High-throughput Measurement of Gut Transit Time Using Larval Zebrafish
In This Article
Summary
The goal of this protocol is to measure the transit time of fluorescently labeled food through the gut of larval zebrafish in a high throughput fashion.
Abstract
Zebrafish are used as alternative model organisms for drug safety testing. The gastrointestinal (GI) tract of zebrafish has genetic, neuronal, and pharmacological similarities to that of the mammals. GI intolerance during clinical testing of drug candidates is common and may pose a serious threat to human health. Testing for GI toxicity in preclinical mammalian models can be expensive in terms of time, test compound, and labor. The high-throughput method presented here may be used to predict GI safety issues. Compared to mammalian models, this method allows for more expedient assessment of test compound effects on GI transit while using low quantities of compound. In this method, larval zebrafish (7 days post fertilization) are fed food containing a fluorescent label. After feeding, each larval fish is placed into a well of a 96-conical-bottom-well plate and dosed with test compound (dissolved in water) or the vehicle. As gut transit occurs, fecal matter accumulates on the bottom of the wells, and the rate at which this happens is monitored by measuring fluorescence from the bottom of the well repeatedly over time using a plate spectrophotometer. The fluorescence from larvae in a given treatment group are averaged and these values are graphed along with standard error, for each measurement time, yielding a curve representing average transit of food over time. Effects on gut transit time are identified by comparing the area under the curve for each treatment group to that of the vehicle-treated group. This method detected changes in zebrafish GI transit time induced by drugs with known clinical GI effects; it can be employed to interrogate dozens of treatments for GI effects per day. As such, safer compounds can be quickly prioritized for mammalian testing, which expedites discovery and proffers 3Rs advancement.
Introduction
Zebrafish (Danio rerio) are used to model vertebrate biology and predict drug toxicity and/or efficacy; new applications in these fields emerge each year. The advantages of zebrafish over mammalian models include their fecundity, small size, and transparency through organogenesis. Zebrafish are used to predict drug candidate acute toxicity, as well as to assess compound impact on organ function, e.g., cardiac, ocular, gastrointestinal (GI)1,2. Zebrafish development and physiology are similar to those of mammals in many ways3 and 80% of genes that are associated with human disease have a zebrafish homolog4.
The GI tract of zebrafish has similar physiology to the mammalian GI tract but has a simpler architecture5. Zebrafish have no stomach; the anterior intestinal bulb acts as a food depot. Gene expression in zebrafish intestine has many similarities to that of the mammal5. Like mammals, the enteric nervous system of zebrafish controls gut motility, and intestinal innervation mirrors that of other vertebrates6,7. Based on these similarities, functional disorders of the human intestine have been studied in zebrafish, using methods derived from mammalian models8.
GI intolerance during clinical testing of drug candidates is common and may pose a serious threat to human health. A review of programs in a major pharmaceutical company during 2005–2010 revealed GI safety as the principal cause for 9% of clinical study terminations9. Testing for GI toxicity in preclinical mammalian models can be expensive in terms of time, compound, and labor. A predictive high-throughput assay for GI transit can provide flexibility to compound toxicity testing, and deliver 3Rs impact. A novel method offering such an assay is presented by the protocol described herein. This high-throughput assay could be employed early in drug development to prioritize candidates, and contribute to the reduction of GI safety testing in larger species.
Protocol
All methods described here have been approved by the Institutional Animal Care and Use Committee (IACUC) of AbbVie.Abbvie operates under the National Institutes of Health Guide for Care and Use of Laboratory Animals in a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC). No animal health concerns were observed in these studies.
1. Breed Adult Zebrafish and Collect Embryos
- House and breed adult zebrafish using general husbandry and breeding practices. For example, see Westerfield10.
- Prepare embryo medium by dissolving dehydrated sea salt (see Table of Materials) in deionized water at a concentration of 60 mg/L.
- Collect fertilized eggs from the adult breeding chamber, rinse well with embryo medium and house embryos in about 50 mL of embryo medium in 10 cm Petri dishes (50 embryos per dish) at 28 ± 1 °C on a 14:10 h light:dark cycle.
- Remove non-surviving embryos after 24 h and supplement with surviving embryos so that each dish contains 50 embryos per dish.
2. Train Larvae to Feed Using Non-dyed Food
- On the 4th day post-fertilization (4 dpf), feed the larvae in each Petri dish 2 mg of powdered larval fish food (see Table of Materials) by sprinkling the food on top of the water.
- Allow the larvae 3–4 h to feed and then transfer them to a clean (no food) Petri dish containing about 50 mL fresh embryo medium.
- To aid in transferring as little food as possible, rinse each larva in a Petri dish containing about 50 mL fresh embryo medium before the final transfer to the new dish.
- Repeat feeding and rinsing of the larvae on 5 dpf and 6 dpf.
3. Prepare Fluorescent Food on the 6 dpf and Feed to Larvae on the 7 dpf
- Prepare food containing a fluorescent label (hereafter referred to as fluorescent food, according to methods of Field et al.3). Briefly, mix 300 µL of fluorescent label (see Table of Materials), 100 µL of deionized water, and 200 mg of powdered larval food on a 10 cm watch glass.
- Spread the resulting paste into a thin layer on the watch glass. Allow the paste to dry at room temperature in the dark for >8 h.
- Scrape the dried mixture off the watch glass, crush to powder, and store at room temperature in the dark. The fluorescent food is now ready to be fed to the larvae.
- On the 7 dpf, feed the fluorescent food to larvae in the same manner as done for previous feedings, that is provide 2 mg fluorescent food per dish (see steps 2.1–2.2.1).
NOTE: Ensure that the food is finely ground to a powder. Rubbing fluorescent food that is wrapped in weighing paper between thumb and forefinger is a useful method for ensuring finely ground food.
4. Prepare Concentrated Dosing Solutions of Test Compounds
- Dissolve each test compound in embryo medium to a concentration that is 2x the target dose. If testing of a dose range is desired, prepare multiple concentrated dosing solutions of the appropriate concentrations for the desired doses.
- Prepare enough of each concentrated dosing solution so that 24 larvae can be treated. Each larva will require 100 µL dosing solution (the final volume per well is 200 µL), so at the very least, 2.4 mL dose solution is needed for each treatment group; 2.5 mL would be an appropriate volume.
- If a solvent (i.e., dimethyl sulfoxide) was used for initial solubilizing of test compound, prepare the appropriate vehicle control dose (i.e., same amount of solvent as the compound treatment but without the compound). And, as was done for each compound-treated group, prepare enough vehicle solution to treat 24 larvae.
5. Transfer Larvae to a 96-well plate and Apply Treatments
- After larvae have been allowed to feed on fluorescent food for 2 h, use a transfer pipette to move them to a rinsing dish, as was done after feeding on prior days.
- After each larva is rinsed, withdraw it along with 100 µL embryo medium, and move it into a well of a 96-well polystyrene conical-bottom multi-well plate (see Table of Materials), dispensing the full 100 µL embryo medium into the well with the larva.
- Once the required numbers of larvae have been transferred to the 96-well-plate, add 100 µL of the 2× concentrated dosing solutions to each well.
NOTE: The use of a 12-channel multichannel pipette facilitates rapid dosing of the larvae (12-at-a-time). To avoid accidentally adding the wrong treatment, keep close track of which larvae have been dosed with which treatment and be sure to change pipette tips between treatments.
6. Measure Initial and Subsequent Fluorescence from Each Well
- After adding the dose solutions, place the 96-well-plate into a plate spectrophotometer capable of exciting and detecting emissions from the fluorescent label.
NOTE: For the yellow-green label used (see the Table of Materials), the appropriate wavelengths of light are exciting at 505 nm and detecting at 515 nm. - Read the fluorescence of the 96-well-plate from the bottom 5 times in immediate succession without shaking the plate. Use the minimum value of the 5 readings from each well as the initial fluorescence from the well.
NOTE: The reason that 5 readings are taken whenever the plate is read is that larvae sometimes swim in the path of the excitation light and the food in their gut emits a very large signal which is unrepresentative of the released feces. Taking the minimum of the 5 reads can help to avoid using artificially high measurements from un-transited food. - Read the fluorescence of the 96-well-plate (as was done for the initial reading) every 20 min for the first 2 h post-dosing, every 30 min for hours 3 and 4 post dosing, and then once every hour for hours 5, 6, 7, and 8 post-dosing.
- Take caution to not disturb (by shaking the 96-well-plate) settled fecal matter between reads and incubate the larvae at 28 ± 1 °C between reads.
- Incubate the larvae over night at 28 ± 1 °C and read the fluorescence from the 96-well-plate the following morning around the same time that the larvae were dosed the day before. Use this measurement as the 24 h post-dose fluorescence.
7. Analyze the Data
NOTE: Here we calculate accumulation per well and group averages for each time point.
- To calculate accumulation per well, using only minimum values from each of the 5 reads, subtract the initial value from each time point's value. Perform this calculation for the initial value as well (this results in an initial accumulation of zero for each well).
- If the accumulation in a well at the 24 h time point is less than 150 relative fluorescent units, exclude that well from further analysis; these low values are most likely due to low or no intake of the fluorescent food by the larvae during feeding, and thus those larvae are not good subjects for measuring transit time.
- For each time point, calculate the average accumulation for wells containing co-treated larvae, as well as standard error of those averages.
- Plot the average accumulation on the Y-axis versus time on the X-axis for each treatment group and compare the areas under those curves (AUCs).
NOTE: Treatments that significantly slow or accelerate transit time will have AUCs significantly smaller or larger, respectively, than the vehicle-treated group.
Representative Results
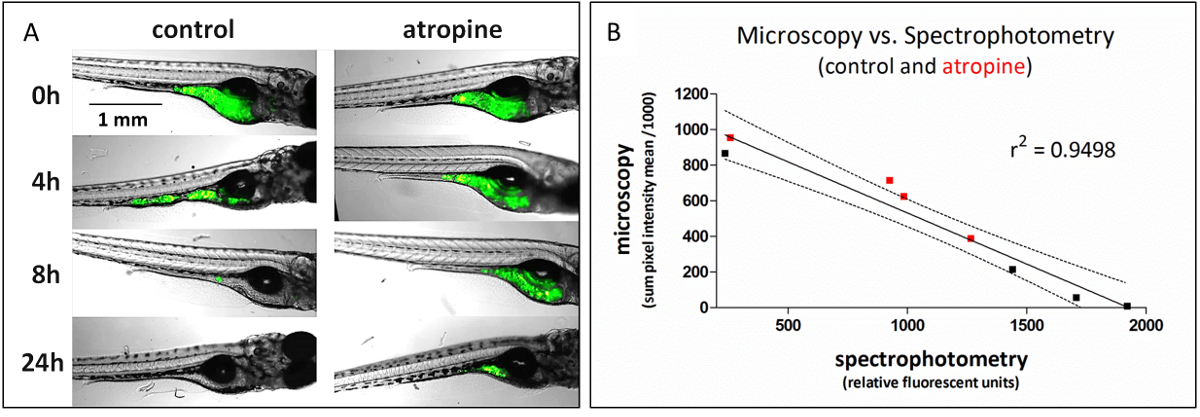
This method, which uses plate-based spectrophotometry to assess GI transit, can be used as a high-throughput replacement of fluorescent microscopy, which is a lower throughput method for assessing the same function (Figure 1). To generate the data in Figure 1, identically treated fish were analyzed for GI transit using either fluorescence microscopy (representative images shown) or spectrophotometry at 4 time points, 0, 4, 8, and 24 h post-dosing; comparison of the data from those experiments gave highly correlated results (linear regression of data r2 = 0.95). The linear regression has a negative slope because microscopy measures the retained fluorescence signal and spectrophotometry measures the transited signal.
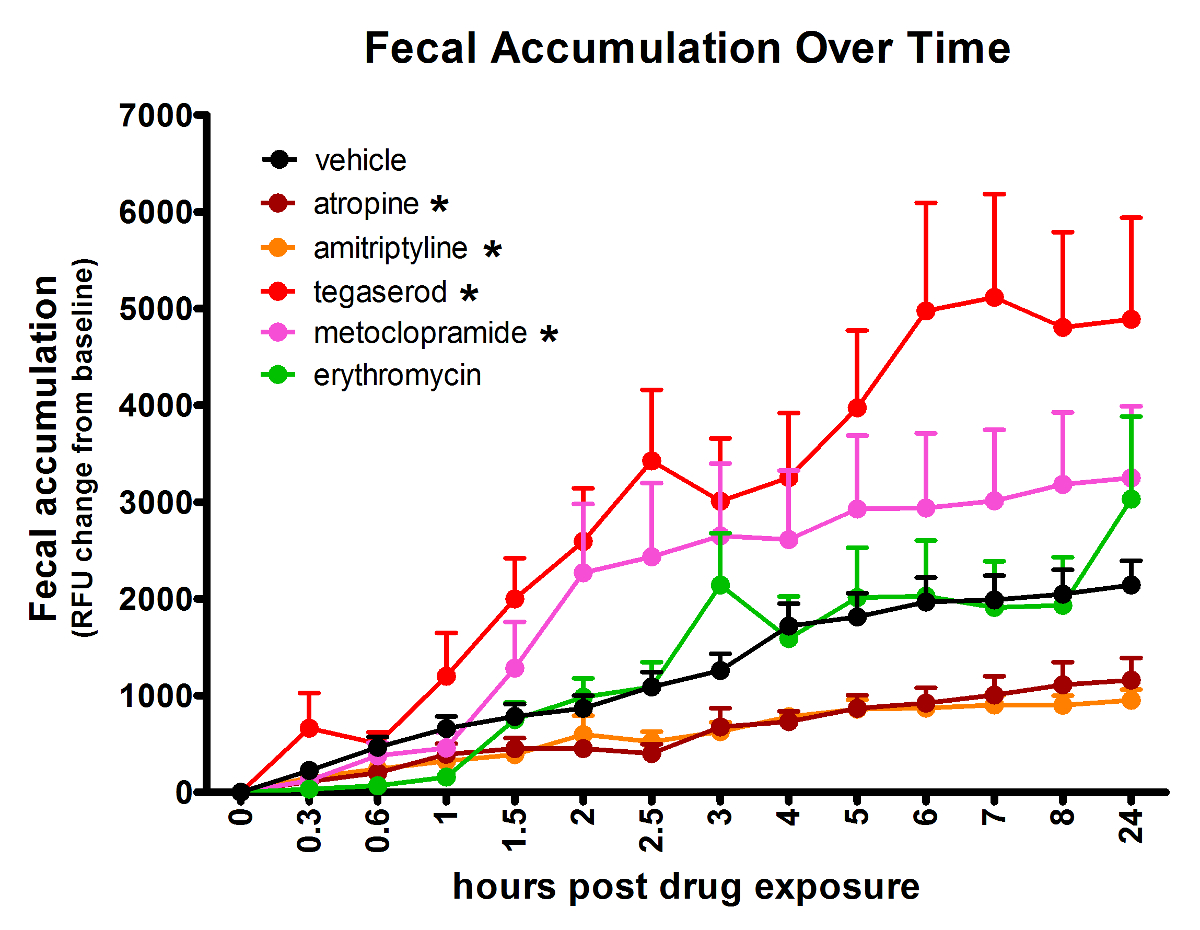
The effects of compounds of disparate mechanisms, with well-established GI activity in humans, can be detected in zebrafish using the spectrophotometry assay (Figure 2). Compared to vehicle-treated controls, atropine (4.2 µM) and amitriptyline (5 µM) slowed GI transit, while tegaserod (3.3 µM) and metoclopramide (33 µM) accelerated transit time. Erythromycin (14 µM), expected to accelerate transit time had no effect as measured by this method. Treatment group sizes were 24 before removing data from larvae with no or very low signal. The AUC for the average signal per time point was compared between compound-treated and vehicle-treated groups using Tukey's Honestly Significant Difference for type-1-error control. Effects were considered significant only when p ≤ 0.05. The concentrations used for the above treatments were the maximum tolerated doses, determined in a prior experiment and defined as the highest dose with no observable adverse effect by gross observation.
The spectrophotometry assay can measure dose-dependent effects of compounds. Figure 3 provides data demonstrating that atropine slows GI transit dose-dependently in zebrafish larvae. The lowest dose of atropine tested, 0.042 µM, had no significant effect, while the two higher concentrations each had significant impact, 0.42 µM having less of an effect than 4.2 µM.
A new assay can be assessed by testing positive and negative controls, that is, compounds known to be active and inactive respectively in the target system (in this case the target system is the mammalian GI transit). For the spectrophotometry assay, 18 active (positive) controls and 6 inactive (negative) controls were tested. Based on these experiments, the spectrophotometry assay has high positive predictive value (90.9%), but low sensitivity (55.6%) and negative predictive value (38.5%). These values are derived from the data presented in Table 1. They reflect, in practice, that if the zebrafish transit time is impacted by a treatment, mammalian transit is likely to be impacted. However, if there is no effect on zebrafish transit time, this is not predictive of mammalian effect.

Figure 1: Fluorescent food transit is detected as a loss of signal from microscopic imaging and a corresponding gain in signal by plate-based spectrophotometry. (A) Representative microscopic images from analysis of atropine (4.2 µM) effect on GI transit time. (B) Average signal quantified from microscopic images is highly (negatively) correlated with signal from voided fecal matter (spectrophotometry) from identically treated fish. Data from atropine-treated fish are in red. This figure copied with permission from Cassar et al.11. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Analyzing fluorescent signal accumulation over time from a multi-well plate allows identification of treatments that change GI transit rate. Asterisk (*) indicates significantly different AUC than vehicle-treated fish. Error bars represent the standard error of the mean signal for larvae in the treatment group per time point. This figure has been reused with permission from Cassar et al.11. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Atropine dose-dependently slows zebrafish gut transit time as reflected by fluorescent spectrophotometry of fecal accumulation over time. Asterisk (*) indicates significantly different AUC than vehicle-treated fish. Error bars represent the standard error of the mean signal for larvae in the treatment group per time point. This figure is reused with permission from Cassar et al.11. Please click here to view a larger version of this figure.
{kind=link}

Table 1: GI activity of 24 compounds in mammals and fish. This table is reused with permission from Cassar et al.11.
Discussion
The novel spectrophotometry method for measuring zebrafish larvae GI transit time, presented here, is an efficient assay that can predict treatment effects on mammalian GI-function. Although the assay has low sensitivity, it has high positive predictivity, which is acceptable for first tier assays employed for paring down the number of candidate treatments based on toxicity12. This method is easier to execute, has higher throughput, and uses less animal handling steps than fluorescent microscopy.
There are technical challenges inherent in this method. Catching individual larvae after feeding the fluorescent food and transferring them into individual wells is challenging at first. However, with practice, a skilled technician can fill a 96-well plate in less than 15 min. If, at a given time point, the plate is accidentally shaken and the fecal matter unsettled from the bottom of the wells before reading, the accumulation will appear to decrease. This can be avoided by moving the plate(s) carefully, without shaking. In our experience, normal movement, including that of the plate spectrophotometer motorized drawer, did not disturb the assay. Plate readers equipped with a heater (i.e., the plate does not have to be returned to incubator between measurements), and located near the assay lab bench could optimize for lower chance of disturbance, but this was not necessary in our experience.
Early attempts at the method did not include feeding on the days before the assay. Without those 'practice' feedings, lower numbers of larvae consumed sufficient fluorescent food during the time allowed before treatment application. In those attempts, variation within treatment groups was greater, and more data was unusable due to low/no signal accumulation over time. Even with practice feeding, some larvae do not consume sufficient amounts of the fluorescent food, excluding data from those larvae decreases variation within groups and increases the ability to identify treatment effects. Starting with larger group sizes (i.e., 24 versus 12) allows for sufficient numbers of larvae contributing useful data to the analysis.
The fluorescent microscopy reveals that food transit was nearly complete by 24 h in atropine-treated larvae (not shown), however the microplate data from the final time point reflect lower final signal in the atropine group, compared to vehicle group. Conversely, higher final microplate fluorescence was associated with treatments that accelerated transit time, even though the vehicle-treated larvae have apparently voided their GI tract before the final time point. Based on the random assignment to treatment of larvae fed fluorescent food, we assume equivalent consumption of the fluorescent food, on average, among treatment groups. Given this, and based on the patterns described above, fluorescence from fecal matter declines with time spent in the larval GI tract or faster transit adds fluorescence to the feces somehow. The actual cause is unknown and has not been interrogated, however the assay still is capable of measuring and comparing transit time among groups.
The employment of this method in detecting toxic GI effects from small molecule compounds is one application. Other possible applications include disease modeling (e.g., irritable bowel syndrome) and novel therapy discovery for such diseases, or for discovering pro-kinetic compounds. Furthermore, coupled with transgenic models, this method could be used to interrogate the role of genes in normal GI transit, as well as transit disorders, including enteric neuron deficiency. The zebrafish larva offers a whole organism platform on a scale that approximates that of cell culturing, but since there are multiple tissues and myriad cell types functioning together in the zebrafish, systems biology questions can be asked and answered using this model.
With advancements in technology, and new applications thereof, efficiency in the conduct of zebrafish toxicity tests will continue to improve. Larval zebrafish handling methods and assays are continuing to improve in terms of higher throughput13,14. The novel method presented here is one example of an improvement that may make zebrafish studies more impactful.
Acknowledgements
Simon Cassar of carpetbones.com designed and created the animated figure used to demonstrate the in-life procedure of the GI transit assay.
Materials
| Name | Company | Catalog Number | Comments |
| Wild type zebrafish breeding pair | Various sources - for example ZIRC (Zebrafish International Resource Center) | ZL-1 | Adult wild type zebrafish of AB lineage |
| 1.7-liter Breeding Tank - Beach Style Design | Tecniplast | 1.7L SLOPED | Breeding tank |
| Instant Ocean sea salt | Intant Ocean | SS15-10 | Dehydrated sea salt |
| First Bites larval fish food | Hikari | 20095 | Powdered larval fish food |
| Yellow-green (505/515) Fluospheres | Invitrogen | F8827 | Fluorescent label |
| V-bottom 96-well polystyrene microplates | Thermo Fisher Scientific | 249570 | Multiwell microplate with V-shaped bottom in each well |
| Atropine | Sigma Aldrich | A0132 | |
| Amitriptyline | Sigma Aldrich | A8404 | |
| Tegaserod | Sigma Aldrich | SML1504 | |
| Metoclopramide | Sigma Aldrich | M0763 | |
| Erythromycin | Sigma Aldrich | E5389 | |
| Spectramax M2e microplate reader | Molecular Devices | Spectramax M2e | A multi-well plate spectrophotometer capable of fluorescent excitation and emission detection. |
| SoftMax Pro | Molecular Devices | SoftMax Pro | Software for spectrophotometer data acquisition |
References
- Berghmans, S., et al. Zebrafish based assays for the assessment of cardiac, visual, and gut function - Potential safety screens for early drug discovery. Journal of Pharmacological and Toxicological Methods. 58 (1), 59-68 (2008).
- Field, H. A., Kelley, K. A., Martell, L., Goldstein, A. M., Serluca, F. C. Analysis of gastrointestinal physiology using a novel intestinal transit assay in zebrafish. Neurogastroenterology and Motility. 21, 304-312 (2009).
- Kanungo, J., Cuevas, E., Ali, S., Paule, M. G. Zebrafish model in drug safety assessment. Current Pharmaceutical Design. 20 (34), 5416-5429 (2014).
- Howe, K., et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 496, 498-503 (2013).
- Shepherd, I., Eisen, J. Development of the zebrafish enteric nervous system. Methods in Cell Biology. 101, 143-160 (2011).
- Holmberg, A., Olsson, C., Holmgren, S. The effects of endogenous and exogenous nitric oxide on gut motility in zebrafish Danio rerio embryos and larvae. The Journal of Experimental Biology. 209, 2472-2479 (2006).
- Olsson, C., Holmber, A., Holmgren, S. Development of enteric and vagal innervation of the zebrafish (Danio rerio) gut. The Journal of Comparative Neurology. 508, 756-770 (2008).
- Fleming, A., Jankowski, J., Goldsmith, P. In vivo analysis of gut function and disease changes in a zebrafish larvae model of inflammatory bowel disease: A feasibility study. Inflammatory Bowel Disease. 16 (7), 1162-1172 (2010).
- Cook, D., et al. Lessons learned from the fate of AstraZeneca's drug pipeline: A five-dimensional framework. Nature Reviews Drug Discovery. 13 (6), 419-431 (2014).
- Westerfield, M. . The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio Rerio). , (2000).
- Cassar, S., Huang, X., Cole, T. A high-throughput method for predicting drug effects on gut transit time using larval zebrafish. Journal of Pharmacolical and Toxicological Methods. 76, 72-75 (2015).
- McKim, J. M. Building a tiered approach to in vitro predictive toxicity screening: A focus on assays with in vivo relevance. Combinatorial Chemistry and High Throughput Screening. 13 (2), 188-206 (2010).
- Bruni, G., Lakhani, P., Kokel, D. Discovering novel neuroactive drugs through high-throughput behavior-based chemical screening in the zebrafish. Frontiers in Pharmacology. 5, 153 (2014).
- Mandrell, D., et al. Automated zebrafish chorion removal and single embryo placement: optimizing throughput of zebrafish developmental toxicity screens. Journal of Laboratory Automation. 17, 66-74 (2012).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved