A Customizable Protocol for String Assembly gRNA Cloning (STAgR)
In This Article
Summary
Here, we present string assembly gRNA cloning (STAgR), a method to easily multiplex gRNA vectors for CRISPR/Cas9 approaches. STAgR makes gRNA multiplexing simple, efficient and customizable.
Abstract
The bacterial CRISPR/Cas9 system has substantially increased methodological options for life scientists. Due to its utilization, genetic and genomic engineering became applicable to a large range of systems. Moreover, many transcriptional and epigenomic engineering approaches are now generally feasible for the first time. One reason for the broad applicability of CRISPR lies in its bipartite nature. Small gRNAs determine the genomic targets of the complex, variants of the protein Cas9, and the local molecular consequences. However, many CRISPR approaches depend on the simultaneous delivery of multiple gRNAs into individual cells. Here, we present a customizable protocol for string assembly gRNA cloning (STAgR), a method that allows the simple, fast and efficient generation of multiplexed gRNA expression vectors in a single cloning step. STAgR is cost-effective, since (in this protocol) the individual targeting sequences are introduced by short overhang primers while the long DNA templates of the gRNA expression cassettes can be re-used multiple times. Moreover, STAgR allows single step incorporation of a large number of gRNAs, as well as combinations of different gRNA variants, vectors and promoters.
Introduction
String assembly gRNA cloning (STAgR) is a method enabling efficient overnight generation of expression vectors containing multiple gRNA expression cassettes in one single cloning step. The STAgR protocol does not depend on specialist expertise or materials and offers multiple options for customization.
The bacterial CRISPR protein Cas9 has a unique capacity. It is able to find and selectively bind only those DNA sequences encoded in naturally occurring crRNAs or engineered gRNAs1. Its utilization as a molecular tool has enabled a large number of approaches that were impossible, unfeasible or only limited to certain cellular models until recently. These include targeted gene mutations2, genome engineering3, epigenome editing4,5,6, transcriptional activation and gene silencing7,8. As far as we know CRISPR is universally deployable, as reports of its application exist for a wide range of target sites, cell types and species. However, many CRISPR applications depend directly or indirectly on the delivery of multiple gRNAs including a series of genomic manipulations (translocations9,10, medium11 and large scale genomic alterations12,13), genome engineering (use of Cas9 nickases14, generation of conditional alleles11,15 or serial mutations16) and tracing strategies17. Moreover, for other approaches (transcriptional engineering18, epigenome engineering19,20) multiple targeting sites are not strictly mandatory, however they reach their maximum effect often only under this condition.
Several useful methods have been described to generate gRNA vectors containing more than one gRNA expression cassette21,22,23,24,25,26,27. Here, we present a protocol for STAgR, string assembly gRNA cloning, which is outstanding in its combination of simplicity and speed (only one overnight cloning step is needed), low cost (as DNA templates can be reused multiple times), customizability (for different gRNA systems and promoters) and effectiveness (it allows the generation of vectors containing high numbers of gRNA cassettes)28.
STAgR can in principle be used to generate expression vectors with few (1–2 gRNAs), several (3–5 gRNAs) and some of the highest number of expression cassettes reported so far (6–8 gRNAs)28. Similarly, STAgR is applicable for any gRNA sequence, backbone or promoter. Since however, STAgR is mainly based on polymerase chain reactions (PCR) and Gibson assembly, gRNAs with high sequence similarities should be generated using an alternative method.
Protocol
1. Design of STAgR Cloning Strategy
- Decide on how many gRNA cassettes will be included in one expression vector and in which order. Determine which promoters and gRNA scaffolds should be used for each of the gRNAs. Design the gRNAs by using any online tool (e.g., www.benchling.com).
2. Design of STAgR Cloning Primers with Overhangs
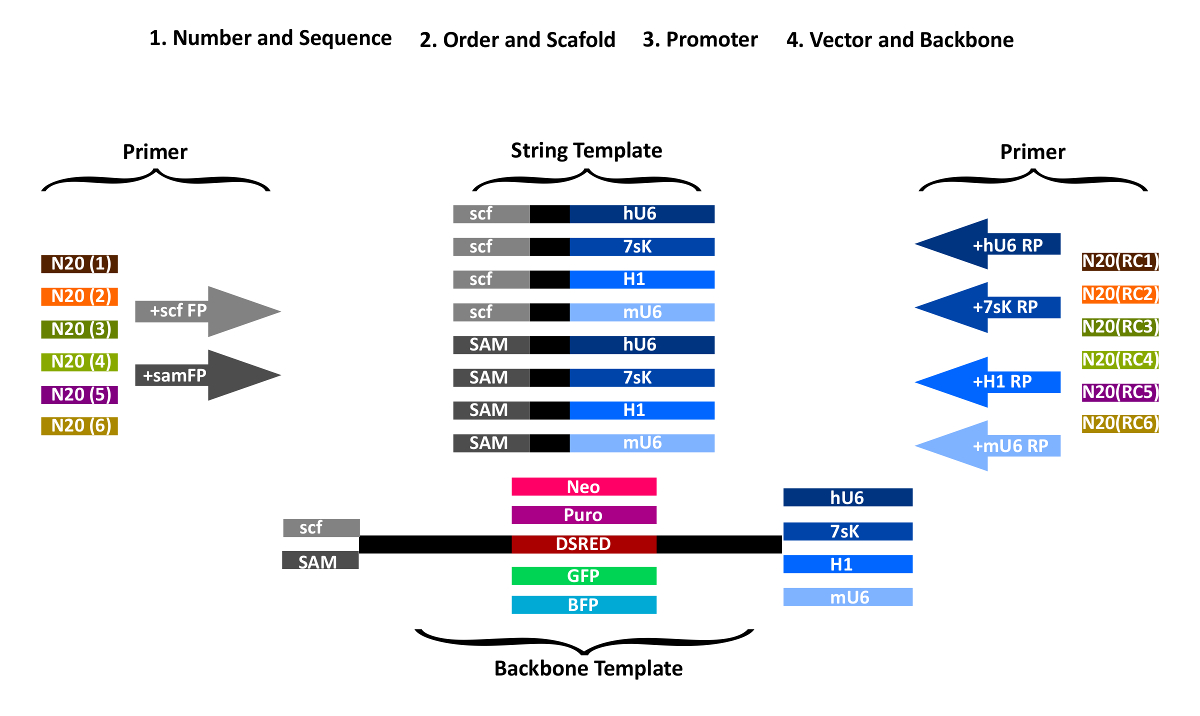
NOTE: The sense protospacer sequence of the last gRNA and the antisense of the first gRNA respectively are added to the PCR primers used for amplification of the vector backbone (Figure 1). The remaining sequences are amplified from the strings, thereby attaching the sense and antisense gRNAs in the desired order. The first string piece is amplified with a forward primer containing the first gRNA N20 sequence as an overhang and a reverse primer with the second gRNA N20 sequence (reverse complement) as an overhang. All the following pieces are generated accordingly.
- Add N20 gRNA sequences to the forward amplification primers for STAgR DNA string as overhangs (Table 1). Add sense gRNA sequences 5' to the forward primer "scf-FP" (for a conventional scaffold) or "sam-FP" (for a MS2 containing scaffold). FP primers anneal to the scaffold/MS2 part of the respective STAgR string (Figure 1).
- Add the reverse complement N20 gRNA sequence to the reverse primer sequences. Choose RP primers depending on the specific promoters and strings used (hU6-RP, mU6-RP, 7sK-RP, H1-RP).
3. Generation of STAgR Cloning Fragments
- To generate the individual cloning fragments for Gibson assembly, perform PCRs by setting up 10 µL of high fidelity (HF) buffer, 1 µL of 10 mM dNTPs, 0.25 µL of overhang-primer (100 µM), 10 ng of the DNA template (string or vector), 0.5 µL HF polymerase, 1.5 µL of dimethyl sulfoxide (DMSO) and add H2O to make a final volume of 50 µL.
NOTE: Use the plasmid "STAgR_Neo" as a template for the backbone PCR to incorporate the gRNA scaffold to the last gRNA, if the SAM Scaffold is chosen, use "STAgR_SAM". For a STAgR plasmid with six gRNA cassettes for example, six PCRs are necessary, five with DNA strings as templates and one with the vector backbone as the template. - Incubate reactions on a thermocycler using the following conditions: 1 cycle of 98 °C for 1 min 30 s; 38 cycles of 98 °C for 10 s, 59 °C (for gRNA scaffold)/ 68 °C (for SAM loop) for 10 s, 72 °C for 30 s (for inserts) / 1 min 30 s (for vectors); 1 cycle of 72 °C for 10 min.
- Remove 5.5 µL from the PCR reaction, add loading dye and analyze the amplified fragments on a 1% agarose gel. Load an appropriate DNA ladder for sizing (100 bp DNA ladder/1 kb DNA ladder) and run the gel in an appropriate gel running buffer (e.g., TLE buffer) at 120 V for 30 min.
- While the gel is running, add 5 µL of the buffer provided with the restriction enzyme and 0.5 µL DpnI (10 units) to the remaining 44.5 µL PCR reaction and incubate for 30 min to 1 h at 37 °C.
- Perform DNA purification using magnetic beads.
- Add 1.8 µL magnetic beads per 1 µL of PCR product and mix by pipetting up and down.Incubate for 2 min at room temperature (RT).
- Separate beads and DNA fragments from residual liquid with a magnet. Wash beads twice with 70% ethanol by rinsing the pellet without resuspending completely.
- Remove all ethanol with a pipette and let the pellet air dry.
- Dissolve the pellet in 15–20 µL H2O by pipetting up and down and separate the beads from the liquid by using a magnet. Transfer the clear supernatant to a new tube.
NOTE: Alternatively, column-based reaction clean-up kits can be used.
- Determine DNA concentrations using a spectrophotometer as described elsewhere29.
4. Gibson Assembly Reaction and Bacterial Transformation
NOTE: The following steps are adapted from Gibson et al.30.
- Prepare a homemade Gibson reaction mix or use a commercial Gibson cloning kit.
- Combine 3 mL of 1 M Tris (Tris(hydroxymethyl)aminomethane)-HCl at pH 7.5, 300 µL of 1 M MgCl, 60 µL of 100 mM dGTP (deoxyguanosine triphosphate), 60 µL of 100 mM dATP (deoxyadenosine triphosphate), 60 µL of 100 mM dTTP (deoxythymidine triphosphate), 60 µL of 100 mM dCTP (deoxycytidine triphosphate), 300 µL of 1 M DTT (dithiothreitol), 1.5 g of PEG-8000 (polyethylene glycol), 300 µL of 100 mM NAD (nicotinamide adenine dinucleotide) and add H2O to obtain 6 mL of 5× isothermal reaction buffer.
NOTE: Aliquoted buffer can be stored at -20 °C for at least 12 months. - For the Gibson assembly master mix, combine 320 µL of 5x isothermal reaction buffer with 697 µL of H2O and add 3 µL of 10 U/µL T5 exonuclease, 20 µL of 2 U/µL DNA polymerase and 160 µL of 40 U/µL Taq DNA ligase.
NOTE: This mix can be aliquoted and stored at -20 °C for at least 12 months.
- Combine 3 mL of 1 M Tris (Tris(hydroxymethyl)aminomethane)-HCl at pH 7.5, 300 µL of 1 M MgCl, 60 µL of 100 mM dGTP (deoxyguanosine triphosphate), 60 µL of 100 mM dATP (deoxyadenosine triphosphate), 60 µL of 100 mM dTTP (deoxythymidine triphosphate), 60 µL of 100 mM dCTP (deoxycytidine triphosphate), 300 µL of 1 M DTT (dithiothreitol), 1.5 g of PEG-8000 (polyethylene glycol), 300 µL of 100 mM NAD (nicotinamide adenine dinucleotide) and add H2O to obtain 6 mL of 5× isothermal reaction buffer.
- Use 7.5 µL of assembly master mix with 2.5 µL of insert and vector. For a 2x STAgR reaction use a molar vector to insert ratio of 1:1 but not more than 0.2 pmol of DNA in total. For more than 2 gRNA expression cassettes, use a vector to insert ratio of 1:3 but do not exceed a total DNA amount of 0.5 pmol.
NOTE: All amplified gRNA expression cassettes should be used in equimolar amounts. Include a plasmid control with the identical amount of vector but no inserts to control for undigested template vector (and/or self-ligation). - Incubate samples at 50 °C for 45 to 60 min. Store samples on ice or at -20 °C for subsequent transformation.
NOTE: The protocol can be paused here. - Directly transform half of the mix into chemically competent bacteria.
- Thaw chemically competent TOP10 E. coli bacteria on ice.
- Add 5 µL of the Gibson mix to 50 µL of competent bacteria. Gently mix by flicking the bottom of the tube. Incubate on ice for 30 min.
- Perform a heat shock by placing the tube in a 42 °C water bath or a heat block for 45 s.
- Put the tubes back on ice. Add 250 µL of SOC medium to the bacteria and let them recover at 37 °C in a shaking incubator for 30–45 min.
- After recovery, plate the transformed bacteria onto 1.5% lysogeny broth (LB) agar plates containing ampicillin (100 µg/ mL) and incubate overnight at 37 °C.
5. Selecting STAgR Clones by Bacterial Colony PCR
- Prepare at least two sets of 200 µL PCR reaction tubes (between 3 and 24) for each construct, which are labeled identically.
- Fill one set with 100 µL LB medium containing 100 µg/ mL ampicillin.
- Use a sterile pipette tip to scratch off the biological material of one bacterial colony and spread it at the bottom of the empty 100 µL PCR reaction tube. Immediately transfer the tip to the second corresponding reaction tube containing the LB medium.
- Swirl the tip around gently to ensure some of the bacteria are transferred to the LB media and incubate at 37 °C for later use.
- Set up a PCR master mix (10 µL per reaction).
- For 10 reactions, combine 10 µL Taq buffer, 2 µL 10 mM dNTPs, 0.5 µL of primer (100 µM), 0.5 µL of Taq polymerase and add H2O to a volume of 100 µL.
- Use the following primers: StAgR_seq_fwd2: ACTGGATCCGGTACCAAGG, StAgR_seq_rev: TTACGGTTCCTGGCCTTTTG.
- Add 10 µL of the PCR master mix to the labeled PCR reaction tubes without the LB media.
- Incubate reactions on a thermocycler using the following conditions: 1 cycle of 94 °C for 5 min, 38 cycles of 94 °C for 30 s, 55 °C for 30 s, 72 °C for 2 min; 1 cycle of 72 °C for 10 min.
NOTE: Elongation time (72 °C step) should be increased with the number of gRNA expression cassettes. Calculate theoretical size of inserts using Table 2 and use 1 min per 1 kb at 72 °C. - Add loading dye and analyze the amplified fragments on a 1% agarose gel. Load an appropriate DNA ladder for sizing (1kb DNA Ladder) and run the in appropriate gel running buffer (e.g., TLE Buffer) at 120 V for 30 min.
- Calculate the theoretical size of the amplicon with the help of Table 2 by adding up the individual sizes of used promoters, gRNA scaffolds and number of N20 sequences. From the results of the colony PCR, identify the bacterial clones based on the correct band size and whether they are likely to harbor correct vectors.
- Inoculate a 2.5 mL overnight LB culture (with 100 µg/mL ampicillin) with the corresponding 100 µL culture, set up earlier. Incubate at 37 °C for 12 h.
- Extract plasmid DNA using a commercial plasmid mini kit and manufacturer's instructions31.
- Sequence the plasmids using the following primers: StAgR_seq_fwd1 (GAGTTAGGGGCGGGACTATG), StAgR_seq_fwd2 (ACTGGATCCGGTACCAAGG) and StAgR_seq_rev (TTACGGTTCCTGGCCTTTTG) by Sanger Sequencing.
Representative Results
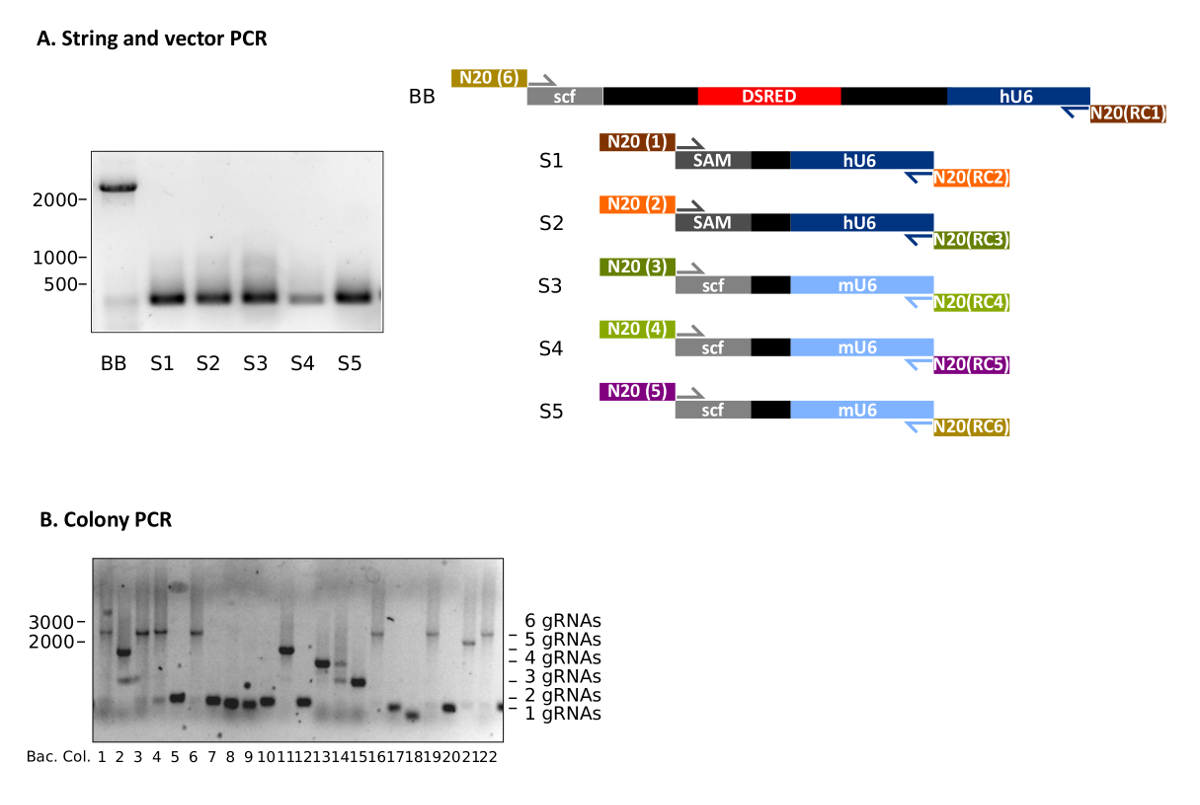
Following the protocol at hand makes the generation of customized multiplexing gRNA vectors feasible (Figure 1). Representative results of STAgR approaches using six different gRNA cassettes are depicted in Figure 2. Figure 2A shows a typical outcome of the PCRs used to obtain the STAgR pieces (protocol 3.1-3.3). The six amplicons (BB, S1-S5) represent linear DNA pieces containing the individual gRNA N20 sequences on their ends. The plasmid backbone (BB) is now extended with the targeting sequences of gRNA1 and gRNA6 and therefore possesses the required overlaps to two other PCRs (S1 and S5) for Gibson assembly (Figure 2A, Table 3). After purification a DNA yield of at least 1 µg for vectors and inserts can be achieved.
After Gibson assembly, bacterial transformation should result in 100 to 700 bacterial colonies. Figure 2B shows a representative analysis of 10 bacterial colonies via colony PCR, following a STAgR protocol with six gRNA cassettes. Gel electrophoresis indicates that three clones show the expected amplicon size for full assembly (Table 2). Other clones likely received STAgR vectors containing one to five gRNA cassettes, whereas one clone (Figure 2B, No.18) is empty.

Figure 1: Overview of STAgR primer design. First gRNA number and scaffolds are chosen, then gRNA order and promoter and last, which type of vector should be used. For each gRNA, a specific forward primer with the gRNA targeting sequence in sense orientation and either the common part of "scf-FP" or "sam-FP" is designed. Similarly, to generate the individual reverse primer the corresponding promoter sequence (RP) is fused with the reverse complement of the N20 sequence of the next gRNA. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Representative results of STAgR approaches using six different gRNA cassettes. (A) Representative example of a PCR of five inserts as well as the vector STAgR_Tomato. (B) Colony PCR of a 6x STAgR reaction using hU6 and mU6 promoters and canonical gRNA as well as SAM scaffolds. 22 bacterial colonies were analyzed, of which 7 showed the expected band indicating complete assembly. Ladder: 1 kb plus, numbers in base pairs (bp). Please click here to view a larger version of this figure.
{kind=link}
| Forward primer for String and Vector | |
| scaffold_fwd | GTTTTAGAGCTAGAAATAGCAAGTT |
| SAM_fwd | GTTTTAGAGCTAGGCCAACATGAGG |
| Reverse primer for String and Vector | |
| hU6_rev | CGGTGTTTCGTCCTTT |
| mU6_rev | CAAACAAGGCTTTTCTCCAAGG |
| hH1_rev | CTGGGAAAGAGTGGTCTCATACAGA |
| h7SK_rev | CCGAGGTACCCAAGCGG |
Table 1: Primer sequences for gRNA scaffolds and promoters.
| Sizes of promoters and scaffolds. | |
| hU6 | 265 bp |
| hH1 | 225 bp |
| mU6 | 316 bp |
| h7SK | 245 bp |
| gRNA scaffold | 83 bp |
| SAMloop + gRNA scaffold | 143 bp |
Table 2: Theoretical sizes of each expression cassette to calculate colony PCR outcome.
| hU6 | hH1 | mU6 | h7SK | STAgR_Neo | STAgR_tdTomato | |
| gRNA Scf | 400 bp | 360 bp | 451 bp | 380 bp | 3722 bp | 4487 bp |
| SAM | 460 bp | 420 bp | 511 bp | 440 bp | - | - |
Table 3: Sizes of promoters and scaffolds.
| hU6 | hH1 | mU6 | h7SK | STAgR_Neo | STAgR_tdTomato | |
| gRNA Scf | 368 bp | 328 bp | 419 bp | 348 bp | +74 bp | +67 bp |
| SAM | 428 bp | 388 bp | 479 bp | 408 bp |
Table 4: Calculated sizes of STAgR pieces after PCR.
Discussion
Here we present a detailed protocol for STAgR allowing the fast and simple generation of gRNA expression plasmids of varying sizes. This protocol can be used for one-step generation of gRNA plasmids with 2–8 gRNA expression cassettes (with or without two MS2 RNA aptamers) into the gRNA expression vector STAgR_Neo, and the use of human U6, mouse U6, human 7sK and human H1 promoters28,32,33. If more than four cassettes are desired, it is recommended to use at least two different promoter/scaffold types to increase efficiency. Other vectors, promoters and promoter combinations as well as more than 8 gRNA cassettes might be feasible as well; however, this has not been tested. Although in our experience the method works reliably and reproducibly, we have determined critical steps and troubleshooting strategies should the expected outcome not immediately occur. In our experience some steps are pivotal for the success of the approach: first and foremost, it is important to use the correct molar ratios of inserts to the vector backbone at the Gibson assembly step (3:1). However, we noticed that for some gRNA sets a 1:1 ratio of inserts to vector is beneficial, even when the gRNA cassette number does exceed two. Secondly, the period of the Gibson assembly reaction should be sufficiently long (at least 45 min recommended).
STAgR can be employed with a large number of modifications. Targeting sequences, gRNA numbers and scaffolds, promoters and vector backbones can be customized and freely combined. However, each combination may have slightly different requirements. In our experience, if medium or high numbers of gRNA cassettes (>4) are desired but not obtained immediately, usually the fastest way to overcome this issue is to change the order of the individual gRNA cassettes in the vector. Similarly, combining different gRNA promoters and scaffolds improves efficiency (and thus lowers the required number of colony PCRs that have to be scored) when many gRNA cassettes are cloned. We found that incorrect clones are usually generated by the spontaneous conjunction of repeated sequences during Gibson assembly. Although this can reduce the efficiency of the method, this also results in the simultaneous generation of vectors with functional gRNA subsets28.
Generation of multiplexing gRNA vectors containing more than one gRNA expression cassette has been reported with a number of different methods21,22,23,24,25,26,27. While some employ simultaneous or sequential cloning of gRNA pairs22,34, others depend on Golden Gate assembly and several sequential cloning steps25,35. A special approach has been used by Xie et al., by employing the endogenous cellular tRNA processing system to cut several gRNAs out of a single progenitor transcript36. The advangetage of this method is the requirement of only one promoter; the drawback is the need of newly synthesized DNA fragments for each gRNA combination. Each gRNA multiplexing method has advantages and disadvantages; STAgR combines simplicity with efficiency, high number of possible gRNA cassettes with low cost.
STAgR (and other multiplexing systems) will likely be instrumental in enabling efficient (and simultaneous) disruption of multiple genes in vivo, as pioneered in zebrafish brains28. Another future application of gRNA multiplexing vectors is the promise for manipulation of complete transcriptional programs in contrast to the induction or repression of single genes. Likely, these approaches will allow transforming cells and organs at will to a much higher degree than it is possible today.
Acknowledgements
The authors would like to thank Prof. Dr. Stephan Beck and Prof. Dr. Jovica Ninkovic for their input, help and support in developing the STAgR method. Tobias Klötzer, Anna Köferle and Valentin Baumann for helpful comments. The work has been supported by DFG (STR 1385/1-1).
Materials
| Name | Company | Catalog Number | Comments |
| Phusion High- Fidelity DNA Polymerase | NEB | M0530L | Polymerase for Gibson Master Mix as well as all PCRs |
| dNTPs | Life Technology | R0191 | dNTPs for all PCRs |
| dATP | Life Technology | R0141 | dNTPs for Gibson Master Mix |
| dCTP | Life Technology | R0151 | dNTPs for Gibson Master Mix |
| dGTP | Life Technology | R0161 | dNTPs for Gibson Master Mix |
| dTTP | Life Technology | R0171 | dNTPs for Gibson Master Mix |
| Agarose | VWR | 443666A | |
| DpnI | NEB | R0176S | |
| AMPure XP Beads | Beckman Coulter | A63881 | for DNA clean up |
| Tris HCL | Roth | 9090.2 | for Gibson Master Mix |
| DTT | Life Technology | R0861 | for Gibson Master Mix |
| PEG 8000 | VWR | RC- 077 | for Gibson Master Mix |
| NAD | Cayman Chemicals Company | 16077 | for Gibson Master Mix |
| T5 Exonuclease | NEB | M0363S | for Gibson Master Mix |
| Taq DNA Ligase | NEB | M0208S | for Gibson Master Mix |
| Ampicilin | SigmaAldrich | PHR1393-1G | |
| DNA Ladder 1kb Plus | Thermo Scientific | SM0311 | |
| Plasmid Mini Kit | Thermo Scientific | K0503 | |
| Plasmid and Primer Sequences | All sequences available at Figshare (https://goo.gl/UGdc3Q). | ||
| EDTA | PanReacAppliChem | 6381-92-6 | for TLE Buffer |
| MgCl2 | SigmaAldrich | M8266 | for Gibson Master Mix |
| Top10 chemically competent bacteria | Thermo Scientific | C404003 | |
| S.O.C. Medium | Thermo Scientific | 15544034 | |
| Yeast- Extract | SigmaAldrich | Y1625 | for lysogenic broth (LB Media) |
| NaCl | SigmaAldrich | S7653 | for lysogenic broth (LB Media) |
| Peptone | SigmaAldrich | 91249 | for lysogenic broth (LB Media) |
| Agar-Agar | SigmaAldrich | 5040 | for lysogenic broth (LB Media) |
| Taq DNA Polymerase | Quiagen | 201203 |
References
- Barrangou, R. Diversity of CRISPR-Cas immune systems and molecular machines. Genome Biol. 16, 247 (2015).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Pulecio, J., Verma, N., Mejia-Ramirez, E., Huangfu, D., Raya, A. CRISPR/Cas9-Based Engineering of the Epigenome. Cell Stem Cell. 21 (4), 431-447 (2017).
- Köferle, A., Stricker, S. H., Beck, S. Brave new epigenomes: the dawn of epigenetic engineering. Genome Med. 7 (1), 59 (2015).
- Stricker, S. H., Koferle, A., Beck, S. From profiles to function in epigenomics. Nature Reviews Genetics. 18 (1), 51-66 (2017).
- Gilbert, L. A., et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 154 (2), 442-451 (2013).
- Maeder, M. L., et al. CRISPR RNA-guided activation of endogenous human genes. Nature Methods. 10 (10), 977-979 (2013).
- Lekomtsev, S., Aligianni, S., Lapao, A., Burckstummer, T. Efficient generation and reversion of chromosomal translocations using CRISPR/Cas technology. BMC Genomics. 17 (1), 739 (2016).
- Jiang, J., et al. Induction of site-specific chromosomal translocations in embryonic stem cells by CRISPR/Cas9. Scientific Reports. 6, 21918 (2016).
- Yang, H., et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 154 (6), 1370-1379 (2013).
- Wang, L., et al. Large genomic fragment deletion and functional gene cassette knock-in via Cas9 protein mediated genome editing in one-cell rodent embryos. Scientific Reports. 5, 17517 (2015).
- Zhang, L., et al. Large genomic fragment deletions and insertions in mouse using CRISPR/Cas9. PLoS ONE. 10 (3), 0120396 (2015).
- Shen, B., et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nature Methods. 11 (4), 399-402 (2014).
- Yang, H., Wang, H., Jaenisch, R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nature Protocols. 9 (8), 1956-1968 (2014).
- Wang, H., et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 153 (4), 910-918 (2013).
- Kalhor, R., Mali, P., Church, G. M. Rapidly evolving homing CRISPR barcodes. Nature Methods. 14 (2), 195-200 (2017).
- Chavez, A., et al. Highly efficient Cas9-mediated transcriptional programming. Nature Methods. 12 (4), 326-328 (2015).
- Bultmann, S., Stricker, S. H. Entering the post-epigenomic age: back to epigenetics. Open Biology. 8 (3), (2018).
- Vora, S., Tuttle, M., Cheng, J., Church, G. Next stop for the CRISPR revolution: RNA guided epigenetic regulators. FEBS Journal. , (2016).
- Maddalo, D., et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 516 (7531), 423-427 (2014).
- Nageshwaran, S., et al. CRISPR Guide RNA Cloning for Mammalian Systems. Journal of Visualized Experiments. , (2018).
- Wrighton, K. H., et al. Synthetic biology: Multiplex genome engineering in eukaryotes. Nature Reviews Genetics. 19 (1), 6-7 (2018).
- Yan, Q., et al. Multiplex CRISPR/Cas9-based genome engineering enhanced by Drosha-mediated sgRNA-shRNA structure. Scientific Reports. 6, 38970 (2016).
- Kabadi, A. M., Ousterout, D. G., Hilton, I. B., Gersbach, C. A. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Research. 42 (19), 147 (2014).
- Cress, B. F., et al. CRISPathBrick: Modular Combinatorial Assembly of Type II-A CRISPR Arrays for dCas9-Mediated Multiplex Transcriptional Repression in E. coli. ACS Synthetic Biology. 4 (9), 987-1000 (2015).
- Port, F., Bullock, S. L. Augmenting CRISPR applications in Drosophila with tRNA-flanked sgRNAs. Nature Methods. 13 (10), 852-854 (2016).
- Breunig, C. T., et al. One step generation of customizable gRNA vectors for multiplex CRISPR approaches through string assembly gRNA cloning (STAgR). PLoS ONE. 13 (4), 0196015 (2018).
- Barbas, C. F., Burton, D. R., Scott, J. K., Silverman, G. J. Quantitation of DNA and RNA. CSH Protoc. 2007, (2007).
- Gibson, D. G. Enzymatic assembly of overlapping DNA fragments. Methods Enzymology. 498, 349-361 (2011).
- Maniatis, T., Fritsch, E. F., Sambrook, J. . Molecular cloning : a laboratory manual. , (1982).
- Koferle, A., et al. CORALINA: a universal method for the generation of gRNA libraries for CRISPR-based screening. BMC Genomics. 17 (1), 917 (2016).
- Koferle, A., Stricker, S. H. A Universal Protocol for Large-scale gRNA Library Production from any DNA Source. Journal of Visualized Experiments. (130), e56264 (2017).
- Vidigal, J. A., Ventura, A. Rapid and efficient one-step generation of paired gRNA CRISPR-Cas9 libraries. Nature Communications. 6, 8083 (2015).
- Sakuma, T., Nishikawa, A., Kume, S., Chayama, K., Yamamoto, T. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Scientific Reports. 4, 5400 (2014).
- Xie, K., Minkenberg, B., Yang, Y. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proceedings of the National Academy of Sciences of the United States of America. 112 (11), 3570-3575 (2015).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved