Large-Scale Multi-Omics Genome-Wide Association Studies (Mo-GWAS): Guidelines for Sample Preparation and Normalization
In This Article
Summary
In this protocol, we present an optimized workflow, which combines an efficient and fast sample preparation of many samples. In addition, we provide a step-by-step guide to reduce analytical variations for high-throughput evaluation of metabolic GWAS studies.
Abstract
Both gas chromatography-mass spectrometry (GC-MS) and liquid chromatography-mass spectrometry (LC-MS) are widely used metabolomics approaches to detect and quantify hundreds of thousands of metabolite features. However, the application of these techniques to a large number of samples is subject to more complex interactions, particularly for genome-wide association studies (GWAS). This protocol describes an optimized metabolic workflow, which combines an efficient and fast sample preparation with the analysis of a large number of samples for legume crop species. This slightly modified extraction method was initially developed for the analysis of plant and animal tissues and is based on extraction in methyl tert-butyl ether: methanol solvent to allow the capture of polar and lipid metabolites. In addition, we provide a step-by-step guide for reducing analytical variations, which are essential for the high-throughput evaluation of metabolic variance in GWAS.
Introduction
Large-scale "omics" approaches have enabled the analysis of complex biological systems1,2,3 and further understanding of the link between genotypes and the resulting phenotypes4. Metabolomics using ultra-high-performance liquid chromatography-mass spectrometry (UHPLC-MS) and GC-MS enabled the detection of a plethora of metabolite features, of which only some are annotated to a certain degree, resulting in a high proportion of unknown metabolites. Complex interactions can be explored by combining large-scale metabolomics with the underlying genotypic variation of a diverse population5. However, handling large sample sets is inherently associated with analytical variations, distorting the evaluation of metabolic variance for further downstream processes. Specifically, major issues leading to analytical variations are based on machine performance and instrumental drift over time6. The integration of batch-to-batch variation is challenging and especially problematic when analyzing large-scale structured plant populations. Multiple normalization procedures were suggested to correct for non-biological variations, e.g., the usage of internal, external, and isotope-labeled internal standards to correct for analytical errors, of which each is inherently associated with known problems and pitfalls7,8,9,10.
In addition to analytical variation, the choice of extraction protocols generally varies depending on the analytical method. Ultimately, it is desired to reduce material and labor costs as well as the necessity of using several aliquots of the same sample for various analytical processes by performing phase separation-based extraction methods. These methods were first introduced using chloroform: methanol/water solvents to fractionate polar and hydrophobic compounds11.
This protocol describes a fast high-throughput pipeline for a multi-omics platform to profile both polar metabolites and lipids in legume species. Further, it shows how those datasets can be appropriately corrected for analytical variation and normalized before integrating genotypic information to detect metabolite quantitative trait loci (QTL) by performing GWAS.
Protocol
1. Experimental design and plant cultivation
NOTE: Set up the experiment depending on the experimental hypothesis, e.g., using a large-scale GWAS population decreases the necessity of multiple replicates, as statistical testing will be performed based on the haplotypes of all the individual SNPs instead of the accession. In contrast, multiple replicates are indispensable in other experimental approaches. The following points must be considered while preparing the experiment.
- Include enough biological replicates, depending on the experimental hypothesis.
- Randomize the biological replicates block-wise to reduce local environmental bias during cultivation, e.g., greenhouse, field.
- Ensure proper maintenance of the plant during growth. Treat plants homogeneously to reduce bias.
2. Preparation of biological plant material
- Harvest preparation
- Label harvesting tubes (20 mL) containing two 5 mm and two 8 mm diameter metal beads for homogenizing. Fill up a dewar with liquid nitrogen.
NOTE: Plants should be in the vegetative stage for fresh leaf and root tissue harvesting.
- Label harvesting tubes (20 mL) containing two 5 mm and two 8 mm diameter metal beads for homogenizing. Fill up a dewar with liquid nitrogen.
- Harvest biological samples by flash-freezing in liquid nitrogen. Harvest as quickly as possible to exclude the influence of circadian oscillation on metabolism during prolonged harvesting durations12,13. Store the harvested fresh leaf and root tissues for further processing at -80 °C.
NOTE: Leaf-cutting to flash-freezing should not take longer than a few seconds as after leaf cleavage, active biological processes would alter metabolic profiles due to wounding. For roots, preclean the roots by washing with water before flash-freezing in liquid nitrogen. Excess water on the root surface should be soaked up with paper tissue. Dried seeds can be stored at room temperature; no freezing in liquid nitrogen is required. - Grind the tissue using a tissue mixer mill.
- Precool the tube holders in liquid nitrogen for a couple of minutes to maintain a low temperature while grinding the tissue.
- Transport the biological samples in a nitrogen-containing dewar after taking them out from the -80 °C freezer.
- Grind the tissues to obtain homogeneous powder; use 25 Hz for 1 min and repeat after freezing in liquid nitrogen if the tissue is not homogeneously ground.
- For grinding dried seeds, place the seeds in a grinding jar with a 15 mm diameter metal bead. Use the same frequency and time as mentioned in 2.3.3.
NOTE: Clean and precooled mortars and pestles can be used if a tissue mixer mill is unavailable. - Precool labeled 2 mL safe-lock microcentrifuge tubes. Weigh 50 mg with an error of ±5 mg of fresh plant material by using an analytical scale. Precool the tools used for transferring plant material in liquid nitrogen. Ensure that plant material stays frozen during the weighing process.
NOTE: Do not expose fresh plant material too long to room temperature as biological processes are activated by increasing temperature, altering metabolic profiles14. - Generate additional quality control (QC) samples by pooling a proportion of each sample and weighing 50 mg with an error of ±5 mg of pooled fresh plant material into precooled 2 mL safe-lock microcentrifuge tubes.
NOTE: At least three QC samples are advised for every 60 samples. The QC samples are essential for the downstream correction, normalization, and analyses.
3. Extraction reagents
- Fresh tissue, e.g., leaves and roots
NOTE: Sample extraction is based on a previously described protocol15. This protocol has been modified based on present needs, e.g., multiple tissues, different internal standards, and large-scale experiments. Additionally, all volumes and instrument settings mentioned below are adjusted to in-house analytical units. Protocol users should adjust these according to their analytical unit and biological samples, based on test samples.- Extraction mixture 1 (EM1): methyl tert-butyl ether (MTBE)/methanol (MeOH) (3:1 v/v)
- Prepare a mixture of MTBE/MeOH in a 3:1 ratio. For 100 mL of extraction solvent, mix 75 mL of MTBE with 25 mL of MeOH in a clean glass bottle.
NOTE: Solvents should be handled carefully in the fume hood with proper safety equipment. - Add 45 µL of 1,2-diheptadecanoyl-sn-glycero-3-phosphocholine (1 mg/mL in chloroform) as an internal standard for the UHPLC-MS based lipid analysis, 400 µL of ribitol (1 mg/mL in water) as internal standard for the GC-MS based analysis, and 125 µL of isovitexin (1 mg/mL in MeOH/water (1:1 v/v)) for UHPLC-MS-based metabolite analysis.
NOTE: The addition of internal standards is necessary for the post-analysis normalization according to analytical needs. As 1 mL of EM1 is needed for each sample, prepare a stock solution according to the experimental sample size, which should be used for the entire experiment. EM1 must be stored at -20 °C. Check for the absence of the used internal standard and overlapping with other compounds in the investigated species. Several standards can be used; the selection of the internal standards in this protocol was based on previous tests using common bean extracts16.
- Prepare a mixture of MTBE/MeOH in a 3:1 ratio. For 100 mL of extraction solvent, mix 75 mL of MTBE with 25 mL of MeOH in a clean glass bottle.
- Extraction mixture 2 (EM2) water/ methanol (MeOH) (3:1 v/v)
- For 100 mL EM2, add 75 mL of double-distilled water and 25 mL of MeOH in a clean glass bottle.
- Add 500 µL of EM2 per sample, and prepare a stock solution according to the experimental sample size, which should be used for the entire experiment. Store EM2 at 4 °C.
- Extraction mixture 1 (EM1): methyl tert-butyl ether (MTBE)/methanol (MeOH) (3:1 v/v)
- Dried seeds
- Extraction mixture 3 (EM3) methanol (MeOH)/ water (7:3 v/v)
- For 100 mL of EM3, add 70 mL of MeOH and 30 mL of double-distilled water in a clean glass bottle. Prepare 1 mL of EM3 for each sample.
- Add 400 µL of ribitol (1 mg/mL in water) as internal standards for the GC-MS-based analysis and 125 µL of Isovitexin (1 mg/mL in MeOH/water (1:1 v/v)) for UHPLC-MS-based metabolite analysis.
NOTE: Prepare a stock solution according to the experimental sample size and use it for the entire experiment. Store EM3 at 4 °C.
- Extraction mixture 3 (EM3) methanol (MeOH)/ water (7:3 v/v)
4. Sample extraction
- Fresh tissue, e.g., leaves and roots
- Prepare three 1.5 mL safe-lock microcentrifuge tubes for each sample. Keep EM1 in a -20 °C liquid cooling system. Transfer the fresh samples from the -80 °C freezer to dry ice or liquid nitrogen for transportation. Add 1 mL of precooled EM1 to each 50 mg aliquot and vortex briefly before keeping on ice.
- Incubate the samples on an orbital shaker at 800 × g for 10 min at 4 °C.
- Sonicate the samples in an ice-cooled sonication bath for 10 min.
- Add 500 µL of EM2 using a multichannel pipette to avoid variation in added volumes.
- Vortex the samples briefly to mix the extraction mixtures before centrifuging at 11,200 × g for 5 min at 4 °C.
- After phase separation occurs, transfer 500 µL of the upper lipid-containing phase to a prelabeled 1.5 mL safe-lock microcentrifuge tube. Remove the rest of the upper phase.
NOTE: Take care while transferring as this upper phase has a high vapor pressure and tends to leak out from the pipette. - Transfer 150 µL and 300 µL of the lower polar and semi-polar metabolite-containing phases in two 1.5 mL safe-lock microcentrifuge tubes used for GC-MS and UHPLC-MS analysis, respectively.
- Concentrate all the extracted fractions by letting the solvents evaporate without heating using a vacuum concentrator and store at -80 °C.
- Dried seeds
- Prepare two 1.5 mL safe-lock microcentrifuge tubes for each sample. Keep EM3 on ice. Place a 5 mm diameter metal bead in the sample aliquots.
- Add 1 mL of EM3 in each 50 mg aliquot and homogenize the samples at 25 Hz for 2-3 min before putting them on ice.
- Sonicate the samples in an ice-cooled sonication bath for 10 min.
- Vortex the samples briefly before centrifuging at 11,200 × g for 5 min at 4 °C.
- Transfer 150 µL and 300 µL of the supernatant in two 1.5 mL safe-lock microcentrifuge tubes used for GC-MS and UHPLC-MS analysis, respectively.
- Concentrate all extracted fractions by letting the solvents evaporate without heating using a vacuum concentrator and store at -80 °C.
NOTE: Based on experience, users are advised to perform step 4.2 for semi-polar metabolites and derivatized metabolite analysis in dried seeds. Perform extraction step 4.1 for dried seed lipid analysis.
5. Analysis of lipids using UHPLC-MS
- Re-suspend the dried lipid fractions in 250 µL of acetonitrile:2-propanol (7:3, vol/vol).
- Sonicate the lipid phase for 5 min, centrifuge at 11,200 × g for 1 min.
- Transfer 90 µL of the supernatant to a glass vial for LC-MS.
- Inject 2 µL of the extracts into the LC-MS.
- Perform lipid fractionation on a reversed-phase C8 column held at 60 °C running with a flow of 400 µL/min with gradual changes of eluent A and B as shown in Table 1. Acquire the mass spectra in positive ionization mode with a mass range of 150-1,500 m/z.
- Include several QC samples in all daily batches and a blank to ensure correction for analytical variation. Randomize samples block-wise in sequential order.
6. Analysis of polar and semi-polar metabolites using UHPLC-MS
- Re-suspend the dried polar phase in 180 µL of UHPLC-grade methanol: water (1:1 v/v).
- Sonicate the polar phase for 2 min, centrifuge at 11,200 × g for 1 min.
- Transfer 90 µL of the supernatant to a glass vial for LC-MS.
- Inject 3 µL of the extracts into the LC-MS.
- Perform metabolite fractionation on a reverse phase C18 column held at 40 °C running with a flow of 400 µL/min with gradual changes of eluent A and B as shown in Table 1. Acquire the mass spectra in a mass range of 100-1,500 m/z in a full MS scan and all ion fragmentation (AIF) induced by high-energy collisional dissociation (HCD) of 40 keV.
NOTE: Use both ionization modes. However, due to limited capacity while running large numbers of samples, run test samples in both ionization modes to determine the preferred ionization mode. - Include several QC samples in all daily batches and a blank to ensure correction for analytical variation. Randomize samples block-wise in sequential order.
- Run a pooled QC in data-dependent MS2 in both negative and positive ionization modes. Use the obtained mass spectra in a later step (8.5) for annotation.
7. Analysis of derivatized Metabolites using GC-MS17,18
NOTE: The analysis of derivatized metabolites is based on a previously described protocol17. Handle all derivatization reagents in the fume hood. Ensure that N-methyl-N-(trimethylsilyl)trifluoracetamide (MSTFA) does not get in contact with water and humidity.
- Derivatization reagent 1 (DR1)
- Dissolve methoxyamine hydrochloride in pyridine to obtain a concentration of 30 mg/mL of DR1. Use 40 µL of DR1 for each sample. Prepare a stock solution according to the sample size, and store at room temperature.
- Derivatization reagent 2 (DR2)
- Dissolve MSTFA with 20 µL of fatty acid methyl esters (FAMEs) per 1 mL of MSTFA. Use 70 µL of DR2 for each sample. Prepare a stock solution according to the sample size. Store MSTFA at 4 °C and the FAMEs at -20 °C.
NOTE: FAMEs include methylcaprylate, methyl pelargonate, methylcaprate, methyllaurate, methylmyristate, methylpalmitate, methylstearate, methyleicosanoate, methyldocosanoate, lignoceric acid methyl ester, methylhexacosanoate, methyloctacosanoate, and triacontanoic acid methylester, which are dissolved in CHCl3 at a concentration of 0.8 µL/mL or 0.4 mg/mL for liquid or solid standards, respectively.
- Dissolve MSTFA with 20 µL of fatty acid methyl esters (FAMEs) per 1 mL of MSTFA. Use 70 µL of DR2 for each sample. Prepare a stock solution according to the sample size. Store MSTFA at 4 °C and the FAMEs at -20 °C.
- Re-dry the pellet from the polar phase (stored at -80 °C) using a vacuum concentrator for 30 min to avoid any interference of H2O originating during the storage with the solvents used for the downstream derivatization.
- Add 40 µL of DR1.
- Shake the samples at 950 × g for 2 h at 37 °C using an orbital shaker, followed by a short spin-down of the liquid.
- Add 70 µL of DR2.
- Shake again at 950 × g for 30 min at 37 °C using an orbital shaker.
- Centrifuge briefly at room temperature before transferring 90 µL into glass vials for GC-MS analysis.
- Inject 1 µL to GC-MS splitless mode, depending on the metabolite concentrations, with a constant helium carrier gas flow of 2 mL/min. Injection temperature is set to 230 °C using a 30-m MDN-35 capillary column.
NOTE: Additional information, e.g., temperature gradient, can be found in Table 1. The mass range is set to 70-600 m/z with 20 scans/min. Include split modes to enable the quantification of putative overloading compounds, saving costs and time for extract re-derivatization in such cases. - Include several QC samples in all daily batches and a blank to ensure correction for analytical variation. Randomize samples properly block-wise in sequential order.
8. Chromatogram processing and compound annotation
- Filter chemical noise by defining intensity thresholds. Include all QC samples while processing the chromatograms.
NOTE: For large-scale data, noise filtering is crucial to decrease computing time and processing power. - Align the chromatograms by defining a retention time shift window. Check the chromatograms from each batch to assess the intra- and inter-batch variation.
- Perform peak detection depending on the peak shape, e.g., height and width for full width at half-maximum (FWHM) calculations.
- Cluster isotopes to reduce redundant signals and filter out singletons.
NOTE: See the Table of Materials for details on software used for chromatogram processing. In-depth protocols on how to process chromatograms using various freely available software tools, e.g., MS-DIAL, MetAlign, MzMine, and Xcalibur19,20,21, are provided. - Use the ddMS2 data of a pooled QC sample for compound annotation. Assess the molecular structure by determining the monoisotopic mass and observing common neutral losses, known charged aglycones, and different types of cleavages, e.g., homolytic or heterolytic16,22.
- For reporting metabolite data, follow the recommendation described in Fernie et al. 201123.
NOTE: Different computational metabolomics approaches can be used to analyze metabolomics data24,25,26.
9. Normalization of large-scale metabolomics dataset
- Check the distribution of the internal standard(s) and normalize by correcting for the response of single or multiple internal standards.
- Correct the peak intensities obtained from the chromatogram over the exact sample weight by dividing the peak intensities by the aliquoted homogenized sample weight from step 2.5.
- Correct for intensity drift across multi-batch series. Perform QC-based correction methods such as locally estimated scatterplot smoothing (LOESS)27 using R.
NOTE: Several tools and packages are available to address the drift of the MS performance during the acquisition of the whole batches28,29. - Ensure normal distribution of traits by data transformation, e.g., Box-Cox transformation30 using the boxcox () function from the R package MASS for carrying out GWAS.
- Perform data scaling, e.g., Pareto scaling, for multivariate analysis to ensure proper weighing of low abundant compounds31.
NOTE: If possible, perform a recovery assay to avoid matrix effects, e.g., ion suppression14.
10. Genome-wide association studies (GWAS)32
- Call single nucleotide polymorphism (SNP) or structural variants (SV) from the sequencing data33,34.
- Filter genotypic data for minor allele frequency (MAF) < 5% and missing rate of >10% to avoid low-frequency bias using Tassel35.
- Calculate best linear unbiased predictions (BLUPs) for each normalized feature over the experimental repetitions to eliminate bias originating from environmental factors (random effects) using the R package Ime436.
- Use BLUPs of each feature individually to perform GWAS using the rMVP package in R37.
NOTE: Each metabolomics feature is viewed here as an individual stand-alone phenotype. - While performing GWAS, correct for population structure using principal component analysis (PCA) and identity by state (IBS) or vanRaden to minimize confounding effects. Furthermore, consider using a mixed linear model (MLM) or a multi-locus mixed model (MLMM), as mixed models contain fixed and random effects.
11. QTL detection
- Check the SNPs showing significant association, taking the Manhattan plots into consideration, for linkage disequilibrium (LD) calculations to determine the underlying genetic region. Perform LD calculations using the R package LD heatmap or Tassel 5.
- Check the associated SNPs for the effect size over the trait by examining the trait levels for statistical changes between haplotypes to find potential causal SNPs, e.g., SNPs leading to an amino acid change in the protein-coding sequence, which could explain the phenotypic variation.
NOTE: As SNP-trait associations do not necessarily yield causal association, it is crucial to determine the genomic region. Compound identity by feature annotation can help immensely in finding the right candidate genes in a specific genomic region. We suggest to combine all detected QTL associated with certain compounds in a pleiotropic map to underline the genetic regions38, as shown in Figure 4. For validation of candidate genes, several approaches can be performed (see the discussion).
Representative Results
Successful metabolomics GWAS experiments should begin with a proper experimental design, followed by sample collection, extraction, data acquisition, and processing, as illustrated in Figure 1. In this protocol, the MTBE method15 was used to extract and analyze hundreds of metabolites belonging to several compound classes. Chromatography depends highly on the properties of the utilized column as well as elution buffer mixtures. Figure 2 shows chromatograms of QC samples, indicating the elution pattern of some major lipid classes in this analytical system. The applied gradients for each platform are given in Table 1. Strong emphasis was placed on handling systemic errors in large-scale experiments. Performing large-scale metabolomics is inherently associated with systemic errors. For demonstration, we analyzed lipidomic data across several common bean species. Supplemental Table 1 provides the extracted raw lipidomic data obtained after chromatogram processing using the software indicated in the Table of Materials. Following this protocol enabled us to circumvent major issues in dealing with omics data, especially while handling large sample sets. The normalization procedure yields in accurate correction of batch-wise analytical errors, as demonstrated in Figure 3. Although increasing the numbers of QC samples would increase the power of the normalization, this is not always feasible due to cost and time constraints. For high-throughput metabolomics GWAS with non-targeted metabolic features, it is essential to illustrate higher numbers of trait-marker association appropriately. A pleiotropic map38 combining multiple GWAS results could be used to highlight the genomic regions to which several traits are linked (Figure 4).

Figure 1: Flowchart of the metabolomics-based GWAS in plants. Several steps starting from the experimental design up to the detection of QTL are shown in the left panel. In the right panel, multiple figures are shown to support several steps mentioned in the left panel. Starting from the right top, (1) a suggested sequence of samples is shown for LC-MS, (2) pre- and post-normalized score plots of PCA, including a representative feature distribution pre- and post-processing, with red indicating QC sample intensities, and (3) a Manhattan plot with significant associations to which LD and haplotype distributions were generated. Abbreviations: GWAS = genome-wide association studies; QTL = quantitative trait loci; PCA = principal component analysis; QC = quality control; LD = linkage disequilibrium; MS = mass spectrometry; LC-MS = liquid chromatography-mass spectrometry; GC-MS = gas chromatography-mass spectrometry; LOESS = locally estimated scatterplot smoothing; MLM/MLMM = mixed linear model/multi-locus mixed-model. Please click here to view a larger version of this figure.
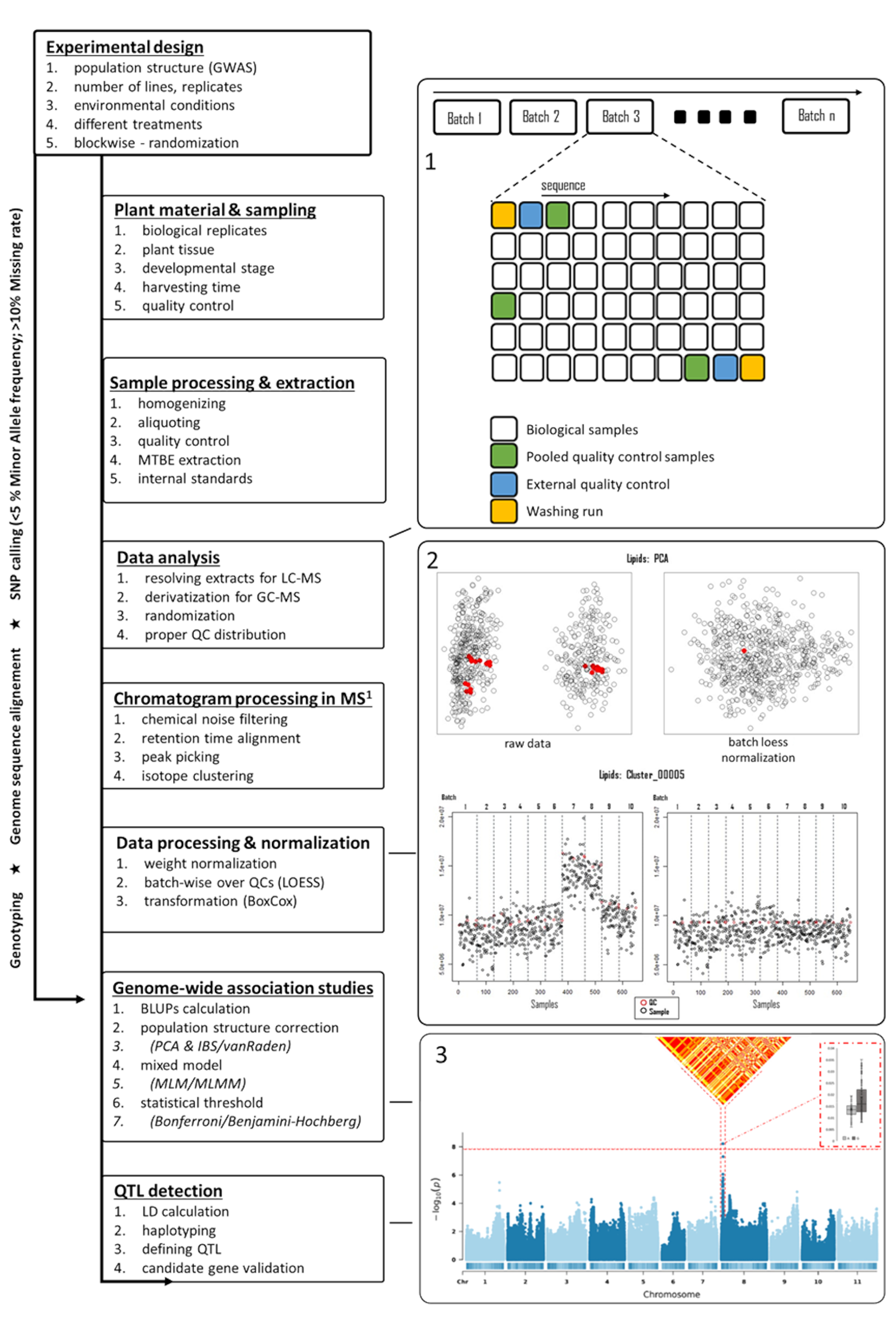{kind=link}

Figure 2: Chromatogram processing. Two QC chromatograms (base peak; lipid data) from different batches demonstrate the batch-wise variation for certain lipid classes in the pooled QC samples. Four major lipid classes are indicated with their respective elution windows in the in-house LC-MS system. The chromatograms were exported from MzMine21. Abbreviations: QC = quality control; LC-MS = liquid chromatography-mass spectrometry. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Correction of systematic error. Principal component analysis of acquired lipidomic data, pre- (left, raw data) and post-correction for systemic errors (right, batch loess). The lower panels illustrate the feature (Cluster_00005) distribution over the samples (n=650) and batches (n=10) pre- (left) and post (right)-correction for analytical variation. Abbreviations: PCA = principal component analysis; QC = quality control; LOESS = locally estimated scatterplot smoothing. Please click here to view a larger version of this figure.
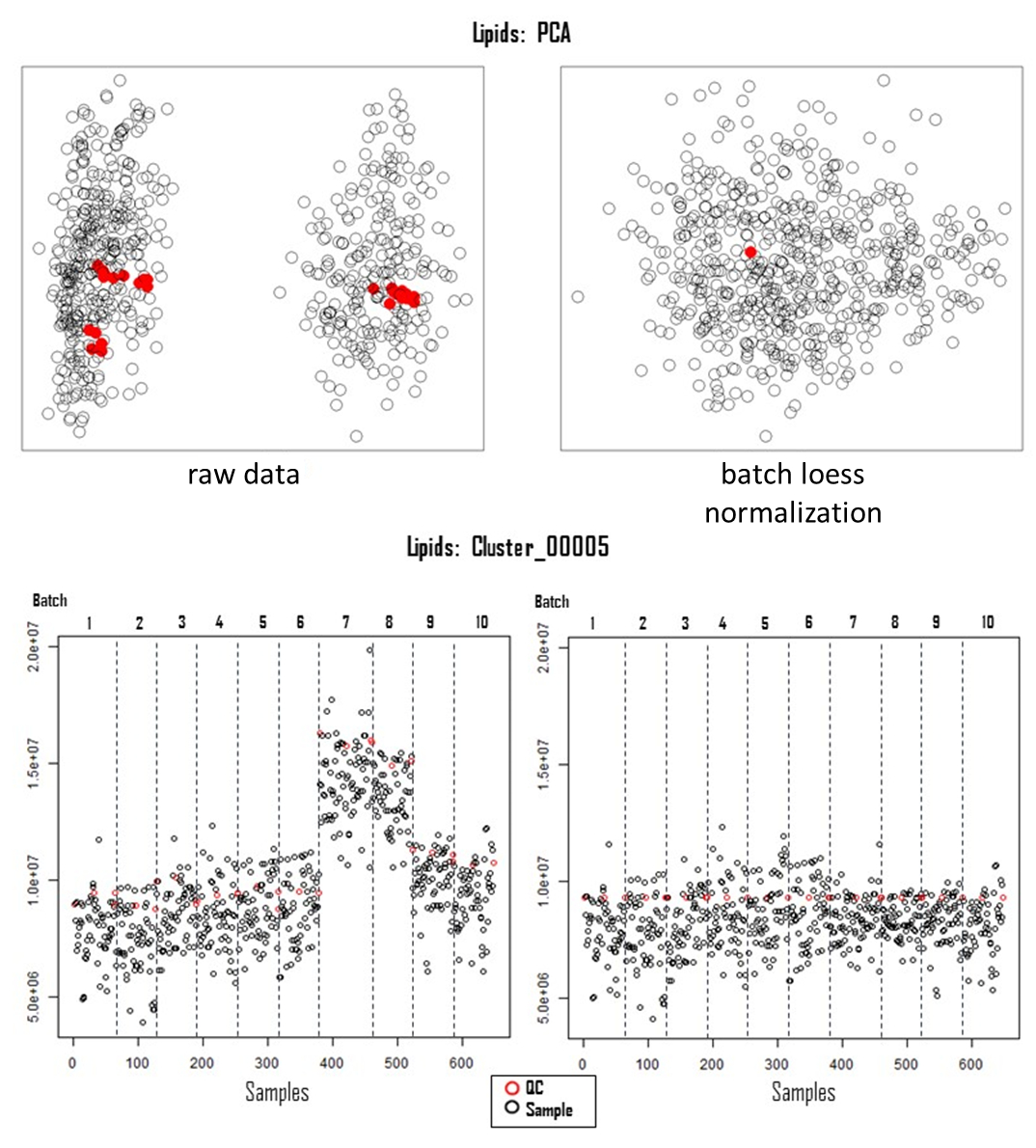{kind=link}

Figure 4: Pleiotropic map illustrating the combined GWAS results. The pleiotropic map highlights regions in the whole genome that are associated with several traits. The numbers on the outer rings indicate the corresponding chromosomes. Each circlet represents an individual trait with its significantly associated SNPs. The colors represent different compound classes (grey = compound class 1; green = compound class 2; purple = compound class 3; yellow = compound class 4). In the case of inter-compound class associations with the same genomic region, genes are highlighted. The inner grey circle shows the sum of all significant SNPs associated with a specific genomic position. The associations shown in this figure are artificially generated only for illustration. Abbreviations: GWAS = genome-wide association studies; SNPs = single-nucleotide polymorphisms. Please click here to view a larger version of this figure.
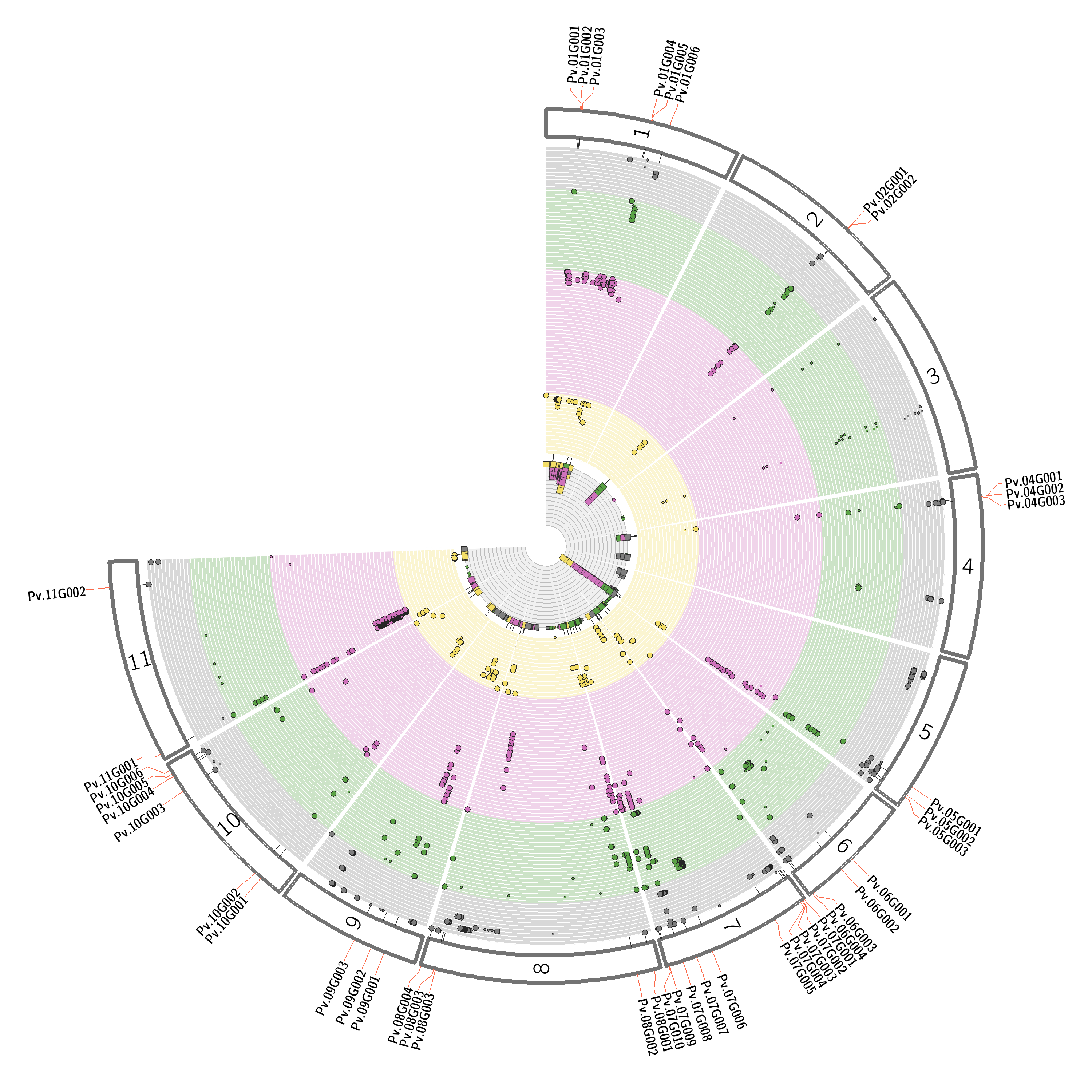{kind=link}
| UHPLC-MS settings for lipids | ||||
| Time [min] | Eluent A to B [%]* | Information | ||
| 0 - 1.00 | 45% A | Eluent A: 1% 1M NH4-Acetate, 0.1% acetic acid in water (UHPLC grade) | ||
| 1.00 - 4.00 | lg 45% - 25% A | Eluent B: 1% 1M NH4-Acetate, 0.1% acetic acid in acetonitrile/2-propanol 7:3 (UHPLC grade) | ||
| 4.00 - 12.00 | lg 25% - 11% A | Flow rate: 400 µL/min | ||
| 12.00 - 15.00 | lg 11% - 0% A | Injection volume: 2 µL | ||
| 15.00 - 19.50 | cw 0% A | |||
| 19.50-19.51 | 0% - 45% A | |||
| 19.51-24.00 | eq 45% | |||
| UHPLC-MS/MS settings for polar and semi-polar metabolites | ||||
| Time [min] | Eluent A and B [%]* | Information | ||
| 0 - 1.00 | 99% A | Eluent A: 0.1% formic acid in water (UHPLC grade) | ||
| 1.00 - 11.00 | lg 99% -60% A | Eluent B: 0.1% formic acid in acetonitrile (UHPLC grade) | ||
| 11.00 - 13.00 | lg 60% - 30% A | Flow rate: 400 µL/min | ||
| 13.00 - 15.00 | lg 30% - 1% A | Injection volume: 3 µL | ||
| 15.00 - 16.00 | cw 1% A | |||
| 16.00 - 17.00 | lg 1% - 99% A | |||
| 17.00 - 20.00 | eq 99% A | |||
| GC-MS settings for derivatized metabolites | ||||
| Time [min] | Temperature [°C] | Information | ||
| 0 - 2.00 | 85 | Carrier gas: Helium | ||
| 2.00 - 18.66 | lg 80 - 330 | Flow rate: 2 mL/min | ||
| 18.66 - 24.66 | cw 330 | Temperature gradient: 15 °C/min | ||
| 24.66 | rapid cooling | Injection volume: 1 µL | ||
Table 1: Gradient settings for each of the analytical platforms7. Abbreviations: lg = linear gradient; cw = column washing; eq = equilibrate; UHPLC-MS = ultra-high-performance liquid chromatography-mass spectrometry; UHPLC-MS/MS = ultra-high-performance liquid chromatography-tandem mass spectrometry; GC-MS = gas chromatography-mass spectrometry. * = percentage value corresponds to eluent A; remaining percentage value corresponds to eluent B.
Supplemental Table 1: Raw lipidomics data. Indicates the peak intensities for each of the detected clusters over each sample. Please click here to download this Table.
Discussion
Both GC-MS and LC-MS are widely used tools for profiling complex mixtures of various metabolite classes. Handling large datasets with these tools is inherently associated with a non-biological variation, e.g., analytical variation, which interferes and biases the interpretation of the results. This protocol presents a robust and high-throughput extraction pipeline for comprehensive metabolic profiling to eliminate variation of non-biological origin and conduct large-scale "omics" studies. The volumes and concentrations used in this protocol were adjusted for legume species in different tissues. However, these parameters can be slightly modified and used for large-scale metabolic samples from other plant species as well.
The previously15 described MTBE-based extractions can be used to analyze derivatized metabolites, semi-polar metabolites, and lipids. This can be expanded for protein and plant hormone extractions39, which were out of the scope of this protocol. Other extraction protocols rely on dichloromethane:ethanol mixtures40,41. Of these extraction protocols, the MTBE:methanol extraction protocol provides a favorable and less hazardous alternative to the existing chloroform-based extraction protocols42 and does not result in a protein pellet as an interphase between the polar and lipid phases. Furthermore, MTBE methods have already been used in several studies for various biological samples43,44,45.
This protocol discusses several crucial steps that might lead to potential variation while handling a large number of samples, e.g., during harvesting12,13, extraction14, as well as randomization46. Furthermore, there are additional issues that have not been discussed in this protocol that must be considered to ensure high-quality metabolomic data, e.g., matrix effect and ion suppression14.
The power of QC-based normalization methods inherently depends on the number of QC samples in each batch. As mentioned earlier, although increasing the number would increase the power, the intra-batch variation of the QCs is relatively marginal compared to inter-batch variation in these analytical systems, as illustrated in Figure 3. Overall, there are other QC-based normalization methods, such as systemic error removal using random forest (SERRF), which have been shown to outperform most of the other normalization methods such as batch-wise-ratio, normalization using an optimal selection of multiple internal standards (NOMIS), and probabilistic quotient normalization (PQN)47. However, SERRF relies on multiple QC samples in each batch, e.g., every tenth sample, which is not feasible while handling large numbers of samples. The main advantage of QC-based normalization over other data-driven or internal standard-based methods is that it retains the essential biological variation while accommodating unwanted technical variation28. Readers may refer to this review on the handling of variation28.
One main issue in GWAS is the rate of false positives, which originate mostly due to the linkage of causal and non-causal sites48,49. Second, the conservative statistical correction approaches, e.g., Bonferroni and FDR, correct for the number of independent tests, which is not equal to the number of assayed SNPs in GWAS due to the linkage between proximate SNPs50,51 Therefore, the actual number of independent tests is often lower. Another way to reduce the conservative statistical threshold would be to reduce the number of tested SNPs used for GWAS based on linkage decay over defined genomic regions52. The GWAS-integrated high-throughput metabolomics platform described in this protocol has a wide range of applications. In particular, it will facilitate improvements in crop breeding by changing the metabolite/lipid composition for industrially and nutritionally desired levels. Overall, metabolomics has provided an in-depth insight into the genetic architecture of a plethora of metabolites and metabolic diversification that occurred during crop domestication over the last decades, indicating the vast potential of metabolomics-associated breeding53. The molecular biological approaches for downstream QTL validation include the generation of CRISPR/Cas9 mutant lines54, T-DNA insertion lines55, stable and/or transient overexpression lines56, VIGS, ex vivo metabolomics approaches57 next to the conventional approach in generating cross F2 populations as well as cross validation in different populations.
By performing the necessary correction for the analytical variations as described above, several integrated approaches can be performed in addition to GWAS, such as metabolite-metabolite, metabolite-lipid correlation analysis, correlation analysis to phenomic data to shed light on more complex traits, and/or co-expression analysis to further unravel the basis of biological systems58.
Acknowledgements
M.B. is supported by the IMPRS-PMPG 'Primary Metabolism and Plant Growth'. A.R.F. and S.A. acknowledge the financial support of the EU Horizon 2020 Research and Innovation Programme, project PlantaSYST (SGA-CSA No. 739582 under FPA No. 664620), and project INCREASE (GA 862862).
Materials
| Name | Company | Catalog Number | Comments |
| Reagents and standards | |||
| 1,2-diheptadecanoyl-sn-glycero-3- phosphocholine (17:0 PC) | Avanti Polar Lipids | 850360P | Internal standard for lipids |
| Chloroform | Supleco | 67-66-3 | FAME solvent |
| Isovitexin | Sigma Aldrich | 38953-85-4 | Internal standard for metabolites |
| Lignoceric Acid Methylester | Sigma Aldrich | 2442-49-1 | FAME |
| Methanol (MeOH) | Biosolve Chemicals | 13684102 | ULC-MS grade |
| Methoxyamin -hydrochlorid | Sigma Aldrich | 593-56-6 | Metabolite deriviatization |
| Methyl laurate | Sigma Aldrich | 111-82-0 | FAME |
| Methyl myristate | Sigma Aldrich | 124-10-7 | FAME |
| Methyl palmitate | Sigma Aldrich | 112-39-0 | FAME |
| Methyl stearate | Sigma Aldrich | 112-61-8 | FAME |
| Methyl tert-butyl ether (MTBE) | Biosolve Chemicals | 13890602 | HPLC grade |
| Methyl-caprat | Sigma Aldrich | 110-42-9 | FAME |
| Methylcaprylat | Sigma Aldrich | 111-11-5 | FAME |
| Methyldocosanoat | Sigma Aldrich | 929-77-1 | FAME |
| Methyleicosanoat | Sigma Aldrich | 1120-28-1 | FAME |
| Methyl-hexacosanoat | Sigma Aldrich | 5802-82-4 | FAME |
| Methyl-octacosanoat | Sigma Aldrich | 55682-92-3 | FAME |
| Methyl-pelargonate | Sigma Aldrich | 1731-84-6 | FAME |
| N-Methyl-N-(trimethylsilyl)trifluoracetamid (MSTFA) | Macherey-Nagel | 24589-78-4 | Metabolite deriviatization |
| Pyridine | Supleco | 110-86-1 | Metabolite deriviatization |
| Ribitol | Supleco | 22566-17-2 | Internal standard for derivatized metabolites |
| Triacontanoic Acid Methyl Ester | TCI Chemicals | 629-83-4 | FAME |
| Water | Biosolve Chemicals | 23214102 | ULC-MS grade |
| Equipment | |||
| 1.5 mL Safe-lock microcentrifuge tubes | Eppendorf | 3120086 | |
| 2 mL Safe-lock microcentrifuge tubes | Eppendorf | 3120094 | |
| Balance | Sartorius Corporation | 14 557 572 | |
| DB-35ms, 30 m, 0,25 mm, 0,25 µm | Aglient | 123-3832 | Analysis of derivatized metabolites |
| GC-MS system | Leco Pegasus HT TOF-MS (LECO Corporation) | Analysis of derivatized metabolites | |
| Grinding Balls, Stainless Steel | OPS DIAGNOSTICS | GBSS 196-2500-10 | |
| MS system | Exactive, Orbitrap-type, MS (Exactive, Thermo Fisher Scientific) | Analysis of lipids | |
| MS system | Q Exactive Focus (Q Exactive™ Focus Hybrid Quadrupol-Orbitrap™ Massenspektrometer, Thermo Fisher Scientific) | Analysis of metabolites | |
| Refrigerated microcentrifuge | Eppendorf, model 5427R | 22620701 | |
| Reversed Phase (RP) Bridged Ethyl Hybrid (BEH) C8 column (100 mm × 2.1 mm containing 1.7 μm diameter particles) | Waters | 186002878 | Analysis of lipids |
| RP High Strength Silica (HSS) T3 column (100 mm × 2.1 mm containing 1.8 μm diameter particles) | Waters | 186003539 | Analysis of metabolites |
| Shaker | Eppendorf Thermomixer 5436 | 2050-100-05 | |
| Sonicator | USC 300 TH | 142-0084 | |
| Tissue grinding mixer mill | Retsch, Mixer Mill MM 300 | 20.746.0001 | |
| UPLC system | Waters Acquity UPLC system (Waters) | ||
| Vacuum concentrator | Scan Speed Maxi Vac Alpha Evaporators | 7.008.500.002 | |
| Vortex mixer | Vortex-Genie 2, Model G560 | SI-0236 | |
| Software | |||
| MetAlign | Chromatogram processing | ||
| MzMine | Chromatogram processing | ||
| R package "data.table" | |||
| R package "fujiplot" | pleiotrpoic map | ||
| R package "genetics" | |||
| R package "Ime4" | BLUPs calculation | ||
| R package "LDheatmap" | LD plots | ||
| R package "MASS" | transformation | ||
| R package "rMVP" | GWAS | ||
| R version 4.0.4 | |||
| RefinerMS | Chromatogram processing | ||
| RefinerMS Genedata | Expressionist | Chromatogram processing | |
| Tassel 5 | Genotype filtering | ||
| Xcalibur | Thermo Fisher Scientific | OPTON-30965 | Chromatogram processing |
References
- Doerr, A. Global metabolomics. Nature Methods. 14 (1), 32 (2017).
- Fessenden, M. Metabolomics: Small molecules, single cells. Nature. 540 (7631), 153-155 (2016).
- Oliver, S. G., Winson, M. K., Kell, D. B., Baganz, F. Systematic functional analysis of the yeast genome. Trends in Biotechnology. 16 (9), 373-378 (1998).
- Fiehn, O. Metabolomics-the link between genotypes and phenotypes. Plant Molecular Biology. 48 (1), 155-171 (2002).
- Wu, S., et al. Mapping the Arabidopsis metabolic landscape by untargeted metabolomics at different environmental conditions. Molecular Plant. 11 (1), 118-134 (2018).
- Sysi-Aho, M., Katajamaa, M., Yetukuri, L., Orešič, M. Normalization method for metabolomics data using optimal selection of multiple internal standards. BMC Bioinformatics. 8 (1), 93 (2007).
- Chen, M., Rao, R. S. P., Zhang, Y., Zhong, C. X., Thelen, J. J. A modified data normalization method for GC-MS-based metabolomics to minimize batch variation. SpringerPlus. 3 (1), 439 (2014).
- Dunn, W. B., et al. Metabolic profiling of serum using Ultra Performance Liquid Chromatography and the LTQ-Orbitrap mass spectrometry system. Journal of Chromatography B. 871 (2), 288-298 (2008).
- Fiehn, O., et al. Metabolite profiling for plant functional genomics. Nature Biotechnology. 18 (11), 1157-1161 (2000).
- vander Kloet, F. M., Bobeldijk, I., Verheij, E. R., Jellema, R. H. Analytical error reduction using single point calibration for accurate and precise metabolomic phenotyping. Journal of Proteome Research. 8 (11), 5132-5141 (2009).
- Folch, J., Lees, M., Stanley, G. H. S. A simple method for the isolation and purification of total lipides from animal tissues. Journal of Biological Chemistry. 226 (1), 497-509 (1957).
- Fukushima, A., et al. Impact of clock-associated Arabidopsis pseudo-response regulators in metabolic coordination. Proceedings of the National Academy of Sciences of the United States of America. 106 (17), 7251-7256 (2009).
- Kerwin, R. E., et al. Network quantitative trait loci mapping of circadian clock outputs identifies metabolic pathway-to-clock linkages in Arabidopsis. The Plant Cell. 23 (2), 471-485 (2011).
- Tohge, T., et al. From models to crop species: Caveats and solutions for translational metabolomics. Frontiers in Plant Sciences. 2, 61 (2011).
- Salem, M., Bernach, M., Bajdzienko, K., Giavalisco, P. A simple fractionated extraction method for the comprehensive analysis of metabolites, lipids, and proteins from a single sample. Journal of Visualized Experiments:JoVE. (124), e55802 (2017).
- Tohge, T., Fernie, A. R. Combining genetic diversity, informatics and metabolomics to facilitate annotation of plant gene function. Nature Protocols. 5 (6), 1210-1227 (2010).
- Lisec, J., Schauer, N., Kopka, J., Willmitzer, L., Fernie, A. R. Gas chromatography mass spectrometry-based metabolite profiling in plants. Nature Protocols. 1 (1), 387-396 (2006).
- Osorio, S., Do, P. T., Fernie, A. R., Hardy, N. W., Hall, R. D. . Plant Metabolomics: Methods and Protocols. , 101-109 (2012).
- De Vos, R. C. H., et al. Untargeted large-scale plant metabolomics using liquid chromatography coupled to mass spectrometry. Nature Protocols. 2 (4), 778-791 (2007).
- Perez de Souza, ., Alseekh, L., Naake, S., Fernie, T., A, Mass spectrometry-based untargeted plant metabolomics. Current Protocols in Plant Biology. 4 (4), 20100 (2019).
- Pluskal, T., Castillo, S., Villar-Briones, A., Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics. 11 (1), 395 (2010).
- Watson, J. T., Sparkman, D. O. Electron Ionization. Introduction to mass spectrometry: Instrumentation, applications and strategies for data interpretation. , 315 (2007).
- Fernie, A. R., et al. Recommendations for reporting metabolite data. The Plant Cell. 23 (7), 2477 (2011).
- Treutler, H., et al. Discovering regulated metabolite families in untargeted metabolomics studies. Analytical Chemistry. 88 (16), 8082-8090 (2016).
- Wang, M., et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nature Biotechnology. 34 (8), 828-837 (2016).
- Naake, T., Fernie, A. R. MetNet: Metabolite network prediction from high-resolution mass spectrometry data in R aiding metabolite annotation. Analytical Chemistry. 91 (3), 1768-1772 (2019).
- Chambers, J. M. . Statistical models in S. , (1991).
- Misra, B. B. Data normalization strategies in metabolomics: Current challenges, approaches, and tools. European Journal of Mass Spectrometry. 26 (3), 165-174 (2020).
- Livera, A. M. D., et al. Statistical methods for handling unwanted variation in metabolomics data. Analytical Chemistry. 87 (7), 3606-3615 (2015).
- Sakia, R. M. . The Box-Cox transformation technique: a review. 41 (2), 169-178 (1992).
- vanden Berg, R. A., Hoefsloot, H. C. J., Westerhuis, J. A., Smilde, A. K., vander Werf, M. J. Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics. 7, 142 (2006).
- Marees, A. T., et al. A tutorial on conducting genome-wide association studies: Quality control and statistical analysis. International Journal of Methods in Psychiatric Research. 27 (2), 1608 (2018).
- Torkamaneh, D., Laroche, J., Bastien, M., Abed, A., Belzile, F. Fast-GBS: a new pipeline for the efficient and highly accurate calling of SNPs from genotyping-by-sequencing data. BMC Bioinformatics. 18 (1), 5 (2017).
- Zhao, S., Agafonov, O., Azab, A., Stokowy, T., Hovig, E. Accuracy and efficiency of germline variant calling pipelines for human genome data. Scientific Reports. 10 (1), 20222 (2020).
- Bradbury, P. J., et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 23 (19), 2633-2635 (2007).
- Bates, D., Mächler, M., Bolker, B., Walker, S. Fitting linear mixed-effects models using lme4. Journal of Statistical Software. 67 (1), (2015).
- Yin, L., et al. rMVP: A memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genomics, Proteomics & Bioinformatics. , (2021).
- Kanai, M., et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nature Genetics. 50 (3), 390-400 (2018).
- Salem, M. A., et al. An improved extraction method enables the comprehensive analysis of lipids, proteins, metabolites and phytohormones from a single sample of leaf tissue under water-deficit stress. Plant Journal: for Cell and Molecular Biology. 103 (4), 1614-1632 (2020).
- Balcke, G. U., et al. Multi-omics of tomato glandular trichomes reveals distinct features of central carbon metabolism supporting high productivity of specialized metabolites. The Plant Cell. 29 (5), 960-983 (2017).
- Leonova, T., et al. Does protein glycation impact on the drought-related changes in metabolism and nutritional properties of mature pea (Pisum sativum L.) seeds. International Journal of Molecular Sciences. 21 (2), 567 (2020).
- Alfonsi, K., et al. chemistry tools to influence a medicinal chemistry and research chemistry based organisation. Green Chemistry. 10 (1), 31-36 (2008).
- Bozek, K., et al. Organization and evolution of brain lipidome revealed by large-scale analysis of human, chimpanzee, macaque, and mouse tissues. Neuron. 85 (4), 695-702 (2015).
- Delgado, R., Muñoz, Y., Peña-Cortés, H., Giavalisco, P., Bacigalupo, J. Diacylglycerol activates the light-dependent channel TRP in the photosensitive microvilli of Drosophila melanogaster photoreceptors. The Journal of Neuroscience. 34 (19), 6679 (2014).
- Sharma, D. K., et al. UPLC-MS analysis of Chlamydomonas reinhardtii and Scenedesmus obliquus lipid extracts and their possible metabolic roles. Journal of Applied Phycology. 27 (3), 1149-1159 (2015).
- Dunn, W. B., Wilson, I. D., Nicholls, A. W., Broadhurst, D. The importance of experimental design and QC samples in large-scale and MS-driven untargeted metabolomic studies of humans. Bioanalysis. 4 (18), 2249-2264 (2012).
- Fan, S., et al. Systematic error removal using random forest for normalizing large-scale untargeted lipidomics data. Analytical Chemistry. 91 (5), 3590-3596 (2019).
- Larsson, S. J., Lipka, A. E., Buckler, E. S. Lessons from Dwarf8 on the strengths and weaknesses of structured association mapping. PLOS Genetics. 9 (2), 1003246 (2013).
- Platt, A., Vilhjálmsson, B. J., Nordborg, M. Conditions under which genome-wide association studies will be positively misleading. Genetics. 186 (3), 1045-1052 (2010).
- Nyholt, D. R. A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. American Journal of Human Genetics. 74 (4), 765-769 (2004).
- Teo, Y. Y. Common statistical issues in genome-wide association studies: a review on power, data quality control, genotype calling and population structure. Current Opinion in Lipidology. 19 (2), 133-143 (2008).
- Privé, F., Aschard, H., Ziyatdinov, A., Blum, M. G. B. Efficient analysis of large-scale genome-wide data with two R packages: bigstatsr and bigsnpr. Bioinformatics. 34 (16), 2781-2787 (2018).
- Alseekh, S., et al. Domestication of crop metabolomes: desired and unintended consequences. Trends in Plant Science. 26 (6), 650-661 (2021).
- Yano, K., et al. GWAS with principal component analysis identifies a gene comprehensively controlling rice architecture. Proceedings of the National Academy of Sciences of the United States of America. 116 (42), 21262 (2019).
- Wu, S., et al. Mapping the Arabidopsis metabolic landscape by untargeted metabolomics at different environmental conditions. Molecular Plant. 11 (1), 118-134 (2018).
- Ye, J., et al. An InDel in the promoter of Al-ACTIVATED MALATE TRANSPORTER9 selected during tomato domestication determines fruit malate contents and aluminum tolerance. The Plant Cell. 29 (9), 2249-2268 (2017).
- Zhang, W., et al. Genome assembly of wild tea tree DASZ reveals pedigree and selection history of tea varieties. Nature Communications. 11 (1), 3719 (2020).
- Tohge, T., Fernie, A. R. Annotation of plant gene function via combined genomics, metabolomics and informatics. Journal of Visualized Experiments: JoVE. (64), e3487 (2012).
Explore More Articles
This article has been published
Video Coming Soon
SOBRE A JoVE
Copyright © 2024 MyJoVE Corporation. Todos os direitos reservados