A Simple and Efficient Approach to Construct Mutant Vaccinia Virus Vectors
* These authors contributed equally
In This Article
Summary
Vaccinia virus (VV) has been widely used in biomedical research and the improvement of human health. This article describes a simple, highly efficient method to edit the VV genome using a CRISPR-Cas9 system.
Abstract
The CRISPR-associated endonuclease Cas9 can edit particular genomic loci directed by a single guide RNA (gRNA). The CRISPR/Cas9 system has been successfully employed for editing genomes of various organisms. Here we describe a protocol for editing the vaccinia virus (VV) genome in the cytoplasm of VV-infected CV-1 cells using the RNA-guided Cas9. RNA-guided Cas9 induces double-stranded DNA breaks facilitating homologous recombination efficiently and specifically in the targeted site of VV and a transgene can be incorporated into these sites by homologous recombination. By using a site-specific homologous vector with transgene(s), a N1L gene-deleted VV with the red fluorescence protein (RFP) gene incorporated in this region was generated with a successful recombination efficiency 10 times greater than that obtained from the conventional homologous recombination method. This protocol demonstrates successful use of RNA-guided Cas9 system to generate mutant VVs with enhanced efficiency.
Introduction
Vaccinia virus (VV) is an enveloped DNA virus belonging to the poxvirus family and has played a crucial role in one of the greatest achievements in medicine of the eradication of smallpox. In the post-eradication era of smallpox, VV has been developed as a vector for delivering genes for vaccines against HIV and other infectious diseases1-7 by inserting genes from various pathogens into VV vectors. VV has also been extensively used as a vector for cancer immunotherapy8-14 especially for the development of tumor-targeted replicating oncolytic vaccinia virus. In order to create an efficient vaccine vector with improved selectivity for cancer cells, modifications within the viral genome are required, including gene deletions or introduction of therapeutic genes.
With the development of DNA technology and a better understanding of molecular biology and virology, insertion of foreign DNA into VV was originally achieved using homologous recombination (HR) in 1980s 15. This method is still widely used for constructing VV vectors. Introduction of the genetic modification is achieved by using a shuttle vector for HR, which recombines with the VV genome in pre-infected cells. However, this method has proven to be highly inefficient (less than 1% homologous recombination efficiency16) and often results in random insertion of the selection marker into non-targeted regions and/or loss of the marker upon virus expansion.
The efficiency of DNA homologous recombination for inserting exogenous DNA at genomic loci can be dramatically enhanced in the presence of double-strand breaks (DSBs) 17. Therefore, the technology that can induce DSBs at target loci holds great potential for genome engineering of VV.
The recently developed CRISPR-Cas9 system shows promise for triggering DSBs in any VV gene region. CRISPR-Cas9 is an RNA-guided nuclease involved in adaptive immunity against invading phages and other foreign genetic materials18-20. There are three CRISPR-Cas systems in a range of microbial species21. The type II CRISPR-Cas system is widely used for editing the genome of eukaryotic cells and large viruses. It consists of the RNA-guided Cas9 endonuclease (from Streptococcus pyogenes), a single guide RNA (sgRNA) and the trans-activating crRNA (tracrRNA) 22-24. The Cas9/sgRNA complex recognizes the complementary 20-nucleotide genomic sequence preceding a 5'-NGG-3' Protospacer adjacent motif (PAM) sequence in mammalian cells22, 23. It has been successfully used for effective generation of genetically modified cells, viruses and animal models22-32.
The CRISPR-Cas9 system has proven to be an efficient tool for genome targeting in combination with homologous recombination in the cytoplasm of VV infected CV-1 cells to generate mutant vaccinia viruses33, 34. In order to extend the potential application of this method, we present detailed information about this system's methodology. The described protocol can be used to create a mutant VV with a particular gene deletion and/or arm the mutant virus with a therapeutic transgene.
Protocol
1. Preparation of Target RNA and Cas9 Constructs and Repair Donor Vector
- Cloning of gRNAs
- Design a guide-RNA target sequence targeting the N1L gene of vaccinia virus following the principle stated previously25. Align the guide-RNA target sequence against the genome of the vaccinia virus (in this case, Lister strain of vaccinia virus) to rule out any off-target gRNA target sequences.
- Synthesize and clone gRNA oligos into a gRNA cloning vector as described previously25. Confirm the sequence of each individual gRNA in the resulting vector by Sanger sequencing33. Name the resulting gRNA construct as gRNA N233.
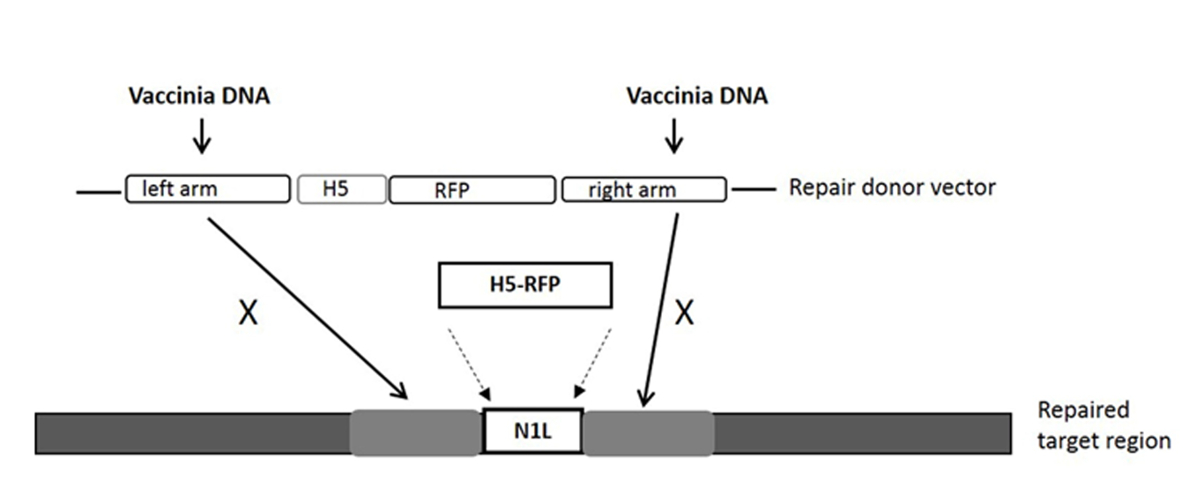
NOTE: The gRNA target site is within the N1L gene, and is in the region between the left arm and right arm that the repair donor vector covers (see Figure 1).
- Clone the Cas9 gene lacking the nuclear localization signal (NLS) into the pST1374 vector and designate the construct as pST-Cas933.
NOTE: Any mammalian expression cloning vector can be used for the construction of the Cas9 expression vector. - Construction of repair donor vector for HR
- Use the N1L gene of VV as the target region for modification33. Ensure that the lengths of the left and right arms are about 500-600 bp each, and flanking the target region.
NOTE: The left arm/right arm can have up to 50 bp overlap with the target region (See Figure 1). - Clone the N1L region repair donor vector as described previously33, 34 and designate the repair donor vector as pT-N1L.
NOTE: See Figure 1 for the repair donor vector and its target region. Other regions of the VV may be targeted using region (gene)-specific gRNA(s) and region-specific repair donor vector. Any cloning vector can be used for the construction of the repair donor vector.
- Use the N1L gene of VV as the target region for modification33. Ensure that the lengths of the left and right arms are about 500-600 bp each, and flanking the target region.
- Expansion of plasmid
- Transform the plasmid into chemically competent E. coli according to the manufacturer's instructions. Purify the plasmid using a plasmid extraction kit referring to the protocol supplied by the manufacturer.
2. Seeding Cells (Day 1)
- Maintain CV-1 cells (Monkey kidney fibroblasts) in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 5% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 µg/mL streptomycin, at 37 °C with 5% CO2.
- Trypsinize CV-1 cells that are 80-90% confluent
- Remove the cell culture medium from the T-175 flask containing CV-1 cells. Rinse the monolayer with 5 mL of sterile PBS to remove residual serum and aspirate the PBS. Add 2.5 mL 1× Trypsin-EDTA solution to cover the cells and place the flask in 37 °C incubator for approximately 5 min to aid cell detachment.
- Add 10 mL of cell culture medium (see step 2.1) to suspend the cells by gentle pipette action. Count the cell numbers using a hemocytometer.
- Seeding cells in 6-well plates
- Seed 2 × 105 CV-1 cells in 2 mL of cell culture medium into each well of a 6-well plate the day before transfection.
NOTE: An additional centrifugation step to remove trypsin is not required.
- Seed 2 × 105 CV-1 cells in 2 mL of cell culture medium into each well of a 6-well plate the day before transfection.
3. Transfection of Cas9 Plasmid and gRNA Plasmid (Day 2)
- Transfect CV-1 cells (prepared in step 2.2.3.1) with 0.5 µg pST-Cas9 plasmid and 0.5 µg gRNA N2 plasmid using a transfection reagent according to the manufacturer's instructions.
- Incubate the cells for 24 h in an incubator (37 °C with 5% CO2), and then replace the medium with 2 mL of fresh cell culture medium (described in step 2.1).
4. Infection of CV-1 Cells and Shuttle Donor Vector Transfection for HR (Day 3)
- Dilute the backbone VVL15 virus with DMEM cell culture medium to a concentration of 2 × 105 pfu/mL. 24 h post-transfection of Cas9 and gRNA plasmids, add 100 µL of the diluted virus into each well of the transfected CV-1 cells.
- 2 h post-virus infection, transfect 1.0 µg shuttle donor vector into VVL15-infected cells for HR using a transfection reagent according to the manufacturer's instructions. Place the transfected cells in a 37 °C incubator with 5% CO2 for 24 h.
5. Harvest the Recombinant Virus (Day 4)
- After 24 h of transfection with the shuttle donor vector for HR in step 4.2, detach the transfected CV-1 cells using a cell scraper. Collect the cell suspension into a cryovial and store at -80 °C for future use.
NOTE: Wipe off any cell culture medium spillage with disinfectant immediately and then wipe again with 70% ethanol.
6. Purification of the Modified VV (First Round)
- On day 1, seed 15 6-well plates with 3 × 105 CV-1 cells in each well.
- On day 2, thaw the frozen cell suspension from step 5.1 in a water bath at 37 °C for 3 min and vortex the cryovials vigorously for 30 s to obtain cell lysate without removing cell debris.
- For the infection of one 6-well plate of CV-1 cells, dilute 1 µL of cell lysate with 3 mL of DMEM and add 0.5 mL of the diluted cell lysate into each well of the 6-well plate, on top of media already present. Infect all the 6-well plates prepared in step 6.1.
NOTE: Removal of cell debris is not required. - After two days of infection (i.e., on day 4), identify RFP-positive plaques under a fluorescence microscope using 10X objective lens and label the plaques underneath the plate using a marker pen by circling the plaque location.
NOTE: See Figure 3 for representative RFP-positive plaque.- Prepare six labelled cryovials containing 200 µL serum-free DMEM cell culture medium to pick up six different RFP-positive plaques. Aspirate the cell culture medium from the well with labelled plaque(s) (step 6.3). Attach a 200 µL tip to a P200 pipette, set the volume at 30 µL and take ~ 10 µL cell culture medium from one cryovial.
- Hold the pipette push button without releasing the medium and use the tip to scratch the area of one labelled plaque. Release the pipette push button to lift up the detached cells and transfer it into the cryovial containing 190 µL of serum-free DMEM.
- Take another 10 µL medium from the vial with the scratched cells (from last step) and repeat the procedure of picking up the same RFP-positive plaque three times to collect as many scratched cells as possible. Transfer all cells into the same vial and store the vial at -80 °C.
NOTE: Alternatively, freeze the cryovials on dry ice for 10 min if the second round of purification is performed immediately.
7. Purification of the Modified VV (Second Round)
- Seed 3 × 105 CV-1 cells into each well of six 6-well plates and culture the cells for 24 h.
- Thaw the frozen cryovials (from step 6.3.3) in 37 °C water bath for 3 min, then vortex the cryovials vigorously for 30 sec to obtain cell lysate.
- Add 0.5 µL of the mixed cell lysate to one well of a 6-well plate. Infect one 6-well plate for each plaque. Incubate the VV-infected 6-well plates at 37 °C with 5% CO2 for 2 days (Second round plaque purification). Then repeat section 6.3.
NOTE: More than six RFP-positive plaques can be picked up; two or more 6-well plates can be used for purification of each plaque.
- Add 0.5 µL of the mixed cell lysate to one well of a 6-well plate. Infect one 6-well plate for each plaque. Incubate the VV-infected 6-well plates at 37 °C with 5% CO2 for 2 days (Second round plaque purification). Then repeat section 6.3.
8. Further Rounds of Plaque Purification
- Carry on a further three to five rounds of purification following the same protocol described in step 7 until all the plaques formed from one positive plaque appear to be RFP-positive under a microscope.
NOTE: This plaque then is considered pure. - Once the plaque is pure (Figure 4, all infected cells expressing RFP), harvest a positive plaque into a cryovial and store at -80 °C as described in section 6.3. Verify the plaque following the protocol described in step 9 and 10.
9. Expand the Purified Plaque(s)
- Seed 3 × 105 CV-1 cells into each well of a 6-well plate one day before virus infection.
- Thaw the frozen cryovial from 8.2 in a 37 °C water bath for 3 min to obtain cell lysate. Add all the lysate (without removing cell debris) into one well of a 6-well plate of CV-1 cells (seeded in step 9.1). Incubate the infected CV-1 cells at 37 °C with 5% CO2 for up to 3 days.
- Expand all the purified plaques (at least three plaques).
NOTE: The infection time varies from 1-3 days depending on the amount of virus in the lysate.
- Expand all the purified plaques (at least three plaques).
- Harvest the VV-infected cells using a cell scraper when more than 50% cells show cytopathic effect observed under a microscope (cells are rounded, easily detached and RFP positive). Collect the detached cells together with cell culture medium into a 15 mL tube.
- Pellet the cells by centrifugation at 300 g for 3 min. Remove the supernatant and keep the cell pellet at -80 °C until further use or use the cell pellet immediately for step 10.
10. Verification of the Modification of Mutant VV Using PCR
- Extract VV DNA from the cell pellet prepared in step 9.3.1 using a DNA extraction kit following the manufacturer's protocol. Elute DNA with 200 µL of double-distilled water.
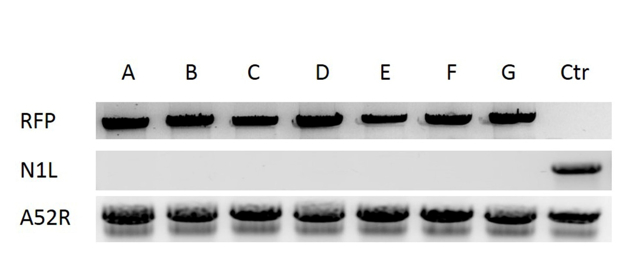
NOTE: Use half of the volume of the pellet to extract DNA and keep the rest for viral production if the clone is verified as containing the correct modification/insertion. - Verification of the deletion of the N1L gene and the insertion of RFP gene into the N1L region.
- Amplify a DNA fragment spanning the N1L gene (L025) and the L026 gene using primers designed against the N1L gene. Amplify the RFP gene using RFP -specific primers. Amplify a control DNA fragment spanning A52R by PCR using A52R-specific primers.
NOTE: See List of Materials for specific primers.- Perform the PCR using a PCR master mix kit. Set up 25 µL of PCR reaction using 2 µL of DNA template, denature the PCR reaction at 94 °C for 2 min, and then run 30 cycles of PCR following these steps: denature the DNA at 94 °C for 15 s, then anneal the primers to the DNA templates at 52 °C for 15 s extend the PCR reaction at 72 °C for 30 s.
- Amplify a DNA fragment spanning the N1L gene (L025) and the L026 gene using primers designed against the N1L gene. Amplify the RFP gene using RFP -specific primers. Amplify a control DNA fragment spanning A52R by PCR using A52R-specific primers.
- Resolve the PCR products using a 1% agarose gel 33. Capture the gel image using a UV gel doc. If the N1L PCR is negative and A52R and RFP PCRs are positive, then the plaque is correctly modified in the N1L region.
Results
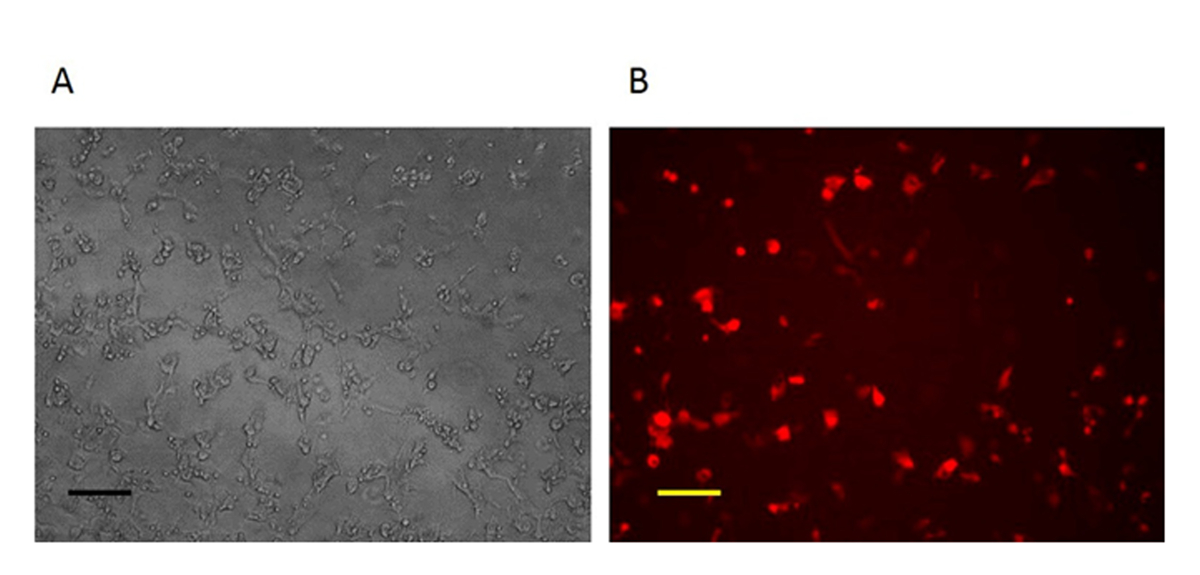
For construction of a new VV vector, the key starting point is to design and construct a donor shuttle vector that can target a particular region of the genome. In this study, the VV N1L gene was used as an example target. The cassette of the donor shuttle vector for the recombination and the targeted region in the VV are shown in Figure 1. In order to enhance the efficiency of the homologous recombination, plasmids expressing Cas9 and a N1L-specific gRNA were co-transfected into the CV-1 cells 48 h prior to performing the homologous recombination33. One day (24 h) post-transfection, RFP was expressed in the CV-1 cells (Figure 2). After infection of CV-1 cells with the whole lysate from the homologous recombination (step 5), RFP-positive and negative plaques were both observed in the CV-1 cells (Figure 3). Following the purification protocol described, a pure plaque could be obtained after 3-5 rounds of purification. As shown in Figure 4, all plaques after infection with cell lysate derived from a pure plaque with the mutant vaccinia virus presented an RFP signal. To verify whether the pure mutant VV had the correct gene modification, PCR was used to confirm the absence of the targeted N1L gene, as shown in Figure 5. PCR of the modified virus showed a positive signal for RFP and an absent signal for N1L (Figure 5 lane A-G), while the PCR results of N1L for the control virus (Ctr) with an intact N1L region is positive. The control DNA fragments of A52R were positive for all recombinant viruses and the Ctr virus. The HR efficiency in RFP-positive plaques is 100% (6/6) in this experiment (it was up to 94% in the previous work34). The method described herein has improved the recombination efficiency by more than 100 fold compared to conventional protocols33, 34.

Figure 1: The cassette of the donor shuttle vector for the homologous recombination, and the targeted region in the VV genome. The purification marker gene RFP is driven by the H5 promoter. The repair donor vector targeting the N1L region results in the RFP gene being incorporated into the N1L region after homologous recombination. 'X' indicates the homologous sequence in the repair donor vector that will replace the same sequence on the vaccinia virus genome. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Imaging of the CV-1 cells one day after homologous recombination. One day after homologous recombination, the cells infected with VV and transfected by repair donor vector are RFP-positive. A: Phase contrast image (original magnification 100X). B: Fluorescence microscopy image (original magnification 100X). Scale bar = 50 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: An RFP-positive plaque in the first round purification of mutant virus. In the first-round purification of the mutant virus, the RFP-positive plaque (circled with red line) with the target region deletion are surrounded by plaques formed by wild type virus (circled with yellow line). A: Phase contrast image (original magnification 100X). B: Fluorescence microscopy image (original magnification 100X). Scale bar = 50 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: The pure mutant VV. After 3-5 rounds of purification, pure plaques were obtained as all the plaques were RFP-positive. A: Phase contrast image (original magnification 100X). B: Fluorescence microscopy image (original magnification 100X). Scale bar = 50 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Verification of the pure mutant VV. DNA was extracted from VV infected CV-1 cells. The deletion of the target region N1L was verified by PCR using N1L primers, the insertion of RFP into N1L region was confirmed by PCR using RFP primers. Lanes A-F correspond to six purified plaques, lane G is a control plaque with N1L deletion verified in previous work33, lane Ctr (control) is the wild type vaccinia virus (with N1L region intact). A52R gene was amplified as the control gene for all the samples. Please click here to view a larger version of this figure.
{kind=link}
Discussion
There are two main goals regarding modification of VV for therapeutic purposes. One is to delete a particular gene, such as the thymidine kinase (TK) gene to attenuate virus for anti-tumor therapeutic use. The other is to arm VV with a desired therapeutic gene (such as GM-CSF) or a marker gene (such as luciferase gene). Achieving either of these involves the deletion of a target region/gene. A direct DNA ligation method35 and an in vitro packaging method36 have been used previously to generate mutant vaccinia viruses with TK gene modifications. However, these methods have limited application regarding mutation of other regions in the genome due to a lack of unique restriction enzyme sites across the genome. The current approach to creating mutant VV involves transfection of a shuttle (donor) vector with a selection marker (RFP or GFP) into CV-1 cells two hours after infection with VV to induce a homologous recombination incorporating the selection marker into the target region. Selection of marker-positive plaques is followed by their purification 24 h post-transfection. The plaque purification process can take up to 10 rounds, last 3-4 weeks and often leads to undesired recombinant viruses with selection markers inserted in off-target regions. DNA double-strand breaks can effectively induce homologous recombination in mammalian cells 37, and by harnessing this mechanism, the efficiency of generating mutant VV can be vastly improved. The gRNA-guided Cas9 system has been successfully employed in genome editing and enhances the efficiency of homologous recombination in eukaryotic organisms22, 25. Recently, in host cells the genomes of adenovirus and type I herpes simplex virus were edited by the gRNA-guided Cas9 system 24. It has been recently demonstrated that CRISPR Cas9 system is a powerful tool for efficiently editing the VV genome and constructing a VV vector for expressing a particular gene.
Both N1L gRNA-guided Cas9 and the traditional homologous recombination (HR) method resulted in successful HR events at the same target site of N1L. Interestingly, however gRNA-guided Cas9 induced HR at much higher levels of efficiency than the traditional HR method. Thus, the use of gRNA-guided Cas9 eliminates the need to purify as many plaques to obtain mutant VVs. In this work, we significantly improved the efficiency and time-frame for generation of mutant VV using the gRNA-guided Cas9 system33. Of note, one critical step for obtaining a high efficiency of homologous recombination is to transfect CV-1 cells with the plasmids expressing Cas9 and the gRNA for 24 h before performing the conventional homologous recombination. The 24 h interval allows Cas9 and gRNA to be expressed at a reasonable level. Furthermore, the gRNA sequence targeting a particular gene may need to be optimized as we found that the different gRNA sequences targeting N1L gene varied in their efficiency33, 34.
The limitation for the CRISPR/Cas9 in editing VV is the low rate of RFP-positive plaques in the first round of recombination. To obtain a sufficient number of RFP-positive plaques containing recombinant virus, usually at least ten 6-well plates are needed, which makes first-round screening tedious. Hence, there is still a need to optimize the protocol to overcome this limitation.
The small size of RFP-positive plaques is another potential problem, which is due to a high volume of lysate used to infect a 6-well plate. To overcome this, a smaller volume of lysate should be used to infect a 6-well plate to obtain larger size RFP-positive plaques. Using a smaller volume of lysate will also enable for better separation of RFP-positive plaques from the RFP-negative ones. Consequently fewer rounds of plaque purification are required to obtain pure RFP-positive plaques.
Off-target effects are always an unwanted issue in gene editing. CRISPR-Cas9 has shown off-target effects. However, with careful design of gRNA, off-target effects can be avoided when editing the VV genomes33, 34. This is the particular benefit of using the gRNA-guided Cas9 system for editing the genome of VV. Given the promises of VV, using the CRISPR/Cas9 system will speed up the development of mutant VVs for biomedical research and in translational medicine. In addition, such a system would also expedite discoveries in cell biology, such as dissection of the signaling pathways used by VV for its actin-based motility.
Disclosures
Authors have no competing financial interests.
Acknowledgements
This work was supported by The MRC DPFS grant (MR/M015696/1) and Ministry of Sciences and Technology of China (2013DFG32080).
Materials
| Name | Company | Catalog Number | Comments |
| Plasmid #41824 | 41824 | Addgene | gRNA cloning vector |
| pST1374 | 13426 | Addgene | Cas9 cloning vector |
| pGEMT easy | A1360 | promega | repair donor vector cloning |
| One Shot TOP10 Chemically Competent E .Coli | C4040-10 | Invitrogen | transformation |
| QIAprep Spin Miniprep Kit | 27106 | Qiagen | plasmid extraction |
| Dulbecco’s Eagle’s Medium (DMEM) | 11965-092 | Life Technologies | cell culture medium |
| fetal bovine serum (FBS) | SH30088.03HI | Hyclone | cell culture serum |
| penicillin, streptomycin | 15070-063 | Thermo Scientific | antibiotics |
| Effectene | 301425 | Qiagen | transfection |
| Thermo Scientific Nalgene Cryogenic vial; 2.0 mL | W-06754-96 | Thermo Scientific | collect virus |
| fluorescence microscope | Olympus BX51 Fluorescence | Olympus | find RFP-positive plque |
| DNeasy Blood & Tissue Kit | 69506 | Qiagen | DNA extraction |
| Extensor Long PCR ReddyMix Master Mix | AB-0794/B | Thermo Scientific | PCR reagent |
| 10cm culture dish | Z688819-6EA | sigma | cell culture |
| 6-well plate | Z707759 | sigma | cell culture |
| cell scraper | C5981 | sigma | scrap cells |
| 1XTrypsin-EDTA solution | T4299 | sigma | trypsinize cells |
| Virkon | 95015661 | Anachem Ltd | desinfectant |
| Falcon tube | CLS430791-500EA | Sigma | hold cell suspension |
| N1L forward 5’-TATCTAGCAATGGACCGT-3’ | Sigma | PCR primer | |
| N1L reverse 5’-CCGAAGGTAGTAGCATGGA-3' | Sigma | PCR primer | |
| A52R forward 5’-ATAGGATTGTGTGCATGC-3’ | Sigma | PCR primer | |
| A52R reverse 5’-TTGCGGTATATGTATGAGGTG-3’ | Sigma | PCR primer | |
| RFP forward 5’- GCTACCGGACTCAGATCCA-3’ | Sigma | PCR primer | |
| RFP reverse 5’-CGCCTTAAGATACATTGATGAG-3’ | Sigma | PCR primer | |
| Argarose | A7431 | Sigma | resolve PCR product |
| G:BOX F3 | G:BOX F3 | Syngene | UV gel doc |
References
- Baroudy, B. M., Venkatesan, S., Moss, B. Incompletely base-paired flip-flop terminal loops link the two DNA strands of the vaccinia virus genome into one uninterrupted polynucleotide chain. Cell. 28, 315-324 (1982).
- Madigan, M. T., Martinko, J. M., Parker, J., Brock, T. D. . Brock biology of microorganisms, 9th edn. , (2000).
- Cooney, E. L., et al. Safety of and immunological response to a recombinant vaccinia virus vaccine expressing HIV envelope glycoprotein. Lancet. 337, 567-572 (1991).
- Mackett, M., Yilma, T., Rose, J. K., Moss, B. Vaccinia virus recombinants: expression of VSV genes and protective immunization of mice and cattle. Science. 227, 433-435 (1985).
- Legrand, F. A., et al. Induction of potent humoral and cell-mediated immune responses by attenuated vaccinia virus vectors with deleted serpin genes. J Virol. 78, 2770-2779 (2004).
- Panicali, D., Davis, S. W., Weinberg, R. L., Paoletti, E. Construction of live vaccines by using genetically engineered poxviruses: biological activity of recombinant vaccinia virus expressing influenza virus hemagglutinin. Proc Natl Acad Sci. 80, 5364-5368 (1983).
- Wiktor, T. J., et al. Protection from rabies by a vaccinia virus recombinant containing the rabies virus glycoprotein gene. Proc Natl Acad Sci. 81, 7194-7198 (1984).
- Gallucci, S., Matzinger, P. Danger signals: SOS to the immune system. Curr Opin Immunol. 13, 114-119 (2001).
- Matzinger, P. The danger model: a renewed sense of self. Science. 296, 301-305 (2002).
- Guo, Z. S., Bartlett, D. L. Vaccinia as a vector for gene delivery. Expert Opin Biol Ther. 4, 901-917 (2004).
- Kwak, H., Horig, H., Kaufman, H. L. Poxviruses as vectors for cancer immunotherapy. Curr Opin Drug Discov Devel. 6, 161-168 (2003).
- Tysome, J. R., et al. Lister strain of vaccinia virus armed with endostatin-angiostatin fusion gene as a novel therapeutic agent for human pancreatic cancer. Gene Ther. 16, 1223-1233 (2009).
- Kirn, D. H., Wang, Y., Liang, W., Contag, C. H., Thorne, S. H. Enhancing poxvirus oncolytic effects through increased spread and immune evasion. Cancer Res. 68, 2071-2075 (2008).
- Park, B. H., et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 9, 533-542 (2008).
- Panicali, D., Paoletti, E. Construction of poxviruses as cloning vectors: insertion of the thymidine kinase gene from herpes simplex virus into the DNA of infectious vaccinia virus. Proc Natl Acad Sci. 79, 4927-4931 (1982).
- Ball, L. A. High-frequency homologous recombination in vaccinia virus DNA. J Virol. 61, 1788-1795 (1987).
- Rouet, P., Smih, F., Jasin, M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol. 14, 8096-8106 (1994).
- Jansen, R., Embden, J. D., Gaastra, W., Schouls, L. M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol. 43, 1565-1575 (2002).
- Brouns, S. J., et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 321, 960-964 (2008).
- van der Oost, J., Jore, M. M., Westra, E. R., Lundgren, M., Brouns, S. J. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem Sci. 34, 401-407 (2009).
- Jinek, M., et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 343, 1247997 (2014).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339, 819-823 (2013).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337, 816-821 (2012).
- Bi, Y., et al. High-efficiency targeted editing of large viral genomes by RNA-guided nucleases. PLoS Pathog. 10, 1004090 (2014).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339, 823-826 (2013).
- Yang, L., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 41, 9049-9061 (2013).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 31, 827-832 (2013).
- Ran, F. A., et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 154, 1380-1389 (2013).
- Niu, Y., et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell. 156, 836-843 (2014).
- Yin, H., et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol. 32, 551-553 (2014).
- Wang, H., et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 153, 910-918 (2013).
- Ma, Y., et al. Generating rats with conditional alleles using CRISPR/Cas9. Cell Res. 24, 122-125 (2014).
- Yuan, M., et al. Efficiently editing the vaccinia virus genome by using the CRISPR-Cas9 system. J Virol. 89, 5176-5179 (2015).
- Yuan, M., et al. A marker-free system for highly efficient construction of vaccinia virus vectors using CRISPR Cas9. Mol Ther Methods Clin Dev. 2, 15035 (2015).
- Merchlinsky, M., Moss, B. Introduction of foreign DNA into the vaccinia virus genome by in vitro ligation: recombination-independent selectable cloning vectors. Virology. 190, 522-526 (1992).
- Timiryasova, T. M., Chen, B., Fodor, N., Fodor, I. Construction of recombinant vaccinia viruses using PUV-inactivated virus as a helper. Biotechniques. 31, 534-540 (2001).
- Johnson, R. D., Jasin, M. Double-strand-break-induced homologous recombination in mammalian cells. Biochem Soc Trans. 29, 196-201 (2001).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved