレバーの位置を特定します。このプロトコルに従うには、長さ200 μm、幅28 μm、ばね定数0.08 N / mの三角形カンチレバーを使用します。
- カンチレバーの上部にあるレーザーを、フィードバック制御メカニズムで最適な応答が得られる位置に合わせます。
- スキャニング:
- バイオチップ表面に接触したら、 接触モード または 定量イメージングモードで AFM取得を3〜5つの大きな領域(通常は10 × 10 μm²)から小さな領域(1 x 1 μm²)に開始します。
メモ: スキャンできるさまざまな領域を 図 6D に示します。AFMの特性評価がmm²スポット全体を表していること、および堅牢な分析(各条件に対して最低300台のEVをカウントおよび分析)に適した解像度で十分なEVが視覚化されていることを確認し、計測および形態測定を実行します。
- バイオチップ表面に接触したら、 接触モード または 定量イメージングモードで AFM取得を3〜5つの大きな領域(通常は10 × 10 μm²)から小さな領域(1 x 1 μm²)に開始します。
- AFM画像処理:
- 最初に 高さチャンネルを選択して、JPKデータ処理ソフトウェアでAFM画像を処理します。
- 各線から減算する 多項式フィット を選択して、直線化されたスキャン線を取得します。
- 金粒子 の高さのしきい値 を選択して、表面の粗さを排除します。参照されているソフトウェア( 材料表を参照)の粒子抽出モジュール内で、8.5 nmの高さしきい値を使用して粒子をマークします。グレインが フィルタリングされると、 グレインの数 が表示されます。
注:通常、粗い金基板(RMS は~3 nm)と化学層とリガンド層の存在により、 しきい値 を 8.5 nmに設定する必要があります。 - 高さ、体積、直径など、マークされた粒子のいくつかのプロパティを抽出するには、参照されているソフトウェアで画像を開き、データ処理|穀物 |しきい値でマークします。
- プロパティの機能でフィルターグレインを選択します。新しいウィンドウが表示されたら、次のパラメータを選択します:値 = 最大 z-max、サーフェス = 投影サーフェス A0。 次に、基準 A と B を選択します。
- オープン グレイン分布;表示されるウィンドウで、[ 値 (最大)]、[ ボリューム (ベース: ゼロ)]、および [ 境界 (長さ)] を選択します。表示されるテーブル(.txt形式)には、画像ごとに設定されたしきい値で検出されたすべての粒子 の高さ、体積、 および直径 の値を含む3つの列が表示されます。
- 高さhと直径Dから、式(1)8を使用して各EVの曲率半径Rcを計算し、次に式(2)を使用して体積Vを計算します。
 (1)
(1) (2)
(2) - Vから、式(3)を使用して、各EV(同じ体積の球の直径)の有効直径deffを計算します。
 (3)
(3) - EVのサイズ(測定された高さ、測定された直径、および計算された有効直径)を示すグラフをプロットし、カウントされた各粒子をドットで表します。
注:したがって、特性評価の最後に、NBAプラットフォームは、バイオ検出信号と表現型とEVサブセットの数とサイズの相関を可能にします。

図6:AFMによるバイオチップの特性評価。 SPRi実験後、チップを固定して乾燥させるか、AFM特性評価のために液体に保持しました。(A)機械加工されたスライドガラス(2つの垂直位置決めウェッジ付き、写真に「w」で示されている)は、16個のバイオチップマイクロアレイの局在化を伴うマスクフィッティングを示しています。光露光と透明性により、AFMの特性評価のために設置されると、スライドガラスはAFMチップを目的の場所に配置して特性評価することができます。(B)液体状態でスキャンするために、「マスク」スライド上および一滴の緩衝液の下に設置されたバイオチップ。(C)16個のマイクロアレイのSPRi画像。(D)直径920nmの生体機能化キャリブレーションナノ粒子の免疫捕捉後に光学顕微鏡で画像化された1つのマイクロアレイ。白い四角は、AFMの特性評価を堅牢にするために、関心のある各スポットでAFMによってスキャンされたさまざまな領域のサンプリングを示しています。スケールバー=(C)1ミリメートル、(D)500μm。略語:AFM =原子間力顕微鏡;SPRi = 表面プラズモン共鳴イメージング。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
4. ラマン分光法
注:ラマン分光法では、基板として使用したスライドガラスを、ラマンの特徴が無視できるCaF2のスライドに置き換えます。
- 取得のための光学条件:
- ラマンイメージング顕微鏡の条件を設定します:顕微鏡対物レンズ:50倍、レーザー波長:532 nm、レーザー出力:10 mW、露光時間:500 ms、蓄積数:140、スペクトル範囲:450 cm-1から3,200 cm-1。
注:パワーを使いすぎたり、取得時間が長すぎたりすると、時間の経過とともに不安定なスペクトルによって証明されるように、サンプルが損傷する可能性があります。少量のエネルギーから始めて、信号が弱すぎる場合はこれを増やします。より高いレーザー波長(633 nm、785 nm)を使用して、ラマン測定に有害な蛍光を低減することができます。ただし、強度は波長の4乗とともに減少するため、カメラのスペクトル感度を考慮する必要があります。
- ラマンイメージング顕微鏡の条件を設定します:顕微鏡対物レンズ:50倍、レーザー波長:532 nm、レーザー出力:10 mW、露光時間:500 ms、蓄積数:140、スペクトル範囲:450 cm-1から3,200 cm-1。
- ラマンイメージング:
- まず、蓄積数を減らしたライブスペクトル(10)を観察して、信号対雑音比が最適な領域を見つけます。
注:高周波領域(2,800〜3,000 cm-1)の強い信号は、前に示したように、短い露光時間で表面上のEVの検出を容易にすることができます10。 - ROIを選択したら、取得に利用可能な時間に応じて空間分解能を選択します。
注:空間分解能は回折限界(~500 nm)によって制限されます。 - ラマンマッピングの取得を開始します。
- まず、蓄積数を減らしたライブスペクトル(10)を観察して、信号対雑音比が最適な領域を見つけます。
- データの前処理:
- Python統合開発環境(IDE)(Spyderなど)を使用して、スペクトルを含むファイルを開きます。
- スペクトルのベースラインを差し引いて、蛍光の可能性による干渉を補正します。たとえば、パッケージ "irfpy"23 の "arpls" 関数を使用します。パラメータ「lam」のさまざまな値をテストして、最適なベースライン補正を提供する値を見つけます(通常は103 から107の間)。
- たとえば、スペクトルのすべての強度を2,900 cm-1 の強度で割るか、スペクトルの平均を減算してから標準偏差で割ることによって、スペクトルを正規化します(「標準正規変量」正規化)。
注:この手順は、EVの相対的な分子組成を比較するために必要です。
Access restricted. Please log in or start a trial to view this content.
結果
リガンドグラフトに最適なpH条件の決定
バイオチップの調製に使用されるさまざまなリガンドは、pHとチオレート化学層と相互作用するそれらの可用性の関数としてテストされます(図3)。リガンドは、異なるpH値で酢酸塩バッファーで希釈され、C11C16層で化学的に機能化されたバイオチップに注入されます。溶液を表面にランダムに注入し、各リガンド注?...
Access restricted. Please log in or start a trial to view this content.
ディスカッション
最近最も広く使用されているEV同定法は、EVの起源を確認するためのタンパク質特異的イムノブロッティング、その構造を確認するためのTEM、およびサンプル量中の数とサイズ分布を定量するNTAです3。それにもかかわらず、(生物)医学研究におけるEVへの高い関心と既存の分析ツールの限界により、科学界はEVの特性評価、識別、および定量化のための新しい方法を開発する?...
Access restricted. Please log in or start a trial to view this content.
開示事項
著者は開示する利益相反を持っていません。
謝辞
IVEThコア施設(パリ)のケリー・オベルタンとファビアン・ピコは、ラマンイメージング実験で認められています。Thierry Burnouf(台北医科大学、台湾)とZuzana Krupova(フランス、ヘリンクール出身)は、それぞれ血小板と牛乳のサンプルに由来するEVサンプルを提供したことで認められています。この作業は、ブルゴーニュフランシュコンテ地域とEUR EIPHI大学院(BEGINNERプロジェクト、2021-2024)によってサポートされました。この作業の一部は、CLIPPプラットフォームとFEMTO-ENGINEERINGのRENATECHクリーンルーム施設を使用して行われ、Rabah Zeggariに感謝します。
Access restricted. Please log in or start a trial to view this content.
資料
| Name | Company | Catalog Number | Comments |
| CD41a antibody | Diaclone SAS (France) | 447528 | |
| CP920 | Microparticles GmbH, Germany | 448303 | |
| DXR3xi | Thermo Fisher Scientific | T1502 | |
| EDC | Sigma | A6272 | |
| Ethanolamine | Sigma | P5368-10PAK | |
| Evs derived from platelet concentrates | Collaboration : Pr T. Burnouf (TMU, Taipei) | S2889 | |
| Evs from bovine milk | Collaboration : Dr Z. Krupova (Excilone, Helincourt - France) | 3450 | |
| Glutaraldehyde | Sigma | 56845 | |
| Gwyddion | 853.223.020 | ||
| Magnetron sputtering | PLASSYS | SAB5300165 | |
| mercapto-1-hexadecanoic acid | Sigma | G5882 | |
| Mercapto-1-undecanol | Sigma | O8001 | |
| Mountains SPIP ones | Digital Surf | ||
| NanoWizard 3 Bioscience | Bruker-JPK | ||
| Octyl Glucoside (OG) | Sigma | ||
| Ovalbumine antibody | Sigma | ||
| Phosphate Buffer Saline (PBS) | Sigma | ||
| Rat Albumin Serum (RSA) | Sigma | ||
| Sodium acetate buffer | Sigma | ||
| SPR-Biacore 3000 | GE Healthcare/ Cytiva life sciences | ||
| SPRi Biochip | MIMENTO technology platform | The biochips were produced in-house in the clean room, Besancon | |
| SPRi Plex II | Horiba Scientific | ||
| Sulfo-NHS | Sigma |
参考文献
- Silva, A. K. A., et al. Development of extracellular vesicle-based medicinal products: A position paper of the group "Extracellular Vesicle translatiOn to clinicaL perspectiVEs - EVOLVE France". Advanced Drug Delivery Reviews. 179, 114001(2021).
- Xunian, Z., Kalluri, R. Biology and therapeutic potential of mesenchymal stem cell-derived exosomes. Cancer Science. 111 (9), 3100-3110 (2020).
- Hartjes, T. A., et al. Extracellular vesicle quantification and characterization: Common methods and emerging approaches. Bioengineering. 6 (1), 7(2019).
- Xing, Y., et al. Analysis of extracellular vesicles as emerging theranostic nanoplatforms. Coordination Chemistry Reviews. 424, 213506(2020).
- Wang, T., Xing, Y., Cheng, Z., Yu, F. Analysis of single extracellular vesicles for biomedical applications with especial emphasis on cancer investigations. Trends in Analytical Chemistry. 152, 116604(2022).
- Boireau, W., Elie-Caille, C. Extracellular vesicles: Definition, isolation and characterization. Medecine Sciences: M/S. 37 (12), 1092-1100 (2021).
- Brisson, A. R., et al. Extracellular vesicles from activated platelets: A semiquantitative cryo-electron microscopy and immuno-gold labeling study. Platelets. 28 (3), 263-271 (2017).
- Yuana, Y., et al. Atomic force microscopy: A novel approach to the detection of nanosized blood microparticles. Journal of Thrombosis and Haemostasis. 8 (2), 315-323 (2010).
- Sebaihi, N., de Boeck, B., Yuana, Y., Nieuwland, R., Pétry, J. Dimensional characterization of extracellular vesicles using atomic force microscopy. Measurement Science and Technology. 28 (3), 034006(2017).
- Beekman, P., et al. Immuno-capture of extracellular vesicles for individual multi-modal characterization using AFM, SEM and Raman spectroscopy. Lab on a Chip. 19 (15), 2526-2536 (2019).
- Malenica, M., et al. Perspectives of microscopy methods for morphology characterisation of extracellular vesicles from human biofluids. Biomedicines. 9 (6), 603(2021).
- Verweij, F. J., et al. The power of imaging to understand extracellular vesicle biology in vivo. Nature Methods. 18 (9), 1013-1026 (2021).
- Théry, C., et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles. 7 (1), 1535750(2018).
- Obeid, S., et al. NanoBioAnalytical characterization of extracellular vesicles in 75-nm nanofiltered human plasma for transfusion: A tool to improve transfusion safety. Nanomedicine: Nanotechnology, Biology, and Medicine. 20, 101977(2019).
- Obeid, S., et al. Development of a NanoBioAnalytical platform for «on-chip» qualification and quantification of platelet-derived microparticles. Biosensors and Bioelectronics. 93, 250-259 (2017).
- Ridolfi, A., et al. AFM-based high-throughput nanomechanical screening of single extracellular vesicles. Analytical Chemistry. 92 (15), 10274-10282 (2020).
- Vorselen, D., et al. The fluid membrane determines mechanics of erythrocyte extracellular vesicles and is softened in hereditary spherocytosis. Nature Communications. 9 (1), 4960(2018).
- Hardij, J., et al. Characterisation of tissue factor bearing extracellular vesicles with AFM: Comparison of air-tapping-mode AFM and liquid Peak Force AFM. Journal of Extracellular Vesicles. 2, 21045(2013).
- Jorgensen, M., et al. Extracellular Vesicle (EV) Array: Microarray capturing of exosomes and other extracellular vesicles for multiplexed phenotyping. Journal of Extracellular Vesicles. 2, 20920(2013).
- Remy-Martin, F., et al. Surface plasmon resonance imaging in arrays coupled with mass spectrometry (SUPRA-MS): Proof of concept of on-chip characterization of a potential breast cancer marker in human plasma. Analytical and Bioanalytical Chemistry. 404 (2), 423-432 (2012).
- Czamara, K., et al. Raman spectroscopy of lipids: A review. Journal of Raman Spectroscopy. 46 (1), 4-20 (2015).
- Penders, J., et al. Single particle automated Raman trapping analysis of breast cancer cell-derived extracellular vesicles as cancer biomarkers. ACS Nano. 15 (11), 18192-18205 (2021).
- Baek, S. J., Park, A., Ahn, Y. J., Choo, J. Baseline correction using asymmetrically reweighted penalized least squares smoothing. Analyst. 140 (1), 250-257 (2015).
- Daaboul, G. G., et al. Digital detection of exosomes by interferometric imaging. Scientific Reports. 6, 37246(2016).
- Ertsgaard, C. T., et al. Integrated nanogap platform for sub-volt dielectrophoretic trapping and real-time Raman imaging of biological nanoparticles. Nano Letters. 18 (9), 5946-5953 (2018).
- Maas, S. L., et al. Possibilities and limitations of current technologies for quantification of biological extracellular vesicles and synthetic mimics. Journal of Controlled Release. 200, 87-96 (2015).
Access restricted. Please log in or start a trial to view this content.
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved