
Aby wyświetlić tę treść, wymagana jest subskrypcja JoVE. Zaloguj się lub rozpocznij bezpłatny okres próbny.
Method Article
In vivo Imaging of Fully Active Brain Tissue in Awake Zebrafish Larvae and Juveniles by Skull and Skin Removal
W tym Artykule
Podsumowanie
Here we present a method to image the zebrafish embryonic brain in vivo upto larval and juvenile stages. This microinvasive procedure, adapted from electrophysiological approaches, provides access to cellular and subcellular details of mature neuron and can be combined with optogenetics and neuropharmacological studies for characterizing brain function and drug intervention.
Streszczenie
Understanding the ephemeral changes that occur during brain development and maturation requires detailed high-resolution imaging in space and time at cellular and subcellular resolution. Advances in molecular and imaging technologies have allowed us to gain numerous detailed insights into cellular and molecular mechanisms of brain development in the transparent zebrafish embryo. Recently, processes of refinement of neuronal connectivity that occur at later larval stages several weeks after fertilization, which are for example control of social behavior, decision making or motivation-driven behavior, have moved into focus of research. At these stages, pigmentation of the zebrafish skin interferes with light penetration into brain tissue, and solutions for embryonic stages, e.g., pharmacological inhibition of pigmentation, are not feasible anymore.
Therefore, a minimally invasive surgical solution for microscopy access to the brain of awake zebrafish is provided that is derived from electrophysiological approaches. In teleosts, skin and soft skull cartilage can be carefully removed by micro-peeling these layers, exposing underlying neurons and axonal tracts without damage. This allows for recording neuronal morphology, including synaptic structures and their molecular contents, and the observation of physiological changes such as Ca2+ transients or intracellular transport events. In addition, interrogation of these processes by means of pharmacological inhibition or optogenetic manipulation is feasible. This brain exposure approach provides information about structural and physiological changes in neurons as well as the correlation and interdependence of these events in live brain tissue in the range of minutes or hours. The technique is suitable for in vivo brain imaging of zebrafish larvae up to 30 days post fertilization, the latest developmental stage tested so far. It, thus, provides access to such important questions as synaptic refinement and scaling, axonal and dendritic transport, synaptic targeting of cytoskeletal cargo or local activity-dependent expression. Therefore, a broad use for this mounting and imaging approach can be anticipated.
Wprowadzenie
Over the recent decades, the zebrafish (Danio rerio) has evolved as one of the most popular vertebrate model organisms for embryonic and larval developmental studies. The large fecundity of zebrafish females coupled with the rapid ex utero development of the embryo and its transparency during early embryonic developmental stages are just a few key factors that make zebrafish a powerful model organism to adress developmental questions1. Advances in molecular genetic technologies combined with high resolution in vivo imaging studies allowed for addressing cell biological mechanisms underlying developmental processes2. In particular, in the field of neuronal differentiation, physiology, connectivity, and function, zebrafish has shed light on the interplay of molecular dynamics, brain functions and organismic behavior in unprecedented detail.
Yet, most of these studies are restricted to embryonic and early larval stages during the first week of development as transparency of the nervous system tissue is progressively lost. At these stages, brain tissue is prevented from access by high resolution microscopy approaches becoming shielded by skull differentiation and pigmentation3.
Therefore, key questions of neuronal differentiation, maturation, and plasticity such as the refinement of neuronal connectivity or synaptic scaling are difficult to study. These cellular processes are important in order to define cellular mechanisms driving, for example, social behavior, decision making, or motivation-based behavior, areas to which zebrafish research on several weeks' old larvae has recently contributed key findings based on behavioral studies4.
Pharmacological approaches to inhibit pigmentation in zebrafish larvae for several weeks are barely feasible or may even cause detrimental effects5,6,7,8. Double or triple mutant strains with specific pigmentation defects, such as casper9 or crystal10, have become tremendously valuable tools, but are laborious in breeding, provide few offspring, and pose the danger of accumulating genetic malformations due to excessive inbreeding.
Here, a minimal invasive procedure as an alternative is provided that is applicable to any zebrafish strain. This procedure was adapted from electrophysiological studies to record neuronal activity in living and awake zebrafish larvae. In teleosts, skin and soft skull cartilage can be carefully removed by micro-peeling these layers, because they are not tightly interwoven with the brain vasculature. This allows for exposing brain tissue containing neurons and axonal tracts without damage and for recording neuronal morphology, including synaptic structures and their molecular contents, which in turn include the observation of physiological changes such as Ca2+ transients or intracellular transport events for up to several hours. Moreover, beyond descriptive characterizations, the direct access to brain tissue enables interrogation of mature neuronal functions by means of neuropharmacological substance administration and optogenetic approaches. Therefore, true structure function relationships can be revealed in the juvenile zebrafish brain using this brain exposure strategy.
Protokół
All animal work described here is in accordance with legal regulations (EU-Directive 2010/63). Maintenance and handling of fish have been approved by local authorities and by the animal welfare representative of the Technische Universität Braunschweig.
1. Preparation of artificial cerebro spinal fluid (ACSF), low melting agarose and sharp glass needles
- Prepare the ACSF by dissolving the listed chemicals at following concentrations in distilled water. 134 mM NaCl (58.44 g/mol), 2.9 mM KCl (74.55 g/mol), 2.1 mM CaCl2 (110.99 g/mol), 1.2 mM MgCl2 6x H2O (203.3 g/mol), 10 mM HEPES (238.31 g/mol), and 10 mM d-Glucose (180.16 g/mol).
NOTE: For MgCl2, CaCl2, and KCl, 1 M stock solutions are prepared in desalted sterile water and stored at 4 °C for subsequently preparing fresh ACSF. Glucose, HEPES, and NaCl are dissolved as solid compounds in the fresh ACSF solution. For dissolving chemicals, follow the manufacturer's instructions. - Adjust the pH of the ACSF to 7.8 with 10 M NaOH. Preparation of ACSF requires precise measurement of chemicals and fine adjustment of pH as it replaces the cerebro spinal fluid and maintains the physiological conditions required for neurons to be fully functional else it might cause brain misfunction and neuronal death.
- Store the freshly prepared ACSF at 4 °C for a maximum of 4 weeks. For working conditions, aliquot the required volume of ACSF for the day/experiment and prewarm at 25-28 °C (and optionally oxygenate it, step 2.5)
NOTE: Freshly prepared ASCF is fine for 1 day. If planning to use it over several days, ACSF needs to be sterile filtered. - For later anesthesia of the larvae, prepare a 50 mM stock solution of d-Tubocurarine in distilled water and store the solution at -20 °C as 100 µL aliquots in the freezer until needed.
- To embed the fish, prepare 2.5% low melting (LM) agarose by dissolving 1.25 g LM-agarose (Table of Materials) in 50 mL ACSF and boil until the agarose is completely dissolved.
NOTE: Alternatively, higher or lower concentrations of LM-agarose can be used depending on the experimental set-up. However, if the agarose is too soft, it will not be able to hold the fish in position when opening the skull. - Store the agarose at 37 °C waterbath, to avoid solidification and because this temperature will also not harm the larvae when embedding. After the boiled agarose is cooled down to 37 °C in the waterbath, add the necessary amount of d-Tubocurarine to the aliquoted agarose needed for the day to reach a working concentration of 10 µM. For future use, store the left-over agarose at 4 °C to avoid contamination.
- Prepare sharp and thin glass needles from glass capillaries (Supplementary Figure 1) using a micropipette puller with the following settings.
- Puller I, Capillary type 1: Heat 1: 65.8; Heat 2: 55.1; 2-step pulling
Puller II, Capillary type 2: Heat = 700; Fil = 4; Vel = 55; Del = 130; Pul = 55; 1-step pulling.
NOTE: The units are specific for each puller and glass capillary used here, respectively (see Table of Materials). Other capillaries and pullers can also be used to prepare the glass needles. But the glass needles should not be too thin as they might break when coming in contact with the skull. Capillary: length: 100 mm (4 inches); OD: 1.5 mm; ID: 0.84 mm; filament: Yes
- Puller I, Capillary type 1: Heat 1: 65.8; Heat 2: 55.1; 2-step pulling
2. Anesthesia of larvae and preparations for embedding
- When starting the experiment for the day, transfer the animals that are needed with a plastic Pasteur pipette to a 90 mm diameter Petri dish, which is filled with either Danieau (for larvae which are still kept in a Petri dish with Danieau) or water from the fish facility (for larvae which are older than 7 dpf and are kept in the fish facility).
- When pipetting fish older than 2 weeks, make sure the opening of the pipette is large enough to avoid injuring the fish when transferring them. Do not use a net because it will physically damage especially the younger larvae.
- Add rotifera or artemia nauplii suited for the size of the larvae kept in the Petri dish, to ensure free access to food and maximum health status of the larvae and to reduce stress.
- For embedding, transfer the selected larvae to a 35 mm diameter Petri dish filled with ACSF. Add the necessary volume of d-Tubocurarine to reach a working concentration/effective dose of 10 µM and wait for a few minutes until the larvae are completely immobilized11.
NOTE: When the fish grow older or if a faster full anesthesia is needed (under 5 min), it is possible to increase the concentration of d-Tubocurarine (LD50 for mice is 0.13 mg/kg intravenously12). It is also possible to use a different anesthetic, such as α-bungarotoxin (working concentration: 1 mg/mL), which has the same effect as curare and also keeps the brain fully active13. If a fully active brain is not necessary for the subject of interest, Tricaine in a non-lethal dose (0.02%) is also an option to fully anesthetize the larvae. However, Tricaine blocks sodium-channels, thereby impairing brain activity14. - Prepare the mounting chamber by taking the lid of the 35 mm diameter Petri dish, flip the lid upside down, and place a square glass coverslip (24 x 24 mm) on the bottom of the lid. See Figure 1 (upper part) for a schematic description of these steps. The smoother surface of the glass prevents slipping away of the agarose block, which contains the larvae during the skull opening procedure.
- Aliquot the amount of ACSF needed for the day in an appropriate vial (e.g., 50 mL tube, beaker, Schott bottle, etc.) and oxygenate it with carbogen (5% CO2, 95% O2). If imaging only morphology (e.g., fluorescence patterns) ACSF is still necessary to ensure the integrity of the brain and that cells are not negatively influenced by osmolarity effects but oxygenation of the ACSF is not needed. This step only needs to be performed when full brain activity is necessary for imaging.
NOTE: For optimal oxygen saturation of the medium, add an air stone to the end of the carbogen tube. To guarantee a sufficiently high oxygen level, it is necessary to exchange the ACSF in the imaging chambers with freshly oxygenated ACSF every 20-60 min, depending on the number and age of larvae embedded in the same imaging chamber (e.g., for a single embedded larva ACSF exchange every hour is sufficient. For six larvae older than 14 dpf embedded in parallel, exchanging ACSF every 20 min is necessary) so plan the necessary amount of oxygen saturated ACSF according to the planned experiment.
3. Embedding of the larvae
- Transfer the fully anesthetized larvae with a plastic Pasteur pipette to the (in step 2.4) prepared mounting chamber. Then, carefully remove the excess medium to avoid dilution of the LM-agarose. All the following steps should be performed under a stereo microscope with sufficient magnification.
NOTE: Tilting the mounting chamber can help to fully remove the medium. - Proceed immediately to the next step, by adding a sufficiently large LM-agarose drop on top of the larvae (circa 1 mL, depending on the size of the larvae) to protect the animals from drying out and to reduce unnecessary stress.
- Orient the larvae in position before the agarose solidifies. Ensure that the dorsal part of the larvae is directed upward. Also, make sure to embed the larvae as close to the surface of the agarose as possible.
NOTE: Depending on the size and number of larvae planned to embed at the same time, it is possible to adjust the agarose concentration. For example, for 1-3 larvae that are 30 dpf old, a concentration of 1.8%-2% LM-agarose is recommended. For 1-4 larvae that are 7 dpf old, it is most practicable to use 2.5% LM-agarose, whereas, for 5-8 larvae, 2% is more suited. If a fully active brain is required, it is recommended to only embed three fish at the same time to reduce the time needed to operate the larvae. In general, it is recommended to use lower concentrations (1.8%-2%) the older the larvae get or the more larvae are planned to be embedded at the same time. - If images will be recorded using an inverted microscope, trim the agarose block containing the larvae into a small cuboid shape. This is important for transferring the larvae to the imaging chamber later on. If using an upright microscope, such trimming is not necessary, because the mounting chamber can also be used as the imaging chamber. In Figure 1 (upper part), one can find a schematic description of these steps.
4. Exposing the brain
NOTE: All the following steps should be performed with greatest care to not unnecessarily injure the larvae. If a fully active brain is required for the experiment, keep in mind that with every second that passes, while the fish is still fully mounted in agarose and has an open skull without oxygenated ACSF, the brain will suffer from a lack of oxygen and also dry out. The effects of oxygen deficiency will become even more dramatic, the older the embedded larvae are. Therefore, it is important to perform the surgery not only within the shortest time possible, but also with maximum precision to not evoke mechanical brain damage with the needle. When trained, steps 4.2-4.4 should not take more than 30 s per fish.
- Begin the surgery as soon as the agarose has solidified. First, trim away all of the excess agarose above the brain region of interest to obtain free access to the head and a clear working space. If the dorsal part of the head is already sticking out of the agarose, skip this step.
- Depending on the region of interest, pick a spot to begin with the surgery. Take the glass needle and make a small incision through the skin but without penetrating too deep into the tissue. This will be the starting point for peeling away the overlaying skin.
NOTE: For optimal results, never start directly above the region of interest to reduce the risk of damaging important structures. If necessary, it is possible to even start posterior to the hindbrain and from there work forward until the unwanted area of skin is peeled away. - Continue with very small cuts along the part of skin aiming to remove by barely moving the needle just underneath the surface. Most of the time it is not necessary to move completely around the brain and to cut out a circle-like piece of skin and skull, but rather just make two incisions along the head and then push the skin away to one or the other side. Figure 2 shows a schematic representation of the optimal cutting strategy to obtain free access to the cerebellum.
NOTE: This micro-surgery is a delicate procedure and it will most likely need some training to perfectly remove the skin without damaging the underlying brain. It is also recommended to find out the optimal cutting strategy for the brain region of interest and stick with it for the period of the experiment. - Immediately after removing the skin from all embedded larvae, proceed by pouring (oxygenated) ACSF over the agarose to flood away unwanted skin particles and blood and to keep the brain fully active and protect it from drying out.
NOTE: If a healthy brain is needed for the experiment, it is recommended to go for a maximum of three fish at a time. - If using an upright microscope, start directly with imaging.
- When using an inverted microscope, slide a small spatula underneath the cuboid agarose block (step 3.4).
- Add a small drop of LM-agarose to the bottom of the imaging chamber (e.g., glass-bottom dish) and immediately flip the agarose block containing the larvae with the spatula for 180° and gently push it to the bottom of the imaging chamber, while the liquid agarose drop acts as glue.
- When the agarose has solidified, fill up the imaging chamber with (oxygenated) ACSF, then begin imaging. See Figure 1 (lower part) for a schematic description.
- When full brain activity is required for the experiment, always make sure that ACSF in the imaging chamber has a sufficiently high oxygen level. To ensure this, exchange the medium carefully with freshly oxygenated ACSF when possible every 20-60 min (depending on the number and size of the fish, size and surface of the imaging chamber, and imaging duration).

Figure 1: Schematic procedure for the preparation of open skull zebrafish for in vivo imaging in a stepwise manner. The working instructions for the different steps can be found in the graphic itself. Graphic designed by Florian Hetsch and adapted by Paul Schramm. Please click here to view a larger version of this figure.
{kind=link}

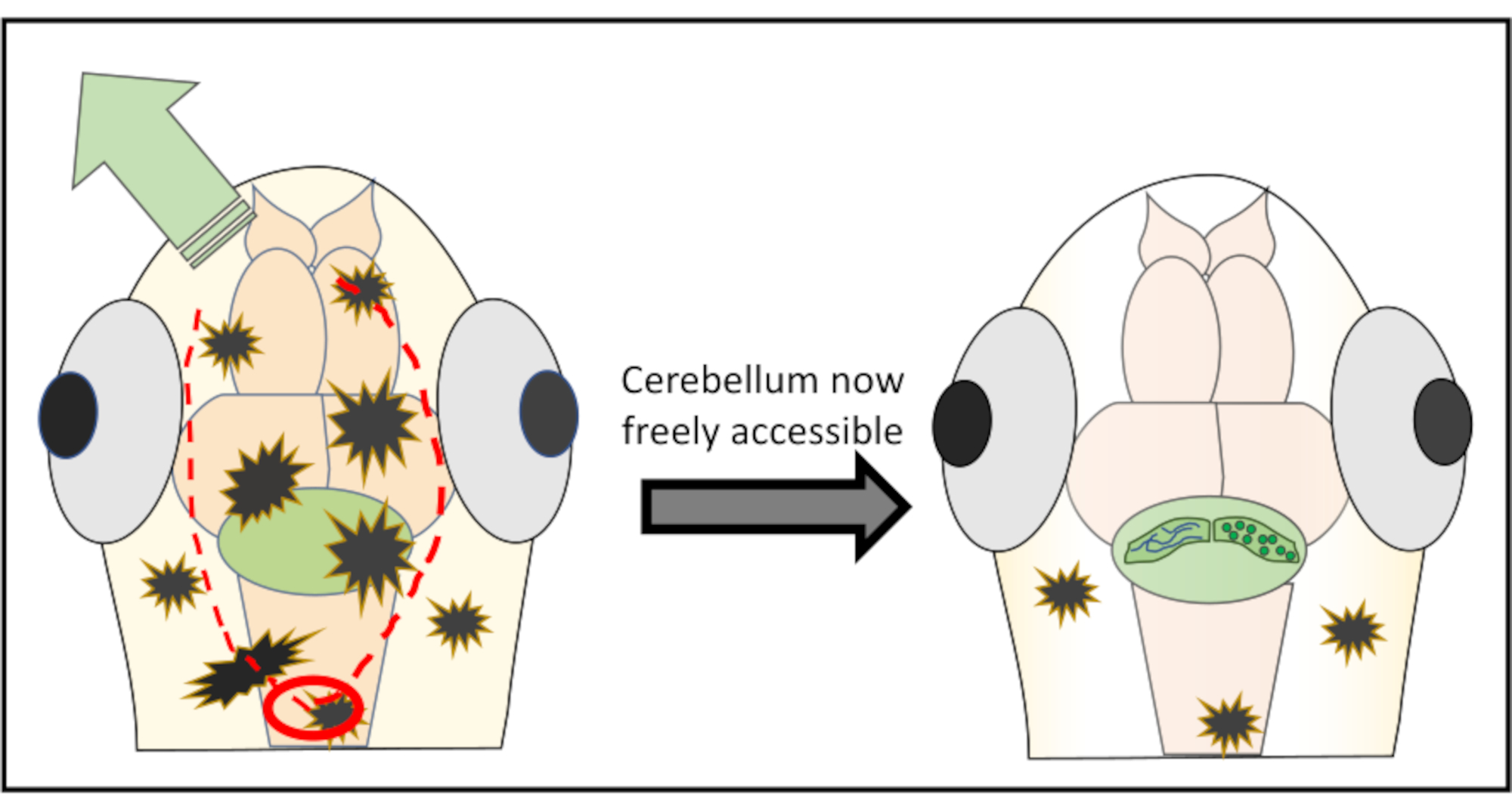
Figure 2: Detailed schematic representation of the micro-surgery performed to remove pieces of skin and skull above the brain region of interest. The red circle marks the spot where the first cut needs to be made. The red-dotted line delineates the optimal path to cut along with the needle to obtain free access to the cerebellum without damaging it. The green arrow marks the direction in which the excessive skin and skull pieces can easily be pushed away. Make sure to never penetrate into the brain tissue during the whole procedure. After successfully peeling away the skin, the brain region of interest (here, cerebellum) will be freely accessible for any kind of high-resolution in vivo imaging. Please click here to view a larger version of this figure.
{kind=link}
Wyniki
Figure 3A,C show a 14 dpf larva of the transgenic line Tg[-7.5Ca8:GFP]bz12[15] with the skull still intact. The pigment cells in the overlaying skin are distributed all over the head and are interfering with the fluorescence signal in the region of interest (here, cerebellum). With the larva in this condition, it is not possible to obtain high resolution images of the brain. F...
Dyskusje
The presented method provides an alternative approach to brain isolation or the treatment of zebrafish larvae with pharmaceuticals inhibiting pigmentation for recording high resolution images of neurons in their in vivo environment. The quality of images recorded with this method is comparable to images from explanted brains, yet under natural conditions.
Furthermore, a loss in intensity of fluorescence is avoided, because there is no need for treatment with fixatives
Ujawnienia
The authors have nothing to disclose.
Podziękowania
We especially thank Timo Fritsch for excellent animal care and Hermann Döring, Mohamed Elsaey, Sol Pose-Méndez, Jakob von Trotha, Komali Valishetti and Barbara Winter for their helpful support. We are also grateful to all the other members of the Köster lab for their feedback. The project was funded in part by the German Research Foundation (DFG, KO1949/7-2) project 241961032 (to RWK) and the Bundesministerium für Bildung und Forschung (BMBF; Era-Net NEURON II CIPRESS project 01EW1520 to JCM) is acknowledged.
Materiały
| Name | Company | Catalog Number | Comments |
| Calcium chloride | Roth | A119.1 | |
| Confocal Laser scanning microscope | Leica | TCS SP8 | |
| d-Glucose | Sigma | G8270-1KG | |
| d-Tubocurare | Sigma-Aldrich | T2379-100MG | |
| Glass Capillary type 1 | WPI | 1B150F-4 | |
| Glass Capillary type 2 | Harvard Apparatus | GC100F-10 | |
| Glass Coverslip | deltalab | D102424 | |
| HEPES | Roth | 9105.4 | |
| Hoechst 33342 | Invitrogen (Thermo Fischer) | H3570 | |
| Imaging chamber | Ibidi | 81156 | |
| Potassium chloride | Normapur | 26764298 | |
| LM-Agarose | Condalab | 8050.55 | |
| Magnesium chloride (Hexahydrate) | Roth | A537.4 | |
| Microscope Camera | Leica | DFC9000 GTC | |
| Needle-Puller type 1 | NARISHIGE | Model PC-10 | |
| Needle-Puller type 2 | Sutter Instruments | Model P-2000 | |
| Pasteur-Pipettes 3ml | A.Hartenstein | 20170718 | |
| Sodium chloride | Roth | P029.2 | |
| Sodium hydroxide | Normapur | 28244262 | |
| Tricain | Sigma-Aldrich | E10521-50G | |
| Waterbath | Phoenix Instrument | WB-12 | |
| 35 mm petri dish | Sarstedt | 833900 | |
| 90 mm petri dish | Sarstedt | 821473001 |
Odniesienia
- Embryology. Zebrafish Development Available from: https://embryology.med.unsw.edu.au/embryology/index.php/Zebrafish_Development (2020)
- Sassen, W. A., Köster, R. W. A molecular toolbox for genetic manipulation of zebrafish. Advances in Genomics and Genetics. Dove Medical Press. 2015 (5), 151-163 (2015).
- Singh, A. P., Nüsslein-Volhard, C. Zebrafish stripes as a model for vertebrate colour pattern formation. Current Biology. 25 (2), 81-92 (2015).
- Kalueff, A. V., et al. Time to recognize zebrafish 'affective' behavior. Brill: Behaviour. 149 (10-12), 1019-1036 (2012).
- Karlsson, J., von Hofsten, J., Olsson, P. -. E. Generating transparent zebrafish: a refined method to improve detection of gene expression during embryonic development. Marine Biotechnology. 3, 522-527 (2001).
- Bohnsack, B. L., Gallina, D., Kahana, A. Phenothiourea sensitizes zebrafish cranial neural crest and extraocular muscle development to changes in retinoic acid and IGF signaling. PloS One. 6, 22991 (2011).
- Elsalini, O. A., Rohr, K. B. Phenylthiourea disrupts thyroid function in developing zebrafish. Development Genes and Evolution. 212, 593-598 (2003).
- Baumann, L., Ros, A., Rehberger, K., Neuhauss, S. C. F., Segner, H. Thyroid disruption in zebrafish (Danio rerio) larvae: Different molecular response patterns lead to impaired eye development and visual functions. Aquatic Toxicology. 172, 44-55 (2016).
- White, R., et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell. 2, 183-189 (2008).
- Antinucci, P., Hindges, R. A crystal-clear zebrafish for in vivo imaging. Scientific Reports. 6, 29490 (2016).
- Burr, S. A., Leung, Y. L. Curare (d-Tubocurarine). Encyclopedia of Toxicology (3rd Edition). , 1088-1089 (2014).
- Gesler, H. M., Hoppe, J. 3,6-bis(3-diethylaminopropoxy) pyridazine bismethiodide, a long-acting neuromuscular blocking agent. The Journal of Pharmacology and Experimental Therapeutics. 118 (4), 395-406 (1956).
- Furman, B. . Alpha Bungarotxin. Reference Module in Biomedical Sciences. , (2018).
- Attili, S., Hughes, S. M. Anaesthetic tricaine acts preferentially on neural voltage-gated sodium channels and fails to block directly evoked muscle contraction. PLoS One. 9 (8), 103751 (2014).
- Namikawa, K., et al. Modeling neurodegenerative spinocerebellar ataxia type 13 in zebrafish using a Purkinje neuron specific tunable coexpression system. Journal of Neuroscience. 39 (20), 3948-3969 (2019).
- Hennig, M. Theoretical models of synaptic short term plasticity. Frontiers in Computational Neuroscience. 7 (45), (2013).
- Wang, Y., et al. Moesin1 and Ve-cadherin are required in endothelial cells during in vivo tubulogenesis. Development. 137, 3119-3128 (2010).
- Hobro, A., Smith, N. An evaluation of fixation methods: Spatial and compositional cellular changes observed by Raman imaging. Vibrational Spectroscopy. 91, 31-45 (2017).
- Knogler, L. D., Kist, A. M., Portugues, R. Motor context dominates output from purkinje cell functional regions during reflexive visuomotor behaviours. eLife. 8, 42138 (2019).
- Hsieh, J., Ulrich, B., Issa, F. A., Wan, J., Papazian, D. M. Rapid development of Purkinje cell excitability, functional cerebellar circuit, and afferent sensory input to cerebellum in zebrafish. Frontier in Neural Circuits. 8 (147), (2014).
- Scalise, K., Shimizu, T., Hibi, M., Sawtell, N. B. Responses of cerebellar Purkinje cells during fictive optomotor behavior in larval zebrafish. Journal of Neurophysiology. 116 (5), 2067-2080 (2016).
- Harmon, T. C., Magaram, U., McLean, D. L., Raman, I. M. Distinct responses of Purkinje neurons and roles of simple spikes during associative motor learning in larval zebrafish. eLife. 6, 22537 (2017).
- Zehendner, C. M., et al. Moderate hypoxia followed by reoxygenation results in blood-brain barrier breakdown via oxidative stress-dependent tight-junction protein disruption. PLoS One. 8 (12), 82823 (2013).
- Dhabhar, F. S. The short-term stress response - mother nature's mechanism for enhancing protection and performance under conditions of threat, challenge, and opportunity. Frontiers of Neuroendocrinology. 49, 175-192 (2018).
Przedruki i uprawnienia
Zapytaj o uprawnienia na użycie tekstu lub obrazów z tego artykułu JoVE
Zapytaj o uprawnieniaThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Wszelkie prawa zastrzeżone