A Cell-based Assay to Investigate Non-muscle Myosin II Contractility via the Folded-gastrulation Signaling Pathway in Drosophila S2R+ Cells
In This Article
Summary
Here we describe a contractility assay using Drosophila S2R+ cells. The application of an exogenous ligand, folded gastrulation (Fog), leads to the activation of the Fog signaling pathway and cellular contractility. This assay can be used to investigate the regulation of cellular contractility proteins in the Fog signaling pathway.
Abstract
We have developed a cell-based assay using Drosophila cells that recapitulates apical constriction initiated by folded gastrulation (Fog), a secreted epithelial morphogen. In this assay, Fog is used as an agonist to activate Rho through a signaling cascade that includes a G-protein-coupled receptor (Mist), a Gα12/13 protein (Concertina/Cta), and a PDZ-domain-containing guanine nucleotide exchange factor (RhoGEF2). Fog signaling results in the rapid and dramatic reorganization of the actin cytoskeleton to form a contractile purse string. Soluble Fog is collected from a stable cell line and applied ectopically to S2R+ cells, leading to morphological changes like apical constriction, a process observed during developmental processes such as gastrulation. This assay is amenable to high-throughput screening and, using RNAi, can facilitate the identification of additional genes involved in this pathway.
Introduction
Studies of embryogenesis conducted with genetic model organisms have proved invaluable to our understanding of how cells are assembled into tissues. Studies of Drosophila melanogaster, in particular, have led to the identification of key genes and biological principles that direct the morphogenesis and development of organisms from fertilization to adulthood1,2,3. In parallel to the genetic research conducted with Drosophila, cultured cell lines derived from fruit fly tissues have also emerged as a powerful system to address a wide range of molecular and cell biological questions4,5,6. Drosophila tissue culture cells have minimal requirements for maintenance, as they are cultured at room temperature and without CO2. As such, they are amenable to high-resolution imaging, and they have a high degree of susceptibility to gene inhibition by RNAi6,7. Many groups have used Drosophila tissue culture cells as a tool to discover genes involved in defining cell shape, cytoskeletal dynamics, viability, phagocytosis, and signal transduction pathways4,8,9,10,11. When employed as a model alongside the whole animal, cultured Drosophila cell lines offer a set of very complementary approaches that accelerate the identification of molecules important for the development and provide a framework in which to determine their mechanistic roles12. Here, we describe a cell-based assay to study the signaling pathways that trigger apical constriction via the folded-gastrulation (Fog) pathway13,14. This cell-based contractility assay allows researchers to investigate both the Fog signaling pathway and the molecular mechanisms that regulate non-muscle myosin II contractility.
Gastrulation of the early Drosophila embryo has been studied for many years as a genetic model for the epithelial morphogenesis and the cellular transition from epithelial to mesenchymal cell identity. One of the key events of gastrulation is the morphogenesis of a subset of epithelial cells along the embryonic ventral midline from columnar to pyramidal in shape15,16,17,18. This simple cell shape change results in the internalization of the presumptive mesodermal cells and is driven by the motor activity of non-muscle myosin II constricting the actin network16,19,20. Decades of genetic research has identified the molecular components of this pathway and has sequentially placed them in the following order: 1) Fog is secreted from the apical domain of the epithelial cells at the ventral midline; 2) Fog binds to the G-protein-coupled co-receptors, Mist and Smog, and signals through a heterotrimeric G-protein complex containing the Gα12/13 subunit Concertina (Cta), which is chaperoned by the non-canonical GEF Ric-8; 3) Cta activates a guanine nucleotide exchange factor, RhoGEF2, which in turn activates the small G-protein Rho1; 4) Rho1 activates Rho kinase (Rok); 5) Rok activates non-muscle myosin II contractility at the apical domain through phosphorylation of the regulatory light chain (RLC), thus producing apical constriction (Figure 1)15,21,22,23,24,25,26,27,28,29. Mutations in several of these components interfere with the normal gastrulation and other morphogenetic movements, including the formation of the wing disc and salivary glands, indicating that this pathway is used at several stages of Drosophila embryogenesis30,31,32. The Fog pathway is one of the best-studied models for epithelial shaping and has provided important insights into how tissue-level morphogenesis is regulated from gene transcription to cytoskeleton-driven cell movements14,15,21.
We developed a cell-based contractility assay that recapitulates many of the cellular responses downstream of Fog that have been observed in developing fly embryos17. We engineered a stable S2 cell line that expresses Fog tagged at its C-terminus with myc under the control of an inducible metallothionine promoter that can be harvested upon the addition of copper sulfate (CuSO4) to the medium. When Fog-conditioned media is applied ectopically to S2 Receptors+ (S2R+) cells, which are a subline of S2 cells distinguished by their differential expression of receptors such as Frizzled and integrin subunits, the cells undergo a reorganization of the cytoskeleton highly reminiscent of apical constriction12,17,27,33. These changes can be observed by phase-contrast microscopy in which Fog treatment leads to the appearance of phase-dark ruffles indicative of a radial increase in non-muscle myosin II contractility, or by fluorescence microscopy where Fog treatment leads to the formation of non-muscle myosin II rings in cells expressing EGFP-tagged RLC34. These rings contain a myosin-phosphorylated regulatory light chain (pRLC) visible via immunostaining23,34,35. This Fog-induced response required Cta, RhoGEF2, Rho, and Rok; thus, using recombinant Fog-Myc and S2R+ cells, we have established a means to investigate Fog-induced constriction in a tissue-culture-based system24,25,34.
Protocol
1. Maintenance of Drosophila Tissue Culture Cells
- Maintain S2R+, S2R+:RLC-EGFP, and S2:Fog-Myc cells at room temperature, preferably between 20 °C and 25 °C, in Shield and Sang M3 insect medium supplemented with 10% fetal bovine serum, 100 units/mL of penicillin, 100 µg/mL streptomycin, and 0.25 amphotericin B at a density no lower than 5 x 105 cells/mL.
NOTE: Lower cell densities lead to death. - Quickly thaw cryo-preserved vials of cells and transfer them to a tissue culture flask. Start with a small tissue culture flask (12.5 cm2 or 25 cm2) in a volume of 4 - 5 mL of cell culture medium until the culture is established and signs of cell death due to thawing have subsided.
NOTE: Signs of cell death due to thawing include non-adherent cells that lack a smooth, rounded shape and small dark bits of cellular debris in the tissue culture medium indicative of necrotic cells. - Slowly scale up the size of the tissue culture by allowing the cells to grow undisturbed for several days (4 - 8 d) to 100% confluency, where there is little to no space between the cells and they begin to pile up on one another. Transfer all the cells from this 100% confluent smaller 12.5-cm2 or 25-cm2 flask to a larger tissue culture flask (of 75 cm2) supplemented with 10 mL of cell culture medium.
NOTE: Maintaining the cells in a 75-cm2 flask will yield enough cells for most experiments. - Passage the cells every 3 - 4 d by pipetting the cell culture medium up and down, using it to dislodge any loosely adhered cells from the tissue culture plastic. Transfer this medium, now containing the resuspended cells, into a fresh flask. Dilute the cells 1:4 with fresh cell culture medium with each passage and repeat as needed.
2. RNAi Treatment of Drosophila S2R+ Cells
NOTE: Refer to Rogers and Rogers6 for detailed instructions on how to produce dsRNA suitable for insect culture.
- Transfer S2R+ cells to a 6-well or 12-well tissue culture plate at a density conducive for the viability, no lower than 5 x 105 cells/mL, using complete Shield and Sang M3 cell culture media for RNAi treatments.
- Working in a tissue culture hood, remove the old cell culture medium by gently tilting the 6-well or 12-well tissue culture plate and aspirating the cell culture medium with a pipette. Quickly replace this old cell culture medium with the 1 mL of fresh cell culture medium by pipetting it into each well.
NOTE: This can be done without substantial loss after the cells have been allowed to loosely adhere for 30 - 45 min prior to exchanging the cell culture medium. - Add 10 µg/mL of dsRNA that has been generated following the detailed protocol in Rogers and Rogers6 by pipetting it directly into the tissue culture medium of each well of cells per RNAi condition.
- Repeat this procedure daily, replacing the cell culture medium with 1 mL of fresh medium and 10 µg/mL of dsRNA per RNAi condition.
NOTE: RNAi experiments are typically carried out for 7 d; however, the exact timing is dependent on protein turnover and efficacy of the dsRNA. RNAi treatments as short as 3 d effectively deplete cells of certain proteins, while other targets require longer treatments6.
3. Fog Production and Harvest
- Scale up the growth of the S2:Fog-Myc cells by allowing them to grow undisturbed for 4 - 8 d to obtain a nearly 100% confluent 75-cm2 flask of cells in 10 mL of cell culture medium (or alternatively, a 150-cm2 flask in 20 mL of cell culture medium) to maximize Fog production.
- Add 50 µL of 100 mM CuSO4 (or 100 µL of 100 mM CuSO4 for a 150-cm2 flask) to the nearly 100% confluent 75-cm2 flask for the induction of the metallothionine promoter. Incubate for 48 h at room temperature.
- Remove the Fog-Myc-containing medium from the tissue culture flask by pipetting and transfer it to a 15-mL or 50-mL conical tube. Centrifuge at 2,500 x g at 4 °C for 10 - 15 min to clear the medium of cells and cellular debris.
- Transfer the cleared cell medium to a new conical tube and maintain it on ice. Working in batches, transfer 5 mL of the cleared medium to 15 mL of 3,000 MW concentrators with regenerated cellulose membranes and centrifuge it at 2,500 x g at 4 °C for 30 min at a time, to concentrate the medium.
NOTE: Alternatively, a concentrator with a larger volume capacity could be used (50 mL). During concentration, a darker colored medium, which is enriched with Fog, will begin to build up at the bottom of the V-shaped filter. - Insert the pipette tip to the bottom of the "V" of the concentrator and remove the darker colored media between each round of centrifugation and transfer it to a fresh 1.7-mL microfuge tube. Maintain the media on ice.
- Using the same concentrator, repeat steps 3.4 and 3.5 by removing the darker colored concentrated Fog-Myc media and replacing it with more precleared medium, until the medium has been concentrated to 1/10th of its original volume (i.e., 1 mL from 10 mL of starting material).
- Store the concentrated Fog-Myc at 4 °C.
NOTE: After storage, be sure to monitor for bacterial contamination, which may result from extended periods of storage. - Perform SDS-PAGE by mixing 10 µL of concentrated Fog-Myc medium with 10 µL of a denaturing SDS-containing buffer to form a lysate. Run the SDS-PAGE gel at 35 mA for approximately 1 h. Transfer the gel to a nitrocellulose membrane (or alternatively, a PVDF membrane) for 1 h at 120 mA. Perform a western blot using any commercially available Myc antibody to detect a 150-kDa band.
NOTE: The concentration of Fog-Myc in the conditioned medium may vary per batch. It is not necessary to determine the exact concentration of Fog-Myc in order to perform the assay, but the efficacy of each batch should be tested prior to performing an experiment. - Following steps 3.1 - 3.2, prepare the control S2-conditioned media by collecting 10 mL of cell culture medium from a nearly 100% confluent 75-cm2 flask of S2 cells (or, alternatively, 20 mL from a 150-cm2 flask) by pipetting. Transfer the control S2 cell culture medium to a fresh 15-mL or 50-mL conical tube.
- Following steps 3.4 - 3.7, clear the lysate of cells and cellular debris by centrifugation (at 2,500 x g at 4 °C for 30 min at a time) and then transfer the cleared cell lysate to concentrators and centrifuge them to concentrate the control medium in batches to 1/10th the original volume.
NOTE: Application of the S2-conditioned media does not lead to substantial cell contractility in the assay. - Store the concentrated S2 control media at 4 °C for several months.
NOTE: After storage, be sure to monitor for the contamination.
4. Preparation of Concanavalin-A-coated Glass-bottomed Dishes and the Plating of S2R+ Cells
- Make 10 mL of the concanavalin A (con A) solution by dissolving 5 mg of con A into 10 mL of water for a final concentration of 0.5 mg/mL. Aliquot this solution into more convenient volumes (500 µL to 1 mL) and store it at -20 °C.
- Prepare 35-mm glass-bottomed dishes by coating the glass portion of each dish with 200 µL of the con A solution and incubate the dishes for approximately 2 min at room temperature in a tissue culture hood. Next, remove all the con A solution (which can be retained and reused for future experiments) and allow the dishes to air-dry completely.
NOTES: Dishes can be prepared in large batches and can be stored for up to 6 months in a dry place such as a lab bench drawer. - Add approximately 2 mL of fresh cell culture media to each glass-bottomed dish. Resuspend the S2R+ cells to be tested by the assay by pipetting the cell culture medium up and down, using it to detach any loosely adhering cells.
- Transfer the resuspended S2R+ cells to the 35-mm glass-bottomed dishes containing the fresh cell culture medium so they are between 50% - 70% confluent. Check the cell density under the tissue culture microscope by focusing up and down through several plains of the cells suspended in the medium to get an estimate of the number of cells.
NOTE: Through trial and error, the appropriate density of cells needed to achieve 50% - 70% confluency can be achieved. - Allow the cells to attach to the con-A-coated glass-bottomed dishes for 45 - 60 min.
NOTE: Fully attached S2R+ cells appear 'fried-egg-like,' flattened and phase-dark.
5. Efficacy Test of the Fog-conditioned Medium, and the Cellular Contractility Assay
- If desired, confirm the yield of Fog-Myc produced and concentrated from the S2:Fog-Myc stable cells lines by SDS-PAGE and western blotting. Add 10 µL of SDS-containing sample buffer to 10 µL of concentrated Fog-Myc and boil it for 5 min. Follow the standard SDS-PAGE and western blotting protocols and blot for anti-Myc to confirm Fog-Myc production.
- Remove all the Shield and Sang M3 medium from the attached S2R+ cells from step 4.3 by gently pipetting, and carefully add back 75 µL of fresh Shield and Sang medium to limit the amount of Fog-conditioned medium used.
- Add 75 µL of Fog-conditioned media to the glass portion of the glass-bottomed dish from step 5.2 to bring the total volume to 150 µL, at a 1:1 ratio of Fog-conditioned media to Shield and Sang M3 media.
NOTE: A total volume of 150 - 200 µL is needed to fully cover the glass portion of a typical glass-bottomed dish; however, this will depend on the dimensions of the glass-bottomed dish used. - Monitor the contractility of the cells by phase-contrast microscopy using 20X or 40X magnification to observe an entire field of cells. Track the morphology of the cells and look for the alternating pattern of phase dark and phase light ruffling indicative of the shape change.
NOTE: The application of Fog typically leads to 30% - 50% of the S2R+ cells undergoing cellular constriction, and after 10 min, the cells begin to relax, returning to their smooth-edged, fried-egg-like morphology. - Monitor the robustness of the cellular contractility. If the cell response is very strong using a 1:1 ratio of Fog-conditioned medium to fresh Shield and Sang medium, reduce the amount of Fog-conditioned medium to conserve the concentrated Fog-Myc medium for future experiments. Use undiluted Fog-conditioned medium if a 1:1 ratio does not lead to robust cellular constriction; generally, a ratio of 1:3 produces enough response to successfully carry out the experiment.
NOTE: Once the appropriate ratio of Fog-conditioned medium to fresh Shield and Sang medium is established per given batch of Fog-conditioned medium, use this ratio for all other experiments. - Monitor non-muscle myosin II dynamics during the cellular contractility assay by using the stable S2R+ cell line that expresses GFP-tagged regulatory light chain (S2R+:Sqh-EGFP).
- Using the established ratio of Fog-conditioned medium to fresh Shield and Sang M3 medium from steps 5.2 - 5.5, remove cell-cultured medium with a pipette and add back fresh Shield and Sang medium in the glass portion of the glassed-bottomed dish. Perfuse Fog-conditioned medium at the predetermined ratio while acquiring an image sequence of the live cells; take care as to not move the glass-bottomed dish during the perfusion.
6. Fixing and Staining of Constricted S2R+ Cells
- Fix S2R+ cells using a 10% paraformaldehyde solution in PEM buffer (100 mM Pipes, 1 mM EGTA, 1 mM MgCl2, pH 6.9).
NOTE: PEM buffer is typically made at 2x stock solution and can be stored for months at 4 °C. - Remove the cell culture medium from Fog-treated S2R+ cells by pipetting and immediately replace it with 1.5 - 2.0 mL of 10% paraformaldehyde solution. Incubate for 15 min at room temperature.
NOTE: Fixed cells can be stored in fixation solution at 4 °C for several days; however, it is important that the initial 15-min fixation is done at room temperature prior to transferring the cells to 4 °C. - Remove the 10% paraformaldehyde solution by pipetting and dispose of this organic waste product properly. Rinse the cells by gently pipetting 1.5 - 2.0 mL of phosphate-buffered saline (PBS) to the 35-mm glass-bottomed dish. Remove the old PBS by pipetting and replace it with 1.5 - 2.0 mL of fresh PBS 2x more to remove residual fixation solution.
NOTE: It is important that the cells do not dry out from this point on in the protocol. - Block the cells with approximately 200 µL of 5% normal goat serum diluted in PBS that has been supplemented with 0.1% non-ionic detergent (PBST) for 20 min at room temperature.
NOTE: Typically, blocking solutions contain serum that matches the organism the secondary antibodies are raised in. - Prepare the primary antibody solution by diluting it in PBST. Use a volume of approximately 200 µL to completely cover the glass portion of the glass-bottomed dish.
- Remove the blocking solution by pipetting and add the primary antibody solution directly to the glass portion of the dish. Incubate for 1 h at room temperature or, alternatively, incubate overnight at 4 °C.
- When incubating overnight at 4 °C, wrap the 35-mm glass-bottomed dish with parafilm to prevent evaporation.
- Remove the primary antibody solution by pipetting and rinse the cells by pipetting 1.5 - 2.0 mL of fresh PBS to the 35-mm glass-bottomed dish. Remove PBS by pipetting and replace it with 1.5 - 2.0 mL of fresh PBS 2x more.
- Prepare a solution of secondary antibody by diluting it in PBST. A volume of 200 µL is adequate to just cover the glass portion of the dish.
- Add the secondary antibody solution directly to the glass portion of the dish. Incubate the cells with secondary antibody solution for 1 h at room temperature or overnight at 4 °C.
- Wrap the 35-mm dish with parafilm if incubating overnight, to prevent evaporation.
- Remove the secondary antibody solution by pipetting and rinse the cells with PBS by pipetting 1.5 - 2.0 mL of fresh PBS to the dish. Remove the old PBS and replace it 2x more, following this same procedure.
- Preserve the fixed and stained cells by adding enough of an anti-fade fluorescent mounting medium to cover the glass portion of the glass-bottomed 35-mm dish.
- Store the samples at 4 °C and away from light.
NOTE: The fluorescent signal will slowly decay over months even if properly stored. For best results, image within a week.
7. Imaging and Quantification of the Cellular Contractility Assay
- Image fixed S2R+ cells using a standard inverted light microscope with phase contrast, DIC, or fluorescence imaging capabilities. Using 20X or 40X magnification, capture 10 - 30 nonoverlapping image fields of cells depending on the cell density, with the goal of acquiring the images of 200 or more cells per condition.
- Using a standard cell counter, count and record the number of contracted (phase-dark, bonneted cells) and noncontracted cells per each field captured.
NOTE: It is best to have two researchers take independent counts of contracted and noncontracted cells per field, or alternatively, the same researcher can count the same image field of cells three times, allowing for an average number of contracted and noncontracted cells per field to be calculated. - Prepare several con-A-coated glass-bottomed dishes per experimental condition as described in step 4.2. Pipette up and down the cell culture medium to dislodge any loosely adherent S2R+:Sqh-EGFP cells and plate them so they are 50% - 70% confluent in the 35-mm glass-bottomed dishes. Allow the cells to attach for 30 - 45 minutes.
- Image the S2R+:Sqh-EGFP cells live using a spinning-disk, swept-field confocal microscope, or by light-sheet microscopy using 60X - 100X magnification. Remove all the Shield and Sang M3 medium from the glass-bottomed dish containing attached S2R+:Sqh-EGFP cells.
- Perfuse the predetermined ratio of Fog-Myc conditioned medium to fresh Shield and Sang M3 medium from steps 5.2 - 5.5 directly onto the glass portion of the glass-bottomed dish using a pipette, careful as to not to disturb the position of the dish on the objective. Acquire images for 12 - 15 min in order to capture the initiation and full contraction of the S2R+:Sqh-EGFP cells.
Note: If staining for non-muscle myosin or myosin-pRLC, the formation of a tight myosin ring at the surface of the cells is also indicative of contractility. If imaging S2R+:Sqh-EGFP cells live, the EGFP-tagged RLC will form similar tight rings (see Figure 3) indicative of contractility. The shape change that occurs will cause the cells to "bonnet", taking up 3D shape; the plane of focus of the microscope may need to be adjusted to track this cell shape change. Under control RNAi conditions, typically, 30% - 50% of the cells undergo constriction upon the addition of Fog. Be aware that some RNAi conditions may lead to constriction in the absence of Fog.
- Perfuse the predetermined ratio of Fog-Myc conditioned medium to fresh Shield and Sang M3 medium from steps 5.2 - 5.5 directly onto the glass portion of the glass-bottomed dish using a pipette, careful as to not to disturb the position of the dish on the objective. Acquire images for 12 - 15 min in order to capture the initiation and full contraction of the S2R+:Sqh-EGFP cells.
Representative Results
S2 and S2:Fog-Myc cells were grown to near confluent conditions in a 150-cm2 tissue culture flask and were treated with 25 - 75 µM CuSO4 over a period of 24 - 48 hours to induce expression of Fog-Myc (Figure 2). Samples from S2 cells (Figure 2A) and S2:Fog-Myc cells (Figure 2B) were removed and Fog-Myc production was monitored by western blot. As expected, we failed to detect Fog-Myc in media harvested from S2 cells under any conditions. However, we did detect Fog in media harvested from the S2:Fog-Myc stable cell line as early as 24 hours after the induction with 75 µM CuSO4. S2R+ cells attached to con-A-coated glass-bottomed dishes expressing an EGFP-tagged regulatory light chain (GFP-RLC) of non-muscle myosin II (Figures 3A and 3B). Live-cell imaging was performed on an inverted microscope equipped with a 100X/1.4 NA objective lens during the perfusion of Fog-conditioned media. Approximately 5 minutes post-treatment with Fog-Myc, the RLC formed rings indicative of non-muscle myosin II constriction. These rings can be observed through immunostaining S2R+ cells with an antibody raised against a synthetic phosphopeptide corresponding to residues surrounding Ser19 of human myosin RLC (Figures 4A and 4B). This antibody cross-reacts with Drosophila RLC.
Here we have a representative example of the cellular contractility assay following a 7-day treatment of cells with control RNAi and dsRNA against Rho1 (Figures 5A - 5E). S2R+ cells were plated on con-A-coated glass-bottomed dishes and treated with Fog-conditioned (+Fog) media or control media (-Fog). The number of contracted cells were then quantified following fixation. The phase-dense ruffles indicative of constriction was prominent in control depleted cells treated with Fog-conditioned media (Figure 5B). In Figure 5C, Rho-depleted cells treated with S2-conditioned medium are a typical example of smooth-edged, fried-egg-like morphology. Note that Rho plays a role in the Fog signaling pathway, as well as in cytokinesis; thus, Rho-depleted cells often fail to properly divide, leading to an increase in cell size and ploidy. Treatment of control depleted cells with Fog-conditioned media typical leads to 30% - 50% of cells undergoing constriction, whereas we failed to observe substantial constriction in Rho-depleted cells.

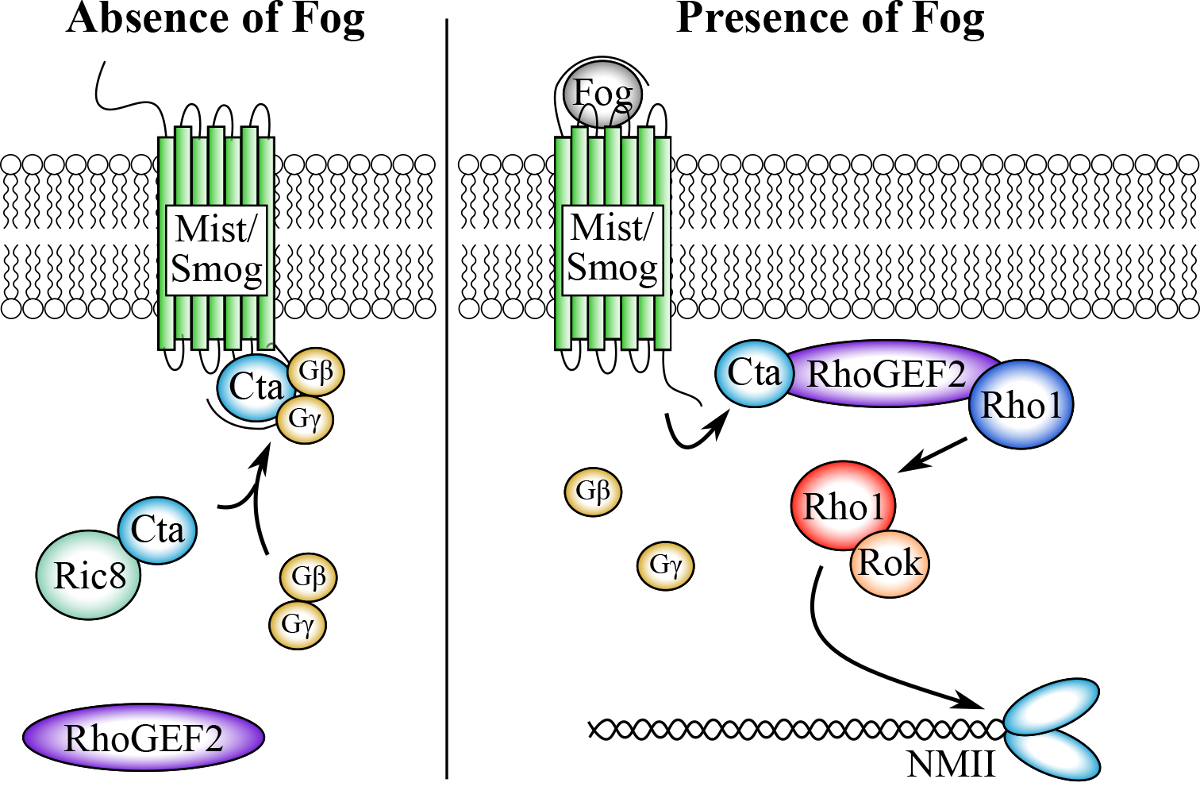
Figure 1:The putative Fog signaling pathway. In the absence of Fog, the Gα12/13 subunit, Concertina (Cta), along with its binding partners Gϒ and Gβ, are inactive and associated with the co-receptors Mist and Smog. Ric-8 chaperones Cta and regulates its subcellular localization. Upon Fog binding, Cta disassociates from Gϒ, and Gβ recruits RhoGEF2 to the cell membrane where it catalyzes the exchange of GDP for GTP in the small GTPase Rho1. GTP-bound Rho1, now active, activates Rho Kinase (Rok), which phosphorylates the RLC of non-muscle myosin, leading to its activation and subsequent cellular contractility. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Time-course of Fog-Myc production. These panels show cell culture medium harvested from a near 100% confluent flask of (A) control S2 cells or (B) S2:Fog-Myc stable cells. The cells were treated with 25 - 75 µM CuSO4 over a period of 24 - 48 hours. The cell culture medium was collected and prepared for SDS-PAGE and western blotting. Fog-Myc was detected by an anti-Myc antibody. Please click here to view a larger version of this figure.
{kind=link}

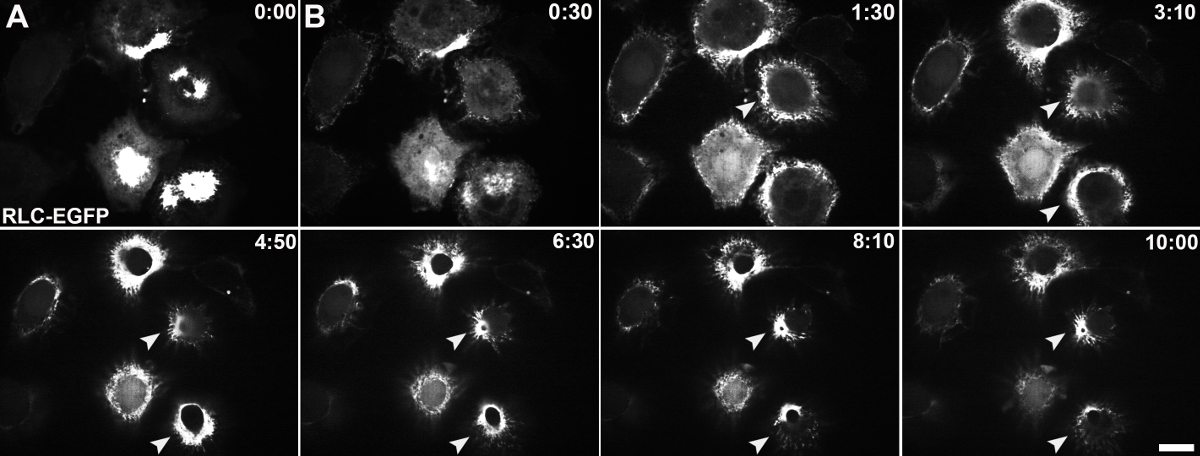
Figure 3: Live-cell imaging of Fog-induced constriction. Stable S2R+ cell lines expressing EGFP-tagged RLC of non-muscle myosin II were imaged by swept-field confocal microscopy during the perfusion of Fog-conditioned media. Time is shown in minutes and seconds. (A) This panel shows an image of a focal plane close to the cell-coverslip interface at time 0:00 before the perfusion of Fog-Myc. (B) This panel shows a time series (0:30 - 10:00) of images taken at a focal plane near the top of the cell. The white arrows indicate the reorganization of non-muscle myosin II as indicated by EGFP-RLC into contractile rings during constriction. The scale bar is 10 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Phosphorylated-RLC localizes to contractile rings following induction of constriction by Fog-Myc. S2R+ cells were fixed and stained for actin using phalloidin (red), phosphorylated RLC using a phosphoserine-19 antibody (green), and DAPI (DNA, blue), following treatment with (A) control or (B) Fog-Myc-conditioned media. Note the reorganization of phosphorylated RLC into rings upon the addition of Fog-Myc. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Representative quantification of the Fog-induced cellular contractility assay. S2R+ cells were treated with (A and B) control RNAi or (C and D) dsRNA against the small GTPase Rho1 for 7 days. Following this time, the cells were plated on con-A-coated glass-bottomed dishes and were treated with (A and C) control media or (B and D) Fog-Myc-conditioned media. The scale bar represents 10 µm. (E) A scatter plot indicates the mean and 95% confidence intervals of the fraction of contracted cells per condition. Individual points represent the fraction of contracted cells per field of view at 40X magnification (10 - 120 cells per field) and the numbers in parentheses indicate the total number of cells counted for both contracted and non-contracted cells over three separate RNAi experiments. Asterisks denote a statistically significant difference between RNAi- and Fog-treated conditions as determined by one-way ANOVA (p < 0.0001) with Tukey's multiple comparison post hoc analysis. Note that there was no statistically significant difference (n.s.) between Rho1 RNAi-treated samples treated with control or with Fog-Myc-conditioned media. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Here, we present a detailed protocol for a cell-based contractility assay using a Drosophila tissue culture cell line (S2R+ cells), which undergoes non-muscle myosin II constriction as a response to Fog signaling. This assay is useful for investigating the Fog pathway, as well as mechanisms that regulate non-muscle myosin II contractility.
Cell-culturing Considerations:
The conditions under which S2R+ cells are maintained is critical to achieve reliable data from this assay. When planning to perform the contractility assay, it best to maintain S2R+ cells in Shield and Sang M3 insect medium. While S2R+ cells can thrive in other insect cell culture media (e.g., SF900 or Schneider's insect medium), they often lose their responsiveness to Fog. S2R+ cells that have a high passage number, generally more than 20 - 25 passages, begin to lose sensitivity to RNAi. It is best to use early-passage cells for all experiments. Another important aspect to RNAi depletion experiments is choosing appropriate controls. Common negative controls include dsRNA targeting EGFP or pBlueScript, neither of which have homology to the fly genome. Targeting proteins in the Fog pathway (Rho, Rok, etc.), which, when depleted, prevent the activation of non-muscle myosin contractility, are also useful controls. S2:Fog-Myc cells can be maintained in an alternative cell culture medium such as SF900 supplemented with 100 units/mL of penicillin, 100 µg/mL streptomycin, and 0.25 amphotericin B. Note that FBS is not required when culturing cells in SF900. Cell density is also a critical component to all aspects of Drosophila tissue culture cells. Unlike mammalian tissue culture cells, Drosophila-derived tissue culture cells thrive best under higher cell densities (densities no lower than 5 x 105 cells/mL). However, when performing this assay, it is critical that cells are not plated on con-A-coated glass-bottomed dishes above 80% confluence, as the quantification of contracted versus non-contracted cells will become extremely tedious.
Critical Aspects and Alternative Approaches of the Cellular Contractility Assay:
The production of Fog, the ligand that triggers non-muscle myosin II contractility, is a key component of this assay. The fog gene encodes a protein of 730 amino acids with a predicted molecular weight of ~78 kDa26. Hydropathy analysis revealed a stretch of 12 hydrophobic residues at Fog's amino-terminus that could function as a secretion signal sequence. In addition, the coding sequence also contains multiple sites for potential N- and O-linked glycosylation, further suggesting Fog is a secreted protein26. In support of this, Fog was localized to secretory vesicles in presumptive epidermal cells undergoing apical constriction16. Fog-Myc expression was induced upon addition of copper sulfate to the medium, and antibodies against Fog or Myc recognized a 150-kD protein from culture medium harvested from induced cultures but not from induced media from S2 cells lacking the Fog-Myc construct. This molecular weight was higher than the predicted 80-kD molecular weight of Fog and suggests that the secretory machinery of S2 cells may glycosylate the protein prior to exocytosis. Due to the potential variability in Fog purification, it is advisable to use the same batch of Fog-conditioned media for all experiments. Making up large batches of concentrated Fog-Myc media will help in maintaining consistency throughout rounds of experiments.
The success of this assay also depends on confidently identifying cells that have undergone constriction. While constricted cells can be observed by fluorescence microscopy, by staining for actin or non-muscle myosin II, the most reliable way to identify and quantify constricted cells is through phase-contrast or DIC microscopy. Accurate counts can be achieved using 20X - 40X magnification on most standard light microscopes. Although the protocol written here uses glass-bottomed dishes, the assay can also be performed using standard 1.5 glass coverslips coated with con A. The addition of Fog can be done in 35 mm of tissue culture on a coverslip placed on parafilm, in order to limit the amount of Fog used for each assay. The quality of fixation is a critical component of the assay, as poorly fixed cells can lead to false positives. Using fresh fixation solution and making sure the cells never dry once fixed will lead to more reliable results. Finally, it is important to count a large number of cells. Typically, only 30% - 50% of untreated or control RNAi-treated cells constrict following the perfusion of Fog. However, there is a basal level of constriction that occurs in S2R+ cells, so a large number of cells is needed to ensure any change in the fraction of constricted cells is due to treatments. Furthermore, the depletion of some proteins involved in the regulation of non-muscle myosin II contractility may lead to hyper-contractility, even in the absence of Fog. The data presented here is from over 3,600 cells that were counted in three successive RNAi treatments using the same batch of Fog-conditioned media.
Applications of the Cellular Contractility Assay:
This contractility assay, when coupled with RNAi screening methods, offers a powerful system to study cell signaling, morphogenesis, and cellular contractility. Previously, it has been used to identify one of two Fog co-receptors21. A targeted screen of 138 G-protein-coupled receptors (GPCRs) is depleted by RNAi in S2R+ cells, and its ability to respond to Fog was assayed as described in the protocol presented here. Of the 138 GPCRs, a single, previously uncharacterized gene, now known as Mist, was uncovered. Further investigation into the function of Mist demonstrated that not only was it required for Fog-induced cellular contractility in S2R+ cells, but it was also essential for gastrulation in developing Drosophila21. Furthermore, this assay was used to demonstrate that Ric-8, a non-canonical GEF, is also a component in the Fog signaling pathway27. A series of epistasis RNAi experiments coupled with the contractility assay demonstrated that Ric-8 interacts with the Gα12/13 subunit Cta and functions to localize it within the cell, which is critical to Fog-induced cellular contractility31.
Drosophila tissue culture is well suited to novices, as the cells are easily maintained at room temperature, do not require CO2 or buffered cell culture medium, and are robust as long as proper cell culture densities are maintained. The cellular contractility assay was successfully performed by the laboratory section of an undergraduate cell biology course, where the students had little to no experience with culturing cells or microscopy. The cellular contractility assay presented here represents a powerful, cell-based tool that can be employed in gene discovery, or to interrogate the Fog signaling pathway, helping us to better understand developmental processes, such as apical constriction and non-muscle myosin II contractility, in general.
Acknowledgements
The authors would like to thank members of the Rogers Lab and the Applewhite lab, Greta Glover, and the students in the Reed College's spring 2018 Cellular Biology course, who have contributed to the development of this protocol. Furthermore, the authors would like to thank members of the Ritz lab for carefully reading and editing this manuscript. Research reported in this publication was supported by the National Science Foundation under award number 716964 to D.A.A. and A.R. and the Reed College Biology Department.
Materials
| Name | Company | Catalog Number | Comments |
| S2R+ cells | Drosophila Genomics Resorce Center | 150 | cell culture line, undergoes apical constriciton |
| S2:Fog-myc cells | Drosophila Genomics Resorce Center | 218 | cell culture line, produces Fog-myc under pMT promoter |
| S2R+ :Sqh-EGFP cells | Drosophila Genomics Resorce Center | 196 | cell culture line, stably expresses Sqh(RLC of non-muscle myosin II) tagged with EGFP |
| Shield and Sang M3 | Sigma-Aldrich | S3652 | cell culture medium |
| SF900 insect medium | Thermofisher Scientific | 12658019 | alternative cell cutlure medium |
| Schneider’s insect medium | Thermofisher Scientific | 21720024 | alternative cell cutlure medium |
| Fetal Bovine Serum | Thermofisher Scientific | 16000044 | media supplement |
| Antibiotic-Antimycotic (100X) | Thermofisher Scientific | 15240112 | antimicrobial/antifugal media supplement |
| Concanavalin A | MP Biomedicals | 150710 | used to coat dishes and coverslip for cellular adhesion |
| Glass bottom plates | Maktek | P35G-1.5-10 | used for assay and for fixing and staining cells |
| Glass cover slips | Corning | 2940-225 | alternative to glass bottom dishes |
| Protein concentrators | Millipore | UFC903008 | 30,000 molecular weight cutoff, used to concentrate media |
| 25 cm2, 75 cm2, 150 cm2 tissue culture flasks | Genessee | 25-204,25-206,25-208,25-210 | used to culture cells |
| 6-well and 12-well tissue culture dishes | Genessee | 25-105, 25-106 | used to culture cells |
| Flourescent mounting medium | Dako | S3023 | to preserve fixed cells |
| 32% Paraformaldehyde | EMS | 15714-S | used to fix cells |
| Pipes | EMS | 19240 | used in PEM buffer for cell fixation |
| Anti-Myc Antibody | Drosophila Hybirdoma Bank | cat# 9E10 | used to dectect Fog-Myc |
| PhosphoSerine-19 antibody | Cell Signaling | 3671 | antibody against phosphorylated regulatory light chain (RLC) serine 19 |
| 488-Phalloidin, 568-Phalloidin | Thermofisher Scientific | A12379, A12380 | to stain for f-actin |
| Hawk-VT multi-point array scanning confocal system | VisiTech International | Used for live cell imaging of S2R+:Sqh cells | |
| Eclipse TE300 Inverted microscope | Nikon | Used for live cell imaging of S2R+:Sqh cells |
References
- Ugur, B., Chen, K., Bellen, H. J. Drosophila tools and assays for the study of human diseases. Disease Model Mechanisms. 9 (3), 235-244 (2016).
- Bier, E. Drosophila, the golden bug, emerges as a tool for human genetics. Nature Reviews Genetics. 6 (1), 9-23 (2005).
- Millburn, G. H., Crosby, M. A., Gramates, L. S., Tweedie, S., FlyBase, C. FlyBase portals to human disease research using Drosophila models. Disease Model Mechanisms. 9 (3), 245-252 (2016).
- Perrimon, N., Mathey-Prevot, B. Applications of high-throughput RNA interference screens to problems in cell and developmental biology. Genetics. 175 (1), 7-16 (2007).
- Currie, J. D., Rogers, S. L. Using the Drosophila melanogaster D17-c3 cell culture system to study cell motility. Nature Protocols. 6 (10), 1632-1641 (2011).
- Rogers, S. L., Rogers, G. C. Culture of Drosophila S2 cells and their use for RNAi-mediated loss-of-function studies and immunofluorescence microscopy. Nature Protocols. 3 (4), 606-611 (2008).
- Applewhite, D. A., Davis, C. A., Griffis, E. R., Quintero, O. A. Imaging of the Cytoskeleton Using Live and Fixed Drosophila Tissue Culture Cells. Methods Molecular Biology. 1365, 83-97 (2016).
- Rogers, S. L., Wiedemann, U., Stuurman, N., Vale, R. D. Molecular requirements for actin-based lamella formation in Drosophila S2 cells. The Journal of Cell Biology. 162 (6), 1079-1088 (2003).
- Yi, C. H., et al. A genome-wide RNAi screen reveals multiple regulators of caspase activation. The Journal of Cell Biology. 179 (4), 619-626 (2007).
- Philips, J. A., Rubin, E. J., Perrimon, N. Drosophila RNAi screen reveals CD36 family member required for mycobacterial infection. Science. 309 (5738), 1251-1253 (2005).
- Nir, O., Bakal, C., Perrimon, N., Berger, B. Inference of RhoGAP/GTPase regulation using single-cell morphological data from a combinatorial RNAi screen. Genome Research. 20 (3), 372-380 (2010).
- Manning, A. J., Peters, K. A., Peifer, M., Rogers, S. L. Regulation of epithelial morphogenesis by the G protein-coupled receptor mist and its ligand fog. Science Signaling. 6 (301), 98 (2013).
- Leptin, M., Grunewald, B. Cell shape changes during gastrulation in Drosophila. Development. 110 (1), 73-84 (1990).
- Leptin, M. Gastrulation movements: the logic and the nuts and bolts. Developmental Cell. 8 (3), 305-320 (2005).
- Martin, A. C., Goldstein, B. Apical constriction: themes and variations on a cellular mechanism driving morphogenesis. Development. 141 (10), 1987-1998 (2014).
- Dawes-Hoang, R. E., et al. folded gastrulation, cell shape change and the control of myosin localization. Development. 132 (18), 4165-4178 (2005).
- Rogers, S. L., Wiedemann, U., Häcker, U., Turck, C., Vale, R. D. Drosophila RhoGEF2 associates with microtubule plus ends in an EB1-dependent manner. Current Biology. 14 (20), 1827-1833 (2004).
- Coravos, J. S., Martin, A. C. Apical Sarcomere-like Actomyosin Contracts Nonmuscle Drosophila Epithelial Cells. Developmental Cell. 39 (3), 346-358 (2016).
- Mason, F. M., Tworoger, M., Martin, A. C. Apical domain polarization localizes actin-myosin activity to drive ratchet-like apical constriction. Nature Cell Biology. 15 (8), 926-936 (2013).
- Fox, D. T., Peifer, M. Abelson kinase (Abl) and RhoGEF2 regulate actin organization during cell constriction in Drosophila. Development. 134 (3), 567-578 (2007).
- Manning, A. J., Rogers, S. L. The Fog signaling pathway: insights into signaling in morphogenesis. Developmental Biology. 394 (1), 6-14 (2014).
- Xie, S., Mason, F. M., Martin, A. C. Loss of Gα12/13 exacerbates apical area dependence of actomyosin contractility. Molecular Biology of the Cell. 27 (22), 3526-3536 (2016).
- Vasquez, C. G., Tworoger, M., Martin, A. C. Dynamic myosin phosphorylation regulates contractile pulses and tissue integrity during epithelial morphogenesis. The Journal of Cell Biology. 206 (3), 435-450 (2014).
- Parks, S., Wieschaus, E. The Drosophila gastrulation gene concertina encodes a G alpha-like protein. Cell. 64 (2), 447-458 (1991).
- Barrett, K., Leptin, M., Settleman, J. The Rho GTPase and a putative RhoGEF mediate a signaling pathway for the cell shape changes in Drosophila gastrulation. Cell. 91 (7), 905-915 (1997).
- Costa, M., Wilson, E. T., Wieschaus, E. A putative cell signal encoded by the folded gastrulation gene coordinates cell shape changes during Drosophila gastrulation. Cell. 76 (6), 1075-1089 (1994).
- Peters, K. A., Rogers, S. L. Drosophila Ric-8 interacts with the Gα12/13 subunit, Concertina, during activation of the Folded gastrulation pathway. Molecular Biology of the Cell. 24 (21), 3460-3471 (2013).
- Jha, A., van Zanten, T. S., Philippe, J. M., Mayor, S., Lecuit, T. Quantitative Control of GPCR Organization and Signaling by Endocytosis in Epithelial Morphogenesis. Current Biology. 28 (10), 1570-1584 (2018).
- Kerridge, S., et al. Modular activation of Rho1 by GPCR signalling imparts polarized myosin II activation during morphogenesis. Nature Cell Biology. 18 (3), 261-270 (2016).
- Myat, M. M., Andrew, D. J. Organ shape in the Drosophila salivary gland is controlled by regulated, sequential internalization of the primordia. Development. 127 (4), 679-691 (2000).
- Kanesaki, T., Hirose, S., Grosshans, J., Fuse, N. Heterotrimeric G protein signaling governs the cortical stability during apical constriction in Drosophila gastrulation. Mechanisms of Development. 130 (2-3), 132-142 (2013).
- Nikolaidou, K. K., Barrett, K. A Rho GTPase signaling pathway is used reiteratively in epithelial folding and potentially selects the outcome of Rho activation. Current Biology. 14 (20), 1822-1826 (2004).
- Yanagawa, S., Lee, J. S., Ishimoto, A. Identification and characterization of a novel line of Drosophila Schneider S2 cells that respond to wingless signaling. Journal Biological Chemistry. 273 (48), 32353-32359 (1998).
- Mason, F. M., Xie, S., Vasquez, C. G., Tworoger, M., Martin, A. C. RhoA GTPase inhibition organizes contraction during epithelial morphogenesis. The Journal of Cell Biology. 214 (5), 603-617 (2016).
- Martin, A. C., Kaschube, M., Wieschaus, E. F. Pulsed contractions of an actin-myosin network drive apical constriction. Nature. 457 (7228), 495-499 (2009).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved