
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
3D細胞培養のための微多孔性アニール粒子足場における粒子分率の制御(英語)
要約
粒状足場内の粒子画分のばらつきを最小限に抑えることで、再現性のある実験が容易になります。この研究では、 in vitro 組織工学アプリケーション向けに、制御された粒子画分を使用して粒状足場を生成する方法について説明します。
要約
ミクロゲルは、微多孔性アニール粒子(MAP)足場の構成要素であり、in vitro 細胞培養と in vivo 組織修復の両方のプラットフォームとして機能します。これらの顆粒状足場では、ミクロゲル間の空隙空間によって生成される生来の多孔性により、細胞の浸潤と遊走が可能になります。空隙率は細胞の生理活性の手がかりであるため、空隙率と粒子画分を制御することはMAP足場の設計にとって重要です。球状ミクロゲルは、制御されたサイズおよび形状のためにマイクロ流体デバイス上で生成し、続いてポリマーネットワークの破砕を防止する方法を用いて凍結乾燥することができる。再水和すると、凍結乾燥されたミクロゲルはMAP足場に制御された粒子画分をもたらす。ミクロゲル凍結乾燥のためのこれらの方法の実施は、高分子拡散および細胞拡散に対する粒子画分の効果を示す再現性のある研究につながった。次のプロトコルは、MAP足場内の粒子画分を制御するためのミクロゲルの製造、凍結乾燥、および再水和、およびin vitroでの3D細胞培養のためのバイオ直交架橋によるミクロゲルのアニーリングをカバーします。
概要
微多孔性アニール粒子(MAP)足場は、ミクロゲル(μgel)ビルディングブロックが相互結合してバルクの多孔質足場を形成する粒状材料のサブクラスです。これらの顆粒状スキャフォールドのユニークなマイクロアーキテクチャにより、連結された球状ミクロゲル間の空隙空間によって生成される生来の多孔性は、加速された細胞浸潤と遊走をサポートします1。MAP足場のミクロゲルビルディングブロックは、化学修飾された合成ポリマーと天然ポリマーの両方から製造できます2。ここで説明する方法は、官能性ノルボルネン(NB)ハンドルで修飾されたヒアルロン酸(HA)骨格からなるミクロゲルの使用を特に強調している。HAポリマーのNB機能ハンドルは、ミクロゲルを形成し、それらを結合してMAP足場を生成するためのクリック化学反応をサポートします3,4。ミクロゲルを一緒に連結(すなわち、アニーリング)するために、酵素1、光ベースの5,6、および無添加クリックケミストリー3,7反応などの多数のスキームが採用されている。この研究では、HA-NBミクロゲルを相互結合するためのテトラジン-ノルボルネン逆電子要求ディールス-アルダー共役を使用して、無添加クリック化学について説明します。
MAPスキャフォールドを製造するために、ユーザーはまず、バッチシステムまたはマイクロ流体デバイス内で逆エマルジョンを使用し、電気流体力学的スプレー、リソグラフィー、または機械的フラグメンテーションを使用してミクロゲルビルディングブロックを生成します2。球状HA−NBミクロゲルの製造は、バッチエマルジョン2およびマイクロ流体液滴生成技術の両方を用いて十分に記載され、以前に報告されている8、9、10、11。この研究では、球状HA-NBミクロゲルを、前述のように、制御されたサイズおよび形状のためのフロー集束マイクロ流体プラットフォーム上に生成した8,9,10。精製後、ミクロゲルは水性懸濁液中に存在し、詰まった状態を誘発するために濃縮されなければならない。詰まると、ミクロゲルはせん断減粘特性を示し、注射可能な空間充填材料として機能することができます1。詰まった状態を誘発する1つの方法は、凍結乾燥、または凍結乾燥を介してミクロゲルを乾燥させ、続いて乾燥生成物を制御された容量12で再水和することである。あるいは、過剰な緩衝液は、ストレーナー上での遠心分離によって、または吸引または吸収材料を使用してミクロゲルペレットから緩衝液を手動で除去することによって、ミクロゲルスラリーから除去することができる。しかしながら、ミクロゲルを乾燥させるために遠心分離を使用すると、顆粒足場を作製するときに非常に可変な範囲の粒子画分および空隙画分を生成することができる12。ミクロゲルを凍結乾燥するための技術は、ポリエチレングリコール(PEG)ミクロゲル13のための70%IPA、ゼラチンメタクリロイル(GelMa)ミクロゲル14のためのフッ素化油、およびHAミクロゲル12のための70%エタノールを用いて記載されている。このプロトコルは、乾燥プロセス中に元のミクロゲルの特性を保持するために、標準的な実験室用試薬である70%エタノールを使用して球状HAミクロゲルを凍結乾燥する方法を強調しています。凍結乾燥HAミクロゲルは、MAP足場12における最終粒子画分を制御するために、ユーザー定義の重量百分率で秤量および再水和することができる。
MAP足場形成の最終ステップは、ミクロゲルをアニーリングしてバルクの多孔質足場1を作成することに依存しています。MAPスキャフォールドは、天然の細胞外マトリックス成分を利用し、バイオ直交アニーリングスキームを採用することにより、in vitro 細胞培養と in vivo 組織修復の両方のための生体適合性プラットフォームとして機能します3。これらのアプローチにより、MAP足場は、組織工学アプリケーションで使用するために、ユーザー定義の粒子画分を備えたHA-NBビルディングブロックから製造できます12。以下のプロトコルは、MAP足場における粒子画分を制御するための凍結乾燥および再水和に続くHA-NBミクロゲルのマイクロ流体製造について説明する。最後に、ミクロゲルをアニーリングするためのステップは、 in vitro 3D細胞培養実験のためのバイオ直交化学を使用して説明されています。
プロトコル
1. マイクロ流体デバイスの作製
- ソフトリソグラフィ
注:このプロトコルは、de Wilsonらによるフローフォーカシングマイクロ流体デバイス設計のデバイス製造について説明しています9。ただし、このプロトコルは、SU-8ウェーハ上の任意のデバイス設計で使用できます。ウェーハはペトリ皿にテープで留めることができ、次いで、PDMSのウェーハ特徴15への付着を防ぐためにシラン化する必要がある。- ポリジメチルシロキサン(PDMS)エラストマーベースと硬化剤( 材料表を参照)を10:1の比率で混合します。ウェーハを~5mmのPDMSで覆うために約100gを準備します。PDMS混合物をウェーハに注ぎ、デシケーターで約30分間脱気します。すべての気泡がなくなったら、60°Cのオーブンに少なくとも2時間入れてPDMSを硬化させます。
- ナイフを使用して、ウェーハにひびが入ることなくデバイスのパラメーターの周りをそっとトレースします。次に、PDMSをウェーハから慎重に剥がします。1 mmの生検パンチ( 材料表を参照)を使用して、入口と出口のチャネルを作成します。
注意: マイクロ流体デバイスを打ち抜くときは優しくしてください。入口または出口チャネルの周りの裂け目または裂け目は、ミクロゲル製造中に漏れを引き起こす可能性があります。 - テープを使用して、機能側のデバイスからほこりを取り除きます。デバイスときれいなスライドガラスを135°Cのホットプレートに少なくとも15分間置き、水分を取り除きます。
- ヒュームフード内で、スライドガラスとデバイス(機能側が露出している)の両方の高い位置にコロナプラズマガン( 材料の表を参照)を約30秒間使用し、次にそれらをすばやく接着します。デバイスとスライドガラスの間の良好なシールを確保するために、静かに圧力をかけます。デバイスを60°Cのオーブンに一晩置き、接着を固定します。
2. ノルボルネン(NB)機能性ハンドルを用いたヒアルロン酸(HA)ミクロゲルのマイクロ流体製造
- HA-NB合成
注:HA-ノルボルネン(HA-NB)合成は、4-(4,6-ジメトキシ-1,3,5-トリアジン-2-イル)-4-メチルモルホリニウムクロリド(DMTMM)から5-ノルボルネン-2-メチルアミン(NMA)へのモル当量1:1.5:2.5の79 kDaのHAナトリウムを用いて、Darlingら3 から適応されました。- 反応物の重さを量ります。20 mg/mLのHAを200 mM MESバッファー(pH ~6)に溶解し、攪拌プレート上のビーカーまたはフラスコで攪拌します。溶解したら、DMTMMをHA溶液に加え、室温で約20分間反応させます。例えば、1 g HA + 1.09 g DMTMM + 845 μL NMAを使用することができます。
- NMAをHA/DMTMMソリューションにドロップワイズで追加します。蒸発を最小限に抑えるために反応容器の開口部にパラフィルムを追加し、反応容器をホイルで覆います。反応を約24時間進行させながら攪拌を続ける。
- 24時間後、200プルーフエタノール(反応量の約10倍)を冷却します。攪拌プレート上で、反応液を冷やしたエタノールに滴下してHA-NBを沈殿させ、200〜300rpmで20分間攪拌を続けます。
- 溶液を50 mLコニカルチューブに移し、5,000 x g で10分間遠心分離します。余分なエタノールを注ぎ、廃棄物として処分します。この時点で、HA-NB製品は円錐管内の白いペレットでなければなりません。デシケーターでHA-NBの掃除機を引き、一晩乾燥させます。
- 分子量12〜14 kDaのカットオフセルロース透析チューブを使用してHA-NBを精製します( 材料の表を参照)。HA-NBを2 M NaCl溶液に溶解し、透析チューブに移します。チューブを結び、クランプで固定します。充填した透析チューブを5Lの超純水の入ったバケツに移し、HA-NBを一晩水に対して透析します。
- 翌日、水を取り除き、1 M NaCl溶液と30分間交換します。NaCl溶液を取り出し、超純水で3日間透析し、毎日交換します。
- 透析した製品を0.2 μmの真空駆動フィルターでろ過し、ろ過した製品を50 mLのコニカルチューブに移します。
- 極低温容器に液体窒素を加え、HA-NBチューブを10分間瞬間凍結します。次に、鉗子で円錐形のチューブを取り外し、キャップをすばやく取り外し、ラボグレードのティッシュで覆います( 材料の表を参照)。組織を輪ゴムで固定し、凍結乾燥容器またはチャンバー( 材料の表を参照)に移し、凍結乾燥します。凍結乾燥生成物を-20°Cで保存する。
注意: 液体窒素は有害物質です。液体窒素を取り扱うときは、適切な個人用保護具を着用してください。 - HA-NBを10 mg/mLのD2Oに溶解し、プロトンNMRで分析することにより、ノルボルネン修飾を定量します(図1A)16。
- 官能化の量を決定するには、まずD2O溶媒ピークを4.8PPMに較正します。HAメチルプロトンのピーク(δ2.05)を積分し、積分を3.0に較正します。次に、δ6.33とδ6.02のペンダントノルボルネン基のピーク(ビニルプロトン、endo)を統合します。これらのピークの積分を対応する数のプロトンに正規化して、平均修飾度3を決定します。
- HA-NBミクロゲル前駆体の調製
- 50 mM HEPESバッファー(pH 7.5)を調製し、0.2 μm真空駆動フィルターを使用してバッファーを滅菌ろ過します。HEPESバッファーを使用して、フェニル(2,4,6,-トリメチルベンゾイル)ホスフィン酸リチウム(LAP)光開始剤およびトリス(2-カルボキシエチル)ホスフィン(TCEP)還元剤のそれぞれ50 mMストックを調製します。LAP溶液を光から遠ざけてください。
- 滅菌蒸留水中でジチオールリンカーおよびRGDペプチドのそれぞれ50mMストックを調製することにより、他のミクロゲル前駆体成分を調製する。HA-NBを秤量し、HEPESバッファーに溶解して10 mg/mLストックを調製します。
注:ユーザーの好みに基づいて、ミクロゲルの内部架橋に異なるジチオールリンカーを使用できます。分解性(すなわち、MMP切断性)および非分解性(ジチオスレイトールまたはDTT)リンカーの両方が 材料表に列挙されている。RGDペプチドは、MAPスキャフォールドにおける細胞接着を促進するためにミクロゲル製剤に含まれていますが、この成分を除去して等量のHEPESバッファーに置き換えることができます。 - 前駆体成分を最終濃度9.9 mM LAP、0.9375 mM TCEP(4チオール/TCEP)、2.8 mMジチオールリンカー、1 mM RGDペプチド、および3.5 wt%(w/v)HA-NBと組み合わせ、HEPESバッファーを追加して目的の最終容量に到達します。容積式ピペットを使用して前駆体をよく混合します。
- P1000ピペットを使用して、混合物全体をゆっくりと引き上げます。チップを1 mLシリンジの端に置き、チップをピペットから取り出します。シリンジプランジャーを引いて混合物をシリンジにロードし、シリンジの端に0.2 μmフィルターを追加し、新しい1.5 mLマイクロ遠心チューブにフィルターをかけます。ろ過した前駆体溶液を遠心分離して、ろ過中に生成された気泡を除去します。
- 再度、P1000ピペットを使用して、気泡が発生しないように注意しながら、ろ過された前駆体をゆっくりと引き上げます。泡がある場合は、先端を軽くたたいて泡が外れ、上に浮かびます。
- チップを1 mLシリンジの端に置き、チップをピペットから取り出します。シリンジを垂直に保ち、前駆体溶液全体がシリンジに入るまでシリンジプランジャーをゆっくりと引きます。鈍い先端の針をシリンジに追加し、前駆体を針の先端に押し込みます。光が当たらないようにシリンジをホイルで包みます。
- ミクロゲルつまみ液の調製
- 重質白色鉱油で5%v / vスパン-80を準備し、よく混ぜます。乾燥して気泡を取り除きます。界面活性剤/油混合物をホイルで包んで室温に保ちます。よく混ぜて乾かしてから使用してください。
- 5 mLシリンジを使用して、プランジャーとフィンガーホールドの間の距離がプリカーサーシリンジの距離とほぼ等しくなるまで、オイル/界面活性剤混合物を吸引します(気泡を最小限に抑えます)。注射器に鈍い針を追加し、針の先端にオイルを押し込みます。
- マイクロ流体デバイスのセットアップ
- 鈍い針を1 mLシリンジに追加し、合成疎水性処理液で満たします( 材料の表を参照)。各入口/出口に溜まるまで、溶液をマイクロ流体デバイスにゆっくりと流します。ベンチトップのデバイスで溶液を約30分間乾燥させてから、出口の真空を引き、余分な溶液を取り除きます。卓上顕微鏡でクランプでデバイスを固定します。
- 15 mLのコニカルチューブをホイルで包み、マイクロゲル収集容器として機能するチューブラックに入れます。クランプ付きのリングスタンドを使用して、UV光プローブを収集チューブの開口部に配置します。UV検出器( 材料表を参照)を使用してUV強度を測定し、20 mW / cm2 が達成されるまでプローブを動かします。後でUVライトをオフにします。
- マイクロ流体デバイスから収集容器まで届く長さでチューブを切断します。チューブの一方の端で、45°の角度を切り取ります。チューブの斜めの端を出口チャネルにそっと挿入します。
注意: チューブをマイクロ流体デバイスに挿入するときは、穏やかにしてください。入口または出口チャネルの周りの裂け目または裂け目は、ミクロゲル製造中に漏れを引き起こす可能性があります。 - 前駆体シリンジと油相シリンジの両方をデュアルシリンジポンプに固定します( 材料の表を参照)。シリンジの先端からマイクロ流体デバイスまで届く長さでさらに2本のチューブを切り取ります。各チューブの一方の端で、45°の角度を切り取ります。チューブ(鈍端)を両方のシリンジチップに慎重に固定します。
- 1 mLシリンジのポンプの設定を変更し、おおよその前駆体容量を含めます。シリンジプランジャーに十分な圧力がかかるまでポンプをゆっくりと前方に押して、オイルと前駆体の両方をチューブの端に押し込み、システムから空気を取り除きます。ステップ5に進む前に、圧力を10〜2.4.6分均等にします。
- チューブの斜めの端をマイクロ流体デバイスの入口チャネルにそっと挿入し、ミクロゲル前駆体溶液を前部入口に、ピンチングオイルを後部入口に挿入します。装置内で流れが始まり、球状のミクロゲルが流れの集束領域で形成され始めるまで、ポンプを少しずつ前方に動かします。0.4 μL/分の流量でポンプを始動し、安定するまで装置を稼働させます。必要に応じて、流量±0.1 μL/minを少しずつ調整して、ミクロゲルの生産を安定させます。
- 図1Bに示すようにミクロゲルの生産が安定したら、収集チューブを新しいチューブと交換し、UVライトをオンにします。定期的に実行をチェックして、実行期間中、ミクロゲルの生産が安定していることを確認します。

図1:ノルボルネン(NB)機能ハンドルを有するヒアルロン酸(HA)ミクロゲルのマイクロ流体生産 。 (A)酸化重水素で実施されたプロトンNMR分析によって決定されたように、HAリピートユニットの約31%がNBで正常に修飾されました。 1δ6.33およびδ6.02(ビニルプロトン、endo)、δ6.26およびδ6.23 ppm(ビニルプロトン、exo)のペンダントノルボルネンのH NMRシフトをHAメチル基δ2.05 ppmと比較し、官能基化を決定した。Anderson et al.12 より、エルゼビアの許可を得て転載。(B)HA-NBマイクロゲルの生成に用いるフロー集束マイクロ流体デバイスの概略図。(C)共焦点顕微鏡からの最大強度投影を使用して、蛍光標識されたμgelを視覚化しました(スケールバー= 500 μm)。(D)マイクロ流体セットアップでの独立した実行からのミクロゲル直径の頻度分布は、使用するデバイスに応じて、ミクロゲルサイズ~50μmまたは~100μmの制御を示しています。(E)ミクロゲル直径は、各独立した実行の平均および標準偏差として報告される。Wilsonら9からWileyの許可を得て転 載。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
3.ミクロゲルの精製と乾燥
- ミクロゲルの精製
- 洗浄バッファー中のミクロゲル洗浄バッファー(300 mM HEPES、50 mM NaCl、50 mM CaCl 2)および2%(w/v)プルロニックF-127界面活性剤溶液を調製します。0.2 μmの真空駆動フィルターを使用して溶液を滅菌します。
- ミクロゲル採取チューブ(5,000 x g)を5分間遠心分離します。滅菌フード内で、上澄み油相を注意深く吸引します。μgelsを1:1で2%プルロニックF-127界面活性剤溶液とボルテックスを組み合わせてよく混合します。遠心分離機(5,000 x g)で5分間、上清洗浄液を吸引します。
- 4倍のミクロゲル容量とボルテックスで洗浄バッファーを加えてよく混合します。混合物を遠心分離(5,000 x g)して5分間、洗浄液を吸引します。界面活性剤がシステムから除去されるまで(すなわち、気泡が残らなくなるまで)、洗浄バッファーで4〜8回の洗浄を完了します。
- HA-NBミクロゲルの蛍光標識
注:蛍光標識テトラジンの社内合成は、2つの塩基触媒によるチオール-マイケル付加反応に依存しており、これらは十分に説明されており、以前に報告されています3。この研究では、Alexa Fluor-488をテトラジンと結合させて、ノルボルネン修飾μgelの標識を行いました。凍結乾燥製品(Alexa Flour 488-Tet)をジメチルホルムアミドに1 mg/mLで溶解し、-20°Cで保存した。- μgelを蛍光標識するには、まず、1 mg/mLストックを滅菌1x PBSで1:14に希釈して、Alexa Fluor 488-Tetの作業溶液を調製します。滅菌フード内で、μgelsを作業溶液(体積で2:1)と混合する。
- 置換ピペットを使用してよく混ぜます。混合物を室温で1時間、または4°Cで一晩インキュベートします。
- 遠心分離機(5,000 x g)し、染色液を吸引します。μgelsを1x PBS(体積比1:1)で2回洗浄し、未反応のAlexa Fluor 488-Tetを除去します。
注:この時点で、蛍光標識されたμgelを共焦点顕微鏡でイメージングして、ミクロゲルのサイズを定量化することができます(図1C-E)9。ミクロゲルサイズを測定する方法は、Roosaらによって徹底的に記載されている17。
- HA-NBミクロゲルの乾燥
- 精製されたμgel(図2A)を容積式ピペットを使用してクライオセーフスクリューキャップチューブに移します。精製されたμgelsに70%エタノール50%(v/v)を加え、置換ピペットでよく混合します。5,000 x gで5分間遠心分離します。
注意: エタノールは非常に可燃性の物質です。
注:クライオセーフスクリューキャップチューブは、μgelsを添加する前に計量し、凍結乾燥後に再度計量してμgelの質量を決定することができます。これは、1 mg未満の量を使用する場合のエラーを最小限に抑えるために推奨されます。使用する前に、スケールが内部で調整または校正されていることを確認してください。 - 上澄み液を吸引し、70%エタノール(50%v / v)と交換します(図2B)。変位ピペットでよく混ぜます。4°Cで一晩インキュベートします。
注:ミクロゲルは、必要に応じて、凍結乾燥の前に4°Cで70%エタノールで保存して長期保存することができます。凍結乾燥ミクロゲルを 図2Cに示す。クライオゲル形成が望まれる場合、他の凍結乾燥媒体をこのステップで使用することができる(図2D)。 - μgelがスクリューキャップチューブの底にあることを確認するために、短時間遠心分離します。極低温容器に液体窒素を加え、μgelsのチューブを加えて瞬間凍結します。
- 5〜10分後、鉗子でμgelのチューブを取り外します。キャップをすばやく取り外し、ラボグレードのティッシュで覆います。組織を輪ゴムで固定し、凍結乾燥容器またはチャンバーに移します。
- 製造元の指示に従って、凍結乾燥機にサンプルをロードします。0.066 Torrおよび-63 °Cで凍結乾燥します。 凍結乾燥されたμgels(lyo-μgels)は室温でしっかりと密封して保管してください。
注:凍結乾燥は、すべての液体がチューブから除去され、乾燥した製品が残ると完了します。有機溶剤は、一般的な凍結乾燥システムのゴム製固定具の寿命を縮める可能性があります。
- 精製されたμgel(図2A)を容積式ピペットを使用してクライオセーフスクリューキャップチューブに移します。精製されたμgelsに70%エタノール50%(v/v)を加え、置換ピペットでよく混合します。5,000 x gで5分間遠心分離します。

図2:HA-NBミクロゲルの乾燥。 (A)水溶液中のμgelの最大強度投影(スケールバー= 100 μm)。(B)精製されたμgelは、選択した凍結乾燥培地中で体積で1:1でインキュベートし、凍結乾燥することができます。(C)乾燥ライオμゲルの最大強度投影(スケールバー= 100 μm)。(d)凍結乾燥後に再懸濁されるミクロゲル。EtOH(70%)は、凍結乾燥プロセス全体を通してμgelの元の特性を保持するために推奨されます。ただし、イソプロピルアルコール(IPA)、水、アセトニトリル(MeCN)などの他の媒体を同じ意味で使用できるため、クライオゲルの形成を容易にすることができます(前述のようにスケールバー= 100または50 μm)。(e)70%EtOHにおける凍結乾燥前(灰色)および凍結乾燥後(緑色)のHA-NBミクロゲル直径の測定を、3つのミクロゲル集団の頻度分布として示す。Anderson et al.12 より、エルゼビアの許可を得て転載。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
4. MAP足場製作
- テトラジンリンカー合成
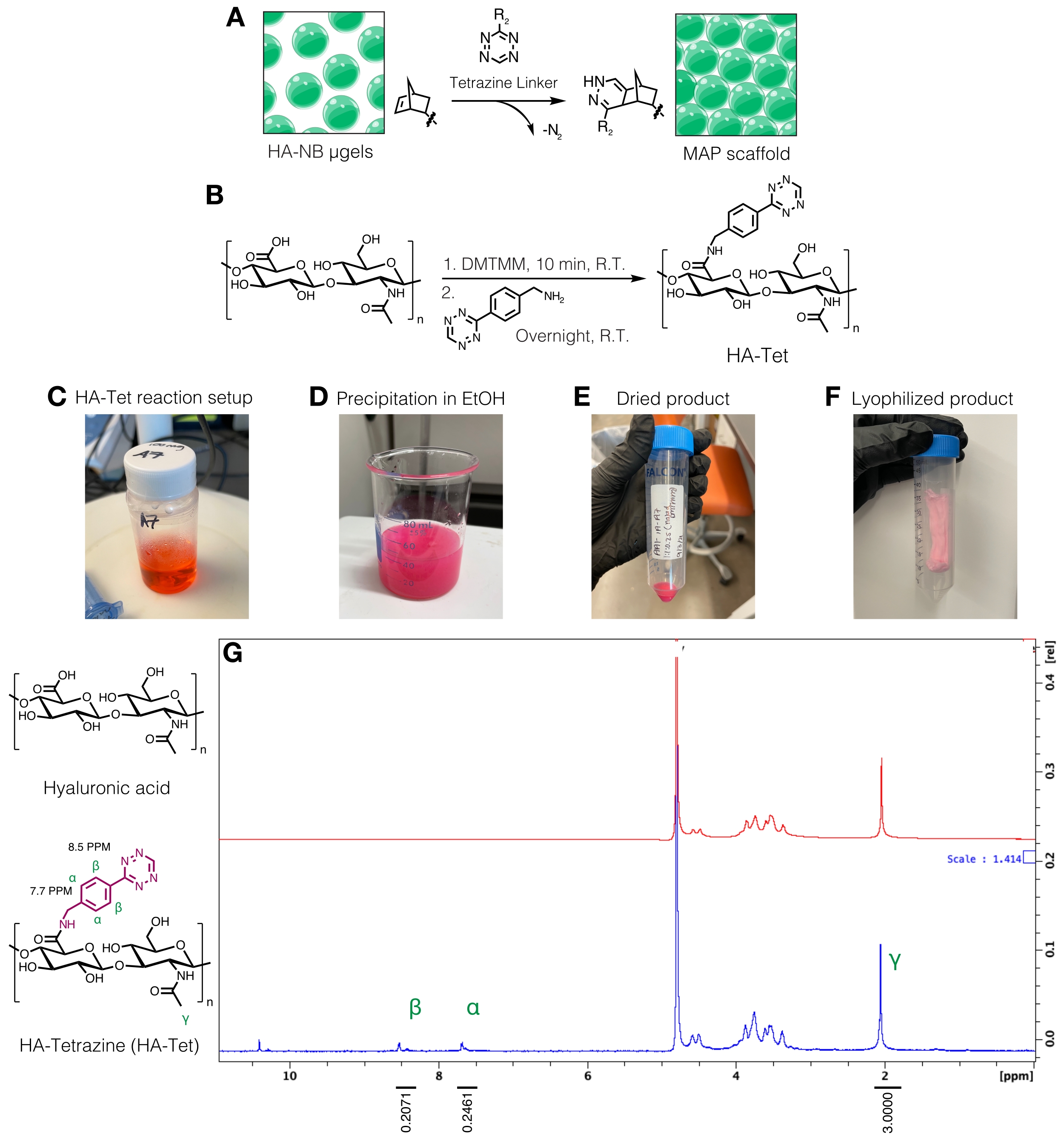
注:テトラジンリンカーは、遊離ノルボルネン基を有するμgelを相互結合するために使用できます(図3A)。HA-テトラジン(HA-Tet)合成手順は、DMTMMからテトラジンアミンへの1:1:0.25モル当量を有する79 kDaのHAナトリウムHAを用いてZhangら18 から適応させた(図3B)12。- 反応物の重さを量ります。20 mg/mLのHAを200 mM MESバッファー(pH ~6)に溶解し、攪拌プレート上のビーカーまたはフラスコで攪拌します。溶解したら、DMTMMをHA溶液に加え、室温で約20分間反応させます。例えば、100mg HA + 72.8mg DMTMM + 14.14mgのテトラジンアミンを使用することができる。
- テトラジンアミンを15 mg/mLで200 mM MESバッファーに溶解し、HA/DMTMM溶液に滴下します。HA-TET反応のセットアップについては、 図3C を参照してください。
- 蒸発を最小限に抑えるために反応容器の開口部にパラフィルムを追加し、反応容器をホイルで覆います。反応を約24時間進行させながら攪拌を続ける。
- 24時間後、200プルーフエタノール(反応量の約10倍)を冷却します。攪拌プレート上で、反応液を冷やしたエタノールに滴下してHA-Tetを沈殿させ(図3D)、20分間攪拌を続けます。
- 溶液を50 mLコニカルチューブに移し、5,000 x g で10分間遠心分離します。余分なエタノールを注ぎ、廃棄物として処分します。デシケーターでHA-Tetの真空を引き、一晩乾燥させます。プロトコルにおけるこのステップにおける乾燥生成物の例を 図3Eに見出すことができる。
- 透析を使用してHA-Tetを精製します。HA-Tetを2 M NaCl溶液に溶解し、分子量カットオフ12〜14 kDaのセルロース透析チューブに移します。充填した透析チューブを5Lの超純水の入ったバケツに移し、HA-Tetを一晩水に対して透析します。
- 翌日、水を取り除き、1 M NaCl溶液と30分間交換します。NaCl溶液を取り出し、超純水で3日間透析し、毎日交換します。
- 透析した製品を0.2 μmの真空駆動フィルターでろ過し、ろ過したHA-Tet製品を50 mLのコニカルチューブに移します。
- 円錐管を液体窒素中で10分間フラッシュ凍結してから、鉗子で円錐管を取り外します。キャップをすばやく取り外し、ラボグレードのティッシュで覆います。組織を輪ゴムで固定し、凍結乾燥容器またはチャンバーに移して凍結乾燥します。凍結乾燥製品(図3F)を-20°Cで保存します。
- HA-Tetを10 mg/mLのD2Oに溶解し、プロトンNMR で 分析することにより、テトラジン修飾を定量します(図3G)16。
- 官能化の量を決定するには、まずD2O溶媒ピークを4.8PPMに較正します。HAメチルプロトンのピーク(δ2.05)を積分し、積分を3.0に較正します。次に、δ8.5(2H)とδ7.7(2H)(芳香族プロトン)のペンダントテトラジン基のピークを積分します。これらのピークの積分を対応する数のプロトンに正規化して、平均修飾度12を決定する。
- ライオ-μgelをインターリンクして特性評価のためのMAP足場を形成
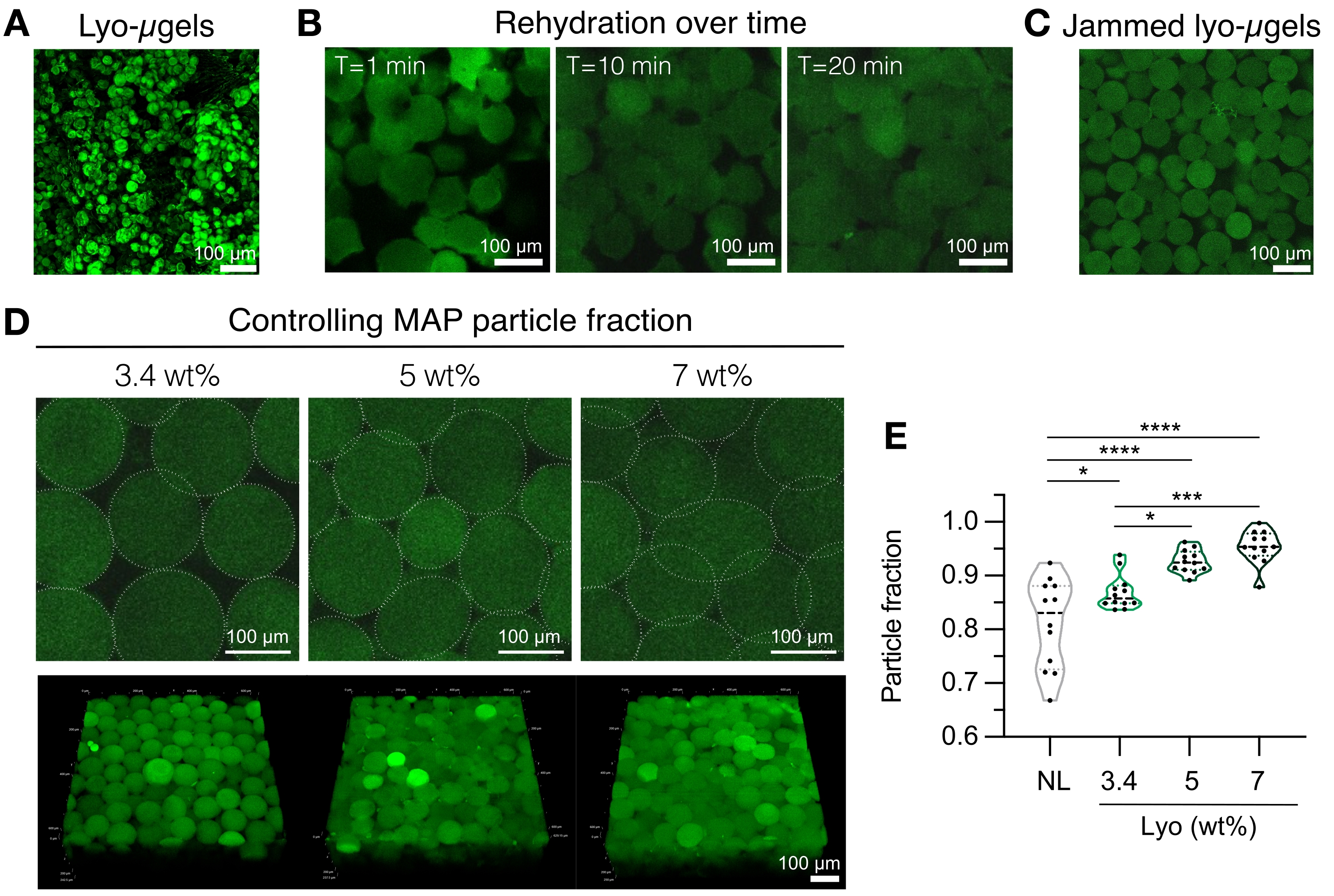
- MAP足場コンポーネント(μgels、HA-Tet、リハイドレーション量)を準備します。ライオμゲルの重量を測定し(図4A)、1x PBSの最終MAP容量の84%に再構成します。ミクロゲルを約20分間膨潤させます(図4B、C)。再水和に使用されるwt%MAPは、最終粒子分率に対するユーザーの好みに基づいて選択できます( 図4D、Eを参照)。
- HA-Tetを選択した濃度で1x PBSに溶解します(以下の注を参照)。
注意: 充填率(wt%MAP経由 )とHA-Tetの濃度の両方を変更すると、バルク足場の機械的特性が変化します。例えば、0.02 mg/mL HA-Tet(アニーリング比2.6 mol Tet:mol HA-NB)で架橋された3.4 wt%のMAP足場は、約700 Paのせん断貯蔵弾性率12を有するMAPスキャフォールドを生成する。 - 変位ピペットを使用して、HA-Tetとlyo-μgelsを組み合わせ、よく混合します。この時点で、混合物は置換ピペット を介して スライドガラス、ウェルプレート、またはユーザーが選択した容器に移すことができます。μgelを37°Cで25分間アニールした後、スパチュラを使用してMAPスキャフォールドを1x PBSで満たされたウェルプレートに移します。特性評価の準備ができるまで、MAPスキャフォールドを1x PBSに保ちます。
- MAP足場粒子率の計算
- 画質を向上させるには、ヘラを使用してMAP足場をガラスカバーガラスに移します。共焦点顕微鏡上の画像MAP足場は、FITC励起および発光用のレーザーを使用します。画像MAPは20倍の対物レンズ上に足場を組み、2.5μmのステップサイズでZ方向に250〜300μm横断するZスタックを取得します。画像のμm/ピクセルキャリブレーションをメモします。
- Zスタック画像を解析ソフトウェアにインポートします( 材料表を参照)。[ 新しいサーフェスを追加] ボタンを選択します。[ 関心領域のみをセグメント化] チェック ボックスをオンにし、青い矢印ボタン [次へ: 関心領域] を選択します。
- 分析対象のボリュームの X、Y、Z ディメンションを追跡しながら、関心領域を定義します。青い矢印ボタン [ 次へ: ソース チャネル] を選択します。
注: X 寸法と Y 寸法はピクセル単位で、Z 寸法はステップ数です。関心領域の推奨Z高さには、少なくとも2つのμgelが含まれている必要があります。 - [ソース チャネル] ドロップダウン リストを使用して、FITC チャネル を選択します。 [平滑 ]の横にあるチェックボックスをオンにして、2.50 μmの表面の詳細を入力します。[しきい値] で [ 絶対強度] を選択し、青い矢印ボタン [ 次へ: しきい値] を選択します。
- FITCチャネルに推奨されるしきい値を使用します。3D 投影を回転してレンダリング品質を評価し、必要に応じて調整します。[ 次へ: サーフェスを分類]を選択します。
注: [ 戻る ] ボタンを使用して、必要に応じて、Z 寸法などのプロセスの前のステップを編集できます。 - [ ボクセルの数 ] が 10.0 であるかどうかを確認し、緑色の二重矢印ボタン [完了: すべての作成手順を実行し、ウィザードを終了します] を選択します。
注: ボリューム レンダリング パラメータをバッチ分析用に保存して、すべてのスキャフォールディングの解析に同じ設定を適用できます。 - データをエクスポートするには、[ 統計 ] タブを選択し、[ 詳細] タブを選択します。2 番目のドロップダウン ボックスを使用して、変数 [ボリューム] を選択します。フロッピーディスクボタン タブ 表示の統計をファイルにエクスポート を選択し 、プロンプトが表示されたらスプレッドシートファイル(.xls)として保存します。
- ファイルを開き、カラムA ボリューム のSUM関数を使用して、関心領域内のμgelの総体積(μm3)を決定します。
- 解析した関心領域の寸法をピクセルからμmに変換します。手順 4.3.1 の画像の μm/ピクセル キャリブレーションを使用して、X 次元と Y 次元を変換します。Z次元(ステップ数)に画像のステップサイズを掛けて、Z寸法をμmに変換します。関心領域の体積 (μm3) を、X、Y、Z 次元を掛けて計算します。
- 足場の粒子分率を決定するには、関心領域(ステップ4.3.8で検出)内のμgelの総体積を、関心領域(ステップ4.3.9で検出)の体積で割ります。

図3:微多孔性アニール粒子(MAP)足場を作製するためのテトラジンリンカーの合成。 (A)HA-NBμgelsをテトラジンリンカーと連結させてMAPスキャフォールドを形成した模式図。(B)HA-TET合成の反応スキーム。(C)HA-Tet反応をセットアップし、一晩反応させた後、(D)エタノール中でHA-Tetを沈殿させた。(E)一旦精製および乾燥した後、HA-Tetを再水和および凍結乾燥して、(F)乾燥した淡いピンク色の生成物を得た。(G)プロトンNMR分析では、HAリピートユニットの11%の修飾に成功しています。Anderson et al.12 より、エルゼビアの許可を得て転載。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図4:MAP足場製造のための凍結乾燥ミクロゲルの再水和。 (A)乾燥したライオμゲルの最大強度投影(スケールバー= 100 μm)。(B)凍結乾燥後、ライオμゲルの再水和には約20分かかることが示されています(スケールバー= 100μm)。(C)Lyo-μgelsは、さまざまなwt%MAPで再水和して、詰まったμgelを生成できます(スケールバー= 100 μm)。(D)リョーμゲルを再水和するときに重量%MAPを増加させると、MAP足場の単一のZスライスと体積投影(スケールバー= 100μm)で示されるように、MAP足場の粒子分率が変化する。(E)これらのユーザー定義のwt%MAPスキャフォールドを使用して、ユニークな粒子画分を達成することができます(NL =非凍結乾燥μgel)。テューキーHSDを用いた一元配置分散分析をサンプルに対して実施し(n = 3)、有意性はp < 0.05(*)、p < 0.01(**)、p < 0.005 (***)、およびp < 0.001(****)で報告された。Anderson et al.12 より、エルゼビアの許可を得て転載。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
5.3D MAPスキャフォールドでの細胞培養
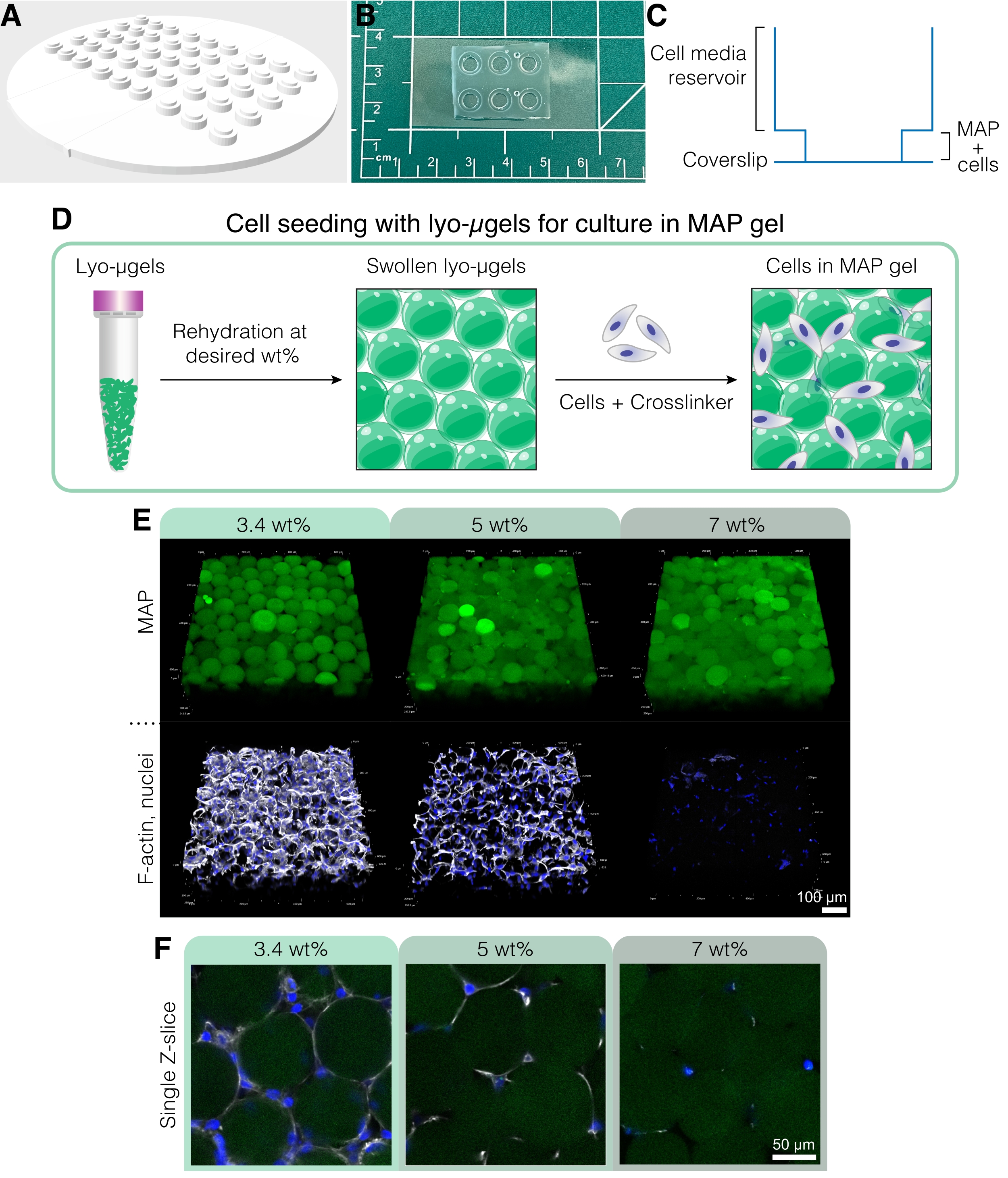
- 細胞培養デバイスの準備
- これらの実験用のカスタム細胞培養デバイスを作成するには(図5A-C)、補足コーディングファイル1にあるCADファイルを使用して、3Dプリンターを使用してネガ型を印刷します。
注:細胞培養装置の寸法は次のとおりです:94.9 mm x 94.9 mm x 4.8 mm、ウェル全体の高さは2.6 mmです。内側ウェルおよび外側ウェルの直径はそれぞれ4mmおよび6mmである。 - ポリジメチルシロキサン(PDMS)エラストマーベースと硬化剤を質量比10:1で混合します。PDMS混合物を大きなプラスチック製のペトリ皿に注ぎ、デシケーターで約30分間、またはすべての気泡が消えるまで脱気します。
- すべての気泡が消えたら、3Dプリントされた金型をPDMSに慎重に配置して、新しい気泡の形成を最小限に抑えます。60°Cのオーブンに少なくとも2時間入れて、PDMSを硬化させます。
- ナイフやカミソリの刃を使って培養装置のパラメータの周りをそっとなぞり、カビを慎重に取り外します。4 mm生検パンチを使用して、ウェルの底からPDMSを除去します。ガラスのカバーガラスに収まるようにデバイスをカットします。
注:細胞培養デバイスはスライドガラスに接着することもできますが、ガラスカバーガラスはサンプルイメージングを改善します。 - テープを使用して、培養装置の底面からほこりを取り除きます。清潔なガラスカバーガラスカバースリップと培養装置(底面を上にして)を135°Cのホットプレートに15分以上置き、水分を取り除きます。
- ヒュームフードで、ガラスカバースリップとデバイスの底面の両方の高い位置にコロナプラズマガンを30秒間使用し、処理された表面をすばやく接着します。培養装置とガラスカバーガラスの間の良好なシールを確保するために、静かに圧力をかけます。
- すべてのデバイスに対して手順5.1.6を繰り返し、60°Cのオーブンに一晩入れてボンドを固定します。 インビトロで使用する前に滅菌するためにデバイスをオートクレーブします。
- これらの実験用のカスタム細胞培養デバイスを作成するには(図5A-C)、補足コーディングファイル1にあるCADファイルを使用して、3Dプリンターを使用してネガ型を印刷します。
- MAPスキャフォールドでの細胞培養
- 目的の粒子画分に基づいてMAP足場コンポーネント(μgel、HA-Tet、培地容量など)を準備します(図4D-Eを参照)。滅菌フード内でライオμゲルを秤量し、選択したwt%MAPに基づいて細胞培地の最終MAP体積の84%に再構成します。μgelを約20分間膨潤させます。
注:これらの方法では、ユーザーはリハイドレーションのためにリョミクロゲル製品を計量する必要があります。少量の場合(1 mg以下)、μgelsを追加および凍結乾燥する前にまずクライオチューブを計量し、次に凍結乾燥後にチューブを再計量して製品の質量を決定し、誤差を最小限に抑えることをお勧めします。 - HA-Tetを最終MAP体積の16%の細胞培地に溶解します。
注:MAPスキャフォールドに播種する細胞を準備するための次の手順は、使用する細胞の種類に応じて変更できます。このプロトコルでは、D1マウス間葉系細胞を、1%ペニシリン-ストレプトマイシン(ペン-ストレプトマイシン)および10%ウシ胎児血清(FBS)を添加したダルベッコ改変イーグル培地(DMEM)で増殖させた( 材料の表を参照)。これらの細胞については、標準的な接着細胞培養プロトコルに従い、組織培養処理された培養容器内で培養液を37°Cおよび5%CO2 に維持する必要があります。 - D1マウス間葉系細胞が70%〜80%のコンフルエントに達したら、培地を吸引し、1x PBSで細胞を洗浄します。組織培養容器の表面を覆うのに十分な量の1%トリプシン-EDTAを加えて、細胞を持ち上げます。37°Cで1〜3分間インキュベートした後、トリプシンEDTAの2倍の容量で1%ペンストレプトと10%FBSを添加したDMEM培地を加えてトリプシン処理を消光します。
- 細胞懸濁液を100 x g で室温で5分間遠心分離し、細胞をペレット化します。上清培地を吸引し、1%ペンストレプトと10%FBSを添加した1 mL DMEM培地に細胞を再懸濁します。
- 細胞懸濁液が十分に混合されていることを確認してから、20 μLを新しいマイクロ遠心チューブに移します。20 μLのトリパンブルー溶液を加え、よく混ぜます。この混合物20 μLを使用して、血球計算盤または細胞計数チャンバースライドを備えた自動セルカウンターを使用して細胞をカウントします。
- 10,000細胞/μL MAPの播種に必要な細胞数を新しいマイクロ遠心チューブに移します。室温で100 x g で5分間遠心分離し、細胞をペレット化します。細胞を吸引することなく、細胞ペレットから上清培地を注意深く吸引する。
- μgelsと架橋剤を置換ピペットでセルペレットに加えます。置換ピペットでよく混合し、ウェルあたり10 μLの混合物を播種します。めっきするときは、円運動でピペットで混合液をウェル内に均等に分配します。
- μgelsをセルインキュベーター内で37°Cで25分間アニールしてから、細胞培地を加えてウェルに充填します(ウェルあたり~50 μLの培地)。3D培養液を37°Cに維持し、必要に応じて培地を交換します。培地交換時に足場が吸引されないように、上部ウェルの尾根に沿ってピペットチップを安定させます。
注:培養ウェルに液体を追加したり、培養ウェルから液体を除去したりするときは、ピペットチップの端をMAP足場の上の棚に置いて、ウェルから足場を妨害または吸引する可能性を最小限に抑えます。 - 目的の時点で、培地を除去し、ウェルあたり50 μLの4%パラホルムアルデヒドを室温で30分間添加してサンプルを修正します。サンプルを50 μLの1x PBSまたは好ましいバッファーで3回洗浄します。プロトコルのこの時点で、ウェルあたり50 μLと作業容量を使用して、免疫蛍光または蛍光染色の標準的な方法に従うことができます。
注:固定および細胞染色のためのこれらの方法は、蛍光染色の使用を具体的に説明しています。ただし、一次抗体および/または二次抗体結合による免疫染色は、ウェルあたりの作業量として50 μLを使用して、製造元の指示に従ってこれらの足場でも実行できます。 - MAP足場の画像細胞を20倍対物レンズを用いて共焦点顕微鏡上で行い、ステップサイズ2.5μmでZ方向に200〜250μm横断するZスタックを得る。DAPI(1x PBS中の0.15%Triton-Xで1:1000に希釈した核染色剤)およびファロイジン-647(1x PBS中の0.15%Triton-Xで1:40に希釈したF-アクチン染色剤)による蛍光染色の例を 図5E、固定 D1細胞をMAPスキャフォールドで3日間培養したFに示します。
注:ガラス表面のプラズマ処理により親水性が向上し、細胞の接着性が向上することが示されています。細胞は細胞培養ウェルの底に沿って広がっているのが観察される可能性がありますが、MAPスキャフォールドの細胞応答を評価するための細胞数または細胞体積の定量に含めるべきではありません。
- 目的の粒子画分に基づいてMAP足場コンポーネント(μgel、HA-Tet、培地容量など)を準備します(図4D-Eを参照)。滅菌フード内でライオμゲルを秤量し、選択したwt%MAPに基づいて細胞培地の最終MAP体積の84%に再構成します。μgelを約20分間膨潤させます。

図5:MAPスキャフォールドでの細胞培養。 (A)細胞培養ウェルを作成するための型は、PDMSで3D印刷および鋳造できます。金型全体の直径は95 mm、大きなウェルは直径6 mm、小さな内側のウェルは直径4 mmです。(B)PDMSでキャストすると、細胞培養デバイスはカバーガラスにプラズマボンディングされ、顕微鏡能力が向上します。(C)細胞培養ウェルの断面は、細胞培地用のリザーバー(~50 μL)と、MAPスキャフォールドに細胞を播種するための小さなリザーバー(~10 μL)を示しています。(D)MAPスキャフォールドに細胞を播種するプロセスは、最初にユーザーの所望の重量でのlyo-μgelsの再水和に依存し、次に細胞およびμgelを相互結合するための架橋剤と混合します。(E)細胞は、変化したwt%MAPを有するMAP足場(緑色)に封入することができる。代表的な画像は、MAPスキャフォールド(スケールバー=100μm)でのD1細胞培養の5日目からのものである。(F)単一のZスライスは、異なるwt%MAP(スケールバー= 50μm)を含む足場における細胞増殖の違いを示す。Anderson et al.12 より、エルゼビアの許可を得て転載。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
結果
このプロトコルの目的は、生体直交架橋スキームを使用した微多孔性アニーリング粒子(MAP)スキャフォールドの調製と、3D細胞培養用の制御された粒子画分を実証することです。まず、HAをノルボルネンペンダント基で修飾し、ミクロゲル形成とインターリンクの両方を使用してMAP足場を形成しました。これらの方法を使用して、HAリピートユニットの約31%がノルボルネン官能的ハンドルで首?...
ディスカッション
HA−NBミクロゲルのマイクロ流体生産は、エマルジョンバッチ生産よりも狭い範囲のサイズ分布を有するミクロゲルを生成することが示されている3、9。このプロトコルに記載されているミクロゲルは、材料分解をサポートするためにMMP切断可能な架橋剤(Ac-GCRDGPQGIWGQDRCG-NH2)を使用して処方されました。しかしながら、HA−NBミクロゲルは、非?...
開示事項
ARAとTSは、この技術に関する仮特許を申請しています。
謝辞
著者らは、国立衛生研究所、国立神経障害および脳卒中研究所(1R01NS112940、1R01NS079691、R01NS094599)、および国立アレルギー感染症研究所(1R01AI152568)に感謝の意を表したい。この作業の一部は、全米ナノテクノロジー調整インフラストラクチャ(NNCI)の一部として国立科学財団(賞番号ECCS-2025064)によってサポートされているノースカロライナリサーチトライアングルナノテクノロジーネットワーク(RTNN)のメンバーであるデューク大学共有材料計装施設(SMIF)で実施されました。著者らは、細胞培養実験用の3Dプリントデバイスの生成を支援してくれたラボの元ポスドクであるルーカスシルマー博士とイーサンニックロウに感謝したいと思います。
資料
| Name | Company | Catalog Number | Comments |
| 1 mL Luer-Lok syringe sterile, single use, polycarbonate | BD | 309628 | |
| 5 mL Luer-Lok syringe sterile, single use, polycarbonate | BD | 309646 | |
| Alexa Fluor 488 C5 maleimide | Invitrogen | A10254 | For synthesis of fluorescently-labeled tetrazine |
| Alexa Fluor 647 Phalloidin | Invitrogen | A22287 | For staining cell culture samples |
| Aluminum foil | VWR | 89107-726 | |
| Biopsy punch with plunger, 1.0 mm | Integra Miltex | 69031-01 | |
| Biopsy punch, 4 mm | Integra Miltex | 33-34 | |
| Blunt needle, 23 G 0.5", Non-Sterile, Capped | SAI Infusion Technologies | B23-50 | |
| Bottle-top vacuum filter, 0.22 μm | Corning | CLS430521 | |
| Calcium chloride | VWR | 1B1110 | For microgel washing buffer |
| Capillary-piston assemblies for positive-displacement pipettes, 1000 μL max. volume | Rainin | 17008609 | |
| Capillary-piston assemblies for positive-displacement pipettes, 25 μL max. volume | Rainin | 17008605 | |
| Capillary-piston assemblies for positive-displacement pipettes, 250 μL max. volume | Rainin | 17008608 | |
| Countess Cell Counting Chamber Slides | Invitrogen | C10228 | |
| Countess II FL Automated Cell Counter | Invitrogen | AMQAF1000 | |
| Centrifuge tube, 15 mL | CELLTREAT | 667015B | |
| Centrifuge tube, 50 mL | CELLTREAT | 229421 | |
| Chloroform, ACS grade, Glass Bottle | Stellar Scientific | CP-C7304 | For synthesis of fluorescently-labeled tetrazine |
| Corona plasma gun, BD-10A High Frequency Generator | ETP | 11011 | |
| CryoTube Vials, Polypropylene, Internal Thread with Screw Cap | Nunc | 368632 | |
| D1 mouse mesenchymal cells | ATCC | CRL-12424 | Example cell line for culture in MAP gels |
| DAPI | Sigma-Aldrich | D9542 | For staining cell culture samples |
| Deuterium oxide, 99.9 atom% D | Sigma-Aldrich | 151882 | For NMR spectroscopy |
| Dialysis tubing, regenerated cellulose membrane, 12-14 kDa molecular weight cut-off | Spectra/Por | 132703 | For purifying HA-NB and HA-Tet |
| Diethyl ether | VWR | BDH1121-4LPC | For synthesis of fluorescently-labeled tetrazine |
| Dimethylformamide | Sigma-Aldrich | 277056 | For synthesis of fluorescently-labeled tetrazine |
| 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM) | TCI-Chemicals | D2919 | For modifying HA |
| Dithiothreitol (DTT) | Thermo Scientific | R0861 | Non-degradable dithiol linker (substitute for MMP-cleavable peptide) |
| Dulbecco's Modified Eagle's Medium (DMEM), high glucose, w/ 4500 mg/L glucose, L-glutamine, sodium pyruvate, and sodium bicarbonate, liquid, sterile-filtered, suitable for cell culture | Sigma-Aldrich | D6429-500ML | For D1 cell culture |
| EMS Paraformaldehyde, Granular | VWR | 100504-162 | For making 4% PFA |
| Ethanol absolute (200 proof) | KOPTEC | 89234-850 | |
| Fetal bovine serum (FBS) | ATCC | 30-2020 | For D1 cell culture |
| Heating Plate | Kopf Instruments | HP-4M | |
| Hemacytometer with coverglass | Daigger Scientific | EF16034F | |
| 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) | Sigma-Aldrich | H3375 | |
| Sodium hyaluronate, 79 kDa average molecular weight, produced in bacteria Streptococcus zooepidemicus, pharmaceutical grade, microbial contamination <100 CFU/g, bacterial endotoxins <0.050 IU/mg | Contipro | N/A | 79 kDa average molecular weight was used for HA-Tet synthesis, but these methods could be adapted for other molecular weights. |
| IMARIS Essentials software package | Oxford Instruments | N/A | Microscopy image analysis software |
| Infusion pump, dual syringe | Chemyx | N/A | |
| Kimwipe | Kimberly-Clark | 34120 | |
| Laboratory stand with support lab clamp | Geyer | 212100 | |
| Liquid nitrogen | Airgas | NI 180LT22 | |
| Lithium Phenyl(2,4,6-trimethylbenzoyl)phosphinate | TCI-Chemicals | L0290 | |
| Lyophilizer | Labconco | N/A | Labconco FreeZone 6 plus has been discontinued, but other lab grade console freeze dryers could be used for this protocol. |
| Methyltetrazine-PEG4-maleimide | Kerafast | FCC210 | For synthesis of fluorescently-labeled tetrazine |
| 2-(4-Morpholino)ethane Sulfonic Acid (MES) | Fisher Scientific | BP300-100 | For modifying HA |
| Micro cover glass, 24 x 60 mm No. 1 | VWR | 48393-106 | |
| Microfluidic device SU8 master wafer | FlowJem | Custom design made either in-house in clean room or outsourced | |
| Mineral oil, heavy | Sigma-Aldrich | 330760 | |
| MMP-cleavable dithiol crosslinker peptide (Ac-GCRDGPQGIWGQDRCG-NH2) | GenScript | N/A | |
| 5-Norbornene-2-methylamine | TCI-Chemicals | 95-10-3 | For HA-NB synthesis |
| Packing tape | Scotch | 3M 1426 | |
| Parafilm | Bemis | PM996 | |
| PEG(thiol)2 | JenKem Technology USA | A4001-1 | For synthesis of fluorescently-labeled tetrazine |
| Penicillin-Streptomycin, 10,000 units/mL | Thermo Fisher Scientific | 15140122 | For D1 cell culture |
| Petri dish, polystyrene, disposable, Dia. x H=150 x 15 mm | Corning | 351058 | |
| Pluronic F-127 | Sigma-Aldrich | P2443 | For washing HMPs |
| Phosphate buffered saline (PBS) 1x | Gibco | 10010023 | |
| RainX water repellent glass treatment | Grainger | 465D20 | Synthetic hydrophobic treatment solution for microfluidic device treatment |
| RGD peptide (Ac-RGDSPGERCG-NH2) | GenScript | N/A | |
| Rubber bands | Staples | 112417 | |
| Sodium chloride | Chem-Impex | 30070 | For dialysis |
| Span 80 for synthesis | Sigma-Aldrich | 1338-43-8 | |
| Sylgard 184 Silicone Elastomer | Electron Microscopy Science | 4019862 | polydimethylsiloxane (PDMS) elastomer for making microfluidic devices and tissue culture devices |
| Syringe filter, Whatman Uniflo, 0.2 μm PES, 13 mm diameter | Cytvia | 09-928-066 | |
| Tetraview LCD digital microscope | Celestron | 44347 | |
| Tetrazine-amine HCl salt | Chem-Impex | 35098 | For HA-Tet synthesis |
| Triethylamine | Sigma-Aldrich | 471283 | For synthesis of fluorescently-labeled tetrazine |
| Tris(2-carboxyethyl)phosphine (TCEP) | Millipore Sigma | 51805-45-9 | |
| Triton X-100 | VWR | 97063-864 | |
| Trypan blue solution, 0.4% | Thermo Fisher Scientific | 15250061 | |
| Trypsin EDTA (0.25%), Phenol red | Fisher Scientific | 25-200-056 | For lifting adherent cells to seed in MAP gels |
| Tygon ND-100-80 Non-DEHP Medical Tubing, Needle Gauge=23, Wall Thickness=0.020 in, Internal diameter = 0.020, Outer diameter = 0.060 in | Thomas Scientific | 1204G82 | |
| UV curing system controller, LX500 LED | OmniCure | 010-00369R | |
| UV curing head, LED spot UV | OmniCure | N/A | |
| UV light meter, Traceable | VWR | 61161-386 | |
| Vacuum dessicator | Bel-Art | 08-594-15C | |
| X-Acto Z Series Precision Utility Knife | Elmer's | XZ3601W |
参考文献
- Griffin, D. R., Weaver, W. M., Scumpia, P. O., Di Carlo, D., Segura, T. Accelerated wound healing by injectable microporous gel scaffolds assembled from annealed building blocks. Nature Materials. 14 (7), 737-744 (2015).
- Daly, A. C., Riley, L., Segura, T., Burdick, J. A. Hydrogel microparticles for biomedical applications. Nature Reviews Materials. 5 (1), 20-43 (2020).
- Darling, N. J., et al. Click by click Microporous Annealed Particle (MAP) scaffolds. Advanced Healthcare Materials. 9 (10), 1901391 (2020).
- Truong, N. F., et al. Microporous annealed particle hydrogel stiffness, void space size, and adhesion properties impact cell proliferation, cell spreading, and gene transfer. Acta Biomaterialia. 94, 160-172 (2020).
- Pfaff, B. N., et al. Selective and improved photoannealing of Microporous Annealed Particle (MAP) scaffolds. ACS Biomaterials Science & Engineering. 7 (2), 422-427 (2021).
- Sideris, E., et al. Particle hydrogels based on hyaluronic acid building blocks. ACS Biomaterials Science & Engineering. 2 (11), 2034-2041 (2016).
- Caldwell, A. S., Campbell, G. T., Shekiro, K. M. T., Anseth, K. S. Clickable microgel scaffolds as platforms for 3D cell encapsulation. Advanced Healthcare Materials. 6 (15), 1700254 (2017).
- Qazi, T. H., et al. Anisotropic rod-shaped particles influence injectable granular hydrogel properties and cell invasion. Advanced Materials. 34 (12), 2109194 (2022).
- Wilson, K. L., et al. Stoichiometric post modification of hydrogel microparticles dictates neural stem cell fate in microporous annealed particle scaffolds. Advanced Materials. 34 (33), 2201921 (2022).
- Muir, V. G., Qazi, T. H., Shan, J., Groll, J., Burdick, J. A. Influence of microgel fabrication technique on granular hydrogel properties. ACS Biomaterials Science & Engineering. 7 (9), 4269-4281 (2021).
- Highley, C. B., Song, K. H., Daly, A. C., Burdick, J. A. Jammed microgel inks for 3D printing applications. Advanced Science. 6 (1), 1801076 (2018).
- Anderson, A. R., Nicklow, E., Segura, T. Particle fraction as a bioactive cue in granular scaffolds. Acta Biomaterialia. 150, 111-127 (2022).
- Pruett, L., Ellis, R., McDermott, M., Roosa, C., Griffin, D. R. Spatially heterogeneous epidermal growth factor release from microporous annealed particle (MAP) hydrogel for improved wound closure. Journal of Materials Chemistry B. 9 (35), 7132-7139 (2021).
- Sheikhi, A., et al. Microengineered emulsion-to-powder technology for the high-fidelity preservation of molecular, colloidal, and bulk properties of hydrogel suspensions. ACS Applied Polymer Materials. 1 (8), 1935-1941 (2019).
- Brower, K., White, A. K., Fordyce, P. M. Multi-step variable height photolithography for valved multilayer microfluidic devices. Journal of Visualized Experiments. (119), e55276 (2017).
- JoVE. Nuclear Magnetic Resonance (NMR) Spectroscopy. JoVE Science Education Database. Organic Chemistry. JoVE. , (2022).
- Roosa, C., et al. Microfluidic synthesis of microgel building blocks for microporous annealed particle scaffold. Journal of Visualized Experiments. (184), e64119 (2022).
- Zhang, H., Dicker, K. T., Xu, X., Jia, X., Fox, J. M. Interfacial bioorthogonal crosslinking. ACS Macro Letters. 3 (8), 727-731 (2014).
- Welzel, P. B., et al. Cryogel micromechanics unraveled by atomic force microscopy-based nanoindentation. Advanced Healthcare Materials. 3 (11), 1849-1853 (2014).
- Plieva, F., Huiting, X., Galaev, I. Y., Bergenståhl, B., Mattiasson, B. Macroporous elastic polyacrylamide gels prepared at subzero temperatures: control of porous structure. Journal of Materials Chemistry. 16 (41), 4065-4073 (2006).
- Rommel, D., et al. Functionalized microgel rods interlinked into soft macroporous structures for 3D cell culture. Advanced Science. 9 (10), 2103554 (2022).
- Kurt, E., Segura, T. Nucleic acid delivery from granular hydrogels. Advanced Healthcare Materials. 11 (3), 2101867 (2021).
- Isaac, A., et al. Microporous bio-orthogonally annealed particle hydrogels for tissue engineering and regenerative medicine. ACS Biomaterials Science & Engineering. 5 (12), 6395-6404 (2019).
- Truong, N. F., Lesher-Pérez, S. C., Kurt, E., Segura, T. Pathways governing polyethylenimine polyplex transfection in Microporous Annealed Particle scaffolds. Bioconjugate Chemistry. 30 (2), 476-486 (2019).
- Koh, J., et al. Enhanced in vivo delivery of stem cells using microporous annealed particle scaffolds. Small. 15 (39), 1903147 (2019).
- Li, F., et al. Cartilage tissue formation through assembly of microgels containing mesenchymal stem cells. Acta Biomaterialia. 77, 48-62 (2018).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved